
Abstract
Early during mammalian embryogenesis, epiblast cells undertake major cell fate decisions, becoming specified towards either the perishable soma or the immortal germline. Despite the importance of these developmental transitions, the transcriptional regulatory mechanisms orchestrating them have remained poorly characterized due to the transient nature and scarcity of the involved cell populations. However, our view of these processes is dramatically changing due to advances in mouse and human embryonic stem cell (ESC) differentiation models that faithfully recapitulate peri-implantation transitions. Recent studies using these models have uncovered enhancers as critical cis-regulators during the maintenance, extinction, or re-establishment of pluripotency. Here, we review the major transcriptional and epigenetic regulators controlling the remodeling of enhancer landscapes during mammalian peri-implantation development. Last but not least, we discuss how a global and mechanistic understanding of enhancer remodeling can provide important insights into somatic reprogramming, the molecular basis of aging, and the implementation of cellular rejuvenation strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Following implantation of the mouse blastocyst, highly relevant and dynamic cellular transitions take place during a developmental time window that we will refer to as peri-implantation development (E4.5–E7.5) (Fig. 1). The pre-implantation epiblast (∼E4.5) displays naïve or ground-state pluripotency, as it can give rise to all embryonic lineages, including the germline [1]. Immediately after implantation, the epiblast progresses to a transient state that has been recently referred to as formative pluripotency (∼E5.5–6.5) and which is characterized by the dismantling of the naïve pluripotency expression program [2]. Subsequently and coinciding with the beginning of gastrulation, the epiblast cells acquire a primed pluripotent state (∼E6.5–7.5) and start expressing somatic germ layer specifiers [2]. Remarkably, while both formative and primed pluripotent cells are able to differentiate into all somatic lineages, only formative cells are competent for primordial germ cell (PGC) induction and specification (∼E6–6.5) [3]. Moreover, PGC specification involves and functionally requires the re-activation of naïve pluripotency genes, including several transcription factors (TFs) with master regulatory functions [4, 5•, 6]. Therefore, a pluripotency cycle maintained by a shared transcriptional regulatory network has been hypothesized to exist between the naïve epiblast and the germline [1].

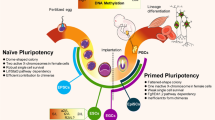
Remodeling of enhancers associated with naïve pluripotency and early germline genes. During mouse peri-implantation transitions, enhancers controlling the expression of naïve pluripotency and early germline genes are active in naïve pluripotent cells (i.e., E4.5 epiblast/2i ESC), as reflected by the presence of H3K4me1/2 and H3K27ac and their overall hypomethylation (empty lollipops). These enhancers are bound by pluripotency TFs (e.g., OCT4, SOX2, NANOG) (activators: green ovals) that recruit co-activators and sustain their active state. Moreover, these enhancers are also bound by transcriptional repressors (e.g., FOXD3, TCF3) (red ovals) that recruit co-repressors and promote the decommissioning of these regulatory sequences. As a result, epiblast cells exit naïve pluripotency and transiently acquire a formative pluripotent state (i.e., E5.5–6.5/EpiLC), in which naïve pluripotency/germline enhancers become decommissioned as they loose H3K27ac and H3K4me2, while retaining H3K4me1 and remaining hypomethylated. Subsequently, these enhancers will undergo two alternative fates: (i) in the germline, these enhancers become re-activated through the binding of PGC and naïve pluripotency master regulators (green ovals), which reinstate the typical chromatin signature of active enhancers (i.e., H3K4me1/2, H3K27ac); (ii) in the primed pluripotent epiblast (E7.5/EpiSC) and in the soma, these enhancers get fully silenced, as they gain H3K9me2/3 and become hypermethylated (filled lollipops). The repressive chromatin environment established around naïve pluripotency/germline enhancers in the soma ensures their stable silencing and represents a major roadblock for somatic reprogramming
As mammalian embryogenesis proceeds, the acquisition of novel cellular identities requires not only the establishment of new gene expression programs but also the extinction of previously existing ones. It is now widely accepted that enhancers play a preponderant role during these developmental events by integrating the regulatory activities of TFs, epigenetic regulators, and signaling pathways [7]. Consequently, developmental transitions are accompanied by dramatic changes in enhancer repertoires, involving not only the activation but also the silencing of a large number of enhancers [8]. The paucity and transient nature of the cell populations involved in peri-implantation development have precluded the molecular characterization of the transcriptional and epigenetic changes that accompany these cellular transitions. Recent advances in ESC culture conditions, together with newly developed differentiation protocol, allow a faithful recapitulation of early embryogenesis, both in mouse and in humans. Chiefly, these in vitro models are genetically tractable and can provide relevant peri-implantation cellular states in numbers compatible with genomic and biochemical approaches. In the next sections, we briefly describe these in vitro models and how they have been used to characterize enhancer dynamics during peri-implantation transitions, giving special emphasis to a core set of enhancers controlling the expression of naïve pluripotency and early germline genes (“pluripotency/germline enhancers”) (Fig. 1).
Enhancer Dynamics During the Transition from Naïve to Primed Pluripotency
Mouse ESC (mESC) have been traditionally grown under “LIF + serum” conditions, which results in heterogeneity and metastability as these cells reversibly switch between naïve and primed pluripotent states [9]. Importantly, these two states can now be independently maintained in culture, as represented by mESC grown under “2i + LIF” (2i ESC, 2i = MEK and GSK3 inhibitors) and epiblast stem cell (EpiSC), respectively [10, 11]. Moreover, the recently described epiblast-like cells (EpiLC) resemble the pre-gastrulating epiblast and display formative pluripotency, a transitional state between naïve and primed pluripotency [2, 6].
The transcriptional regulatory network that maintains the naïve pluripotent state has been extensively characterized. This network consists of a set of “core” (OCT4/POU5F11, SOX2) and “ancillary” (NANOG, ESRRB, KLF4, KLF2, TBX3, PRDM14) TFs that, together with the final effectors of key signaling pathways (LIF, WNT, BMP), sustain naïve pluripotency through cooperative binding and consequent activation of a broad set of enhancers [9, 12, 13]. This involves the recruitment of co-activators (e.g., p300, BRG1) and the establishment of a chromatin signature characteristic of active enhancers (nucleosomal depletion, H3K4me1/2, H3K27ac) [14, 15]. Moreover, at least one of these TFs, PRDM14, ensures naïve pluripotency by repressing, in a polycomb dependent manner, key signaling pathways (i.e., FGF), and epigenetic regulators (i.e., Dnmt3A/B) that promote the transition to primed pluripotency [16].
The establishment of EpiSC and the differentiation of 2i mESC into EpiLC has enabled the characterization of the transition from naïve into formative and then primed pluripotency. During these transitions, core pluripotency TFs (OCT4 and SOX2) remain highly expressed and partner with a different set of TFs, such as OTX2, leading to the cooperative activation of many novel enhancers and their target genes [17•, 18••]. Although some of these enhancers display a primed or poised state in 2i ESC, most of them lack any pre-marking (e.g., H3K4me1, DNAseI hypersensitivity) and are activated de novo in EpiLC [18••]. Overall, the gene expression programs of naïve, formative, and primed pluripotent cells are markedly different, yet they express some genes in common. Remarkably, genes active in both mESC and EpiSC are apparently regulated by different sets of enhancers in each cell type [19•], including a set of EpiSC-specific enhancers referred to as “seed” enhancers that are frequently used at subsequent developmental stages in somatic tissues [10]. Interestingly, it has been recently reported that differentiated somatic cells with the capacity to self-renew (e.g., macrophages) might be able to do so by activating an ESC-like self-renewal gene expression program [20••]. Once again, ESC and differentiated cells utilize distinct sets of enhancers to control such self-renewal program. It would be interesting to determine if “seed” enhancers are preferentially used by self-renewing somatic cells.
As stated above, developmental transitions require not only the induction of new gene expression programs but also the dismantling of previously existing ones. FOXD3 and TCF3/TCF7L1 are transcriptional repressors that promote the exit from naïve pluripotency by mediating the silencing of distinct sets of ground-state regulators (e.g., TCF3 targets: Tfcp2l1, Klf2, Esrrb, Nanog; FOXD3 targets: Prdm14, Tbx3, Pramel6/7) [21, 22••]. This suggests that these two repressors might be independently required to exit naïve pluripotency by dismantling discrete parts of the naïve expression program. FOXD3 executes its repressive function by mediating the decommissioning of a subset of naïve pluripotency enhancers [22••]. Mechanistically, this involves the displacement of pluripotency TFs and co-activators as well as the recruitment of co-repressors (e.g., LSD1, HDAC1/2, NuRD) [22••, 23]. It is currently unknown if TCF3 repressive function involves a similar enhancer decommissioning mechanism, but data from other cellular contexts suggest that this could be certainly the case and could then involve the recruitment of additional co-repressors (e.g., TLE/Groucho) [24]. The repressive activity of these two and most likely other, yet undiscovered, TFs leads to the decommissioning of a large set of enhancers during the transition from naïve (i.e., 2i ESC) to formative pluripotency (i.e., EpiLC,), which involves a progressive loss of H3K27ac and H3K4me2 and retention of H3K4me1 [18••, 22••] (Fig. 1). Interestingly, during this transition, these enhancers start gaining H3K9me2 as part of their silencing mechanism, although they remain hypomethylated despite the raising levels of de novo DNA methyltransferases (i.e., DNMT3A/B) [25•, 26, 27]. As primed pluripotency emerges (i.e., EpiSC), naïve pluripotency enhancers become fully decommissioned, completely loosing not only H3K27ac but also H3K4me1, while gaining even higher levels of H3K9me2 [19•, 25•]. Although not formally shown, since DNMTs display negative and positive crosstalk with H3K4me1/2 and H3K9me2/3, respectively [28, 29], it is likely that these enhancers will become accessible for DNMT3A/B and hypermethylated already in EpiSC. Upon somatic differentiation, the repressive chromatin environment around naïve pluripotency enhancers becomes consolidated, stable, and largely inaccessible [30] (Fig. 1).
The characterization of how enhancer landscapes are remodeled during the transition from naïve to primed pluripotency has been by and large performed using murine in vitro models. Traditional culture conditions for human ESC (hESC) results in a cellular state that, in many aspects, resembles mouse EpiSC and are thus considered to display primed pluripotency [10]. However, after extensive effort, the long-sought conditions to induce naïve pluripotency in hESC were recently reported [31•, 32••]. Perhaps not surprisingly, these two hESC states manifest markedly different transcriptomes and epigenomes as well as enhancer repertoires [32••, 33•]. It remains to be elucidated if mouse and humans use the same or different TFs to control the major enhancer landscape remodeling that characterizes the transition from naïve to primed pluripotency.
Common Enhancers Participate in the Maintenance of Naïve Pluripotency and in Germline Specification
Soon after implantation, epiblast cells become primed towards somatic differentiation and start to express somatic lineage specifiers. However, in response to high levels of BMP4 and WNT3 emanating from the extraembryonic tissues adjacent to the proximo-posterior epiblast, a few epiblast cells revert somatic differentiation and initiate PGC specification [34]. This process is tightly regulated by a group of master regulators (BLIMP1/PRDM1, PRDM14, TFAP2C) that become sequentially activated in PGC precursors (∼E6.25–6.75) [3, 35••, 36]. Importantly, these TFs are all expressed in naïve pluripotent cells, in which, at least PRDM14, is also functionally required to maintain the ground pluripotent state [16]. Likewise, NANOG, a key naïve pluripotency regulator, plays a major role during PGC specification [4, 5•]. The coordinated action of these TFs results in marked transcriptional changes that eventually lead to the establishment of a gene expression program remarkably similar to that of the naïve epiblast [1]. These transcriptional changes include (i) the repression of the incipient somatic program and of genes required for DNA methylation, (ii) the re-activation of the naïve pluripotency network, including highly relevant TFs (e.g., Nanog, Oct4, Sox2) (iii) the silencing of genes that are upregulated during the transition from naïve to formative pluripotency and that characterize the formative pluripotent state (e.g., Otx2, Pou3f1, Foxd3, Tcf3), and (iv) the induction of early germline genes (e.g., Rhox5/6/9, Nanos3) [6, 22••, 34]. As a result, a cluster of approximately 40 PGCs is formed (∼E7.25), which then undergo major epigenetic reprogramming (i.e., global DNA demethylation, imprinting erasure, changes in histone modification profiles) as they proliferate and migrate towards the genital ridges [34].
Historically, uncovering the molecular basis of the transcriptional and epigenetic changes underlying PGC specification has been a daunting task due to the scarcity of these cells. However, the Saitou lab established an in vitro protocol whereby primordial germ cell-like cells (PGCLC) are induced from EpiLC, faithfully recapitulating murine PGC specification [6]. This novel protocol has dramatically improved our understanding of this process, allowing, among other things, to uncover the set of enhancers involved in germline specification [37••]. Analogously to the extensive changes in enhancer usage observed during the transition from naïve to primed pluripotency, PGC specification is also accompanied by a considerable remodeling of enhancer landscapes [37••]. These enhancer changes can be broadly divided into two major categories: (i) enhancers that become rapidly and transiently activated (day 2 PGCLC) and that most likely control the induction of mesodermal genes (e.g., T) directly involved in the earliest steps of PGC specification [38] and (ii) enhancers activated later during PGC specification (day 6 PGCLC) and that presumably promote the re-activation of naïve pluripotency and early germline genes. Notably, this second group of regulatory elements includes multiple enhancers that are originally active in naïve ESC and that become decommissioned in EpiLC [22••, 37••]. This supports the existence of a pluripotency/germline cycle in which a core set of pluripotency/germline enhancers might control the expression of naïve pluripotency and early germline genes in the pre-implantation epiblast and PGCs [1] (Fig. 1).
Intriguingly, upon exit from naïve pluripotency, the initial decommissioning of pluripotency/germline enhancers does not entail a full silencing of these key regulatory elements. Instead, in EpiLC/E6.5 epiblast, these regulatory sequences acquire a transient poised state in which they retain H3K4me1, remain hypomethylated, and are most likely bound by TFs (e.g., FOXD3, OCT4) [18••, 22••, 26, 27] (Fig. 1). The presence of H3K4me1 and TFs is known to be refractory for the enzymatic activity of DNMT3A/B, which could explain why pluripotency/germline enhancers remain hypomethylated in the face of the extensive de novo DNA methylation that occurs in EpiLC and the post-implantation epiblast [28, 39, 40]. We have previously hypothesized that such poised state, imposed at least partially by FOXD3, could then facilitate the re-activation of pluripotency/germline enhancers and their associated genes upon binding of PGC specifiers (e.g., PRDM14, TFAP2C) [22••, 35••, 36]. This could explain why naïve pluripotency and early germline genes are re-activated before the major wave of DNA demethylation that occurs at later stages of PGC specification and that might be critically required to induce genes involved in meiosis and late germline development [41, 42]. As stated above, the poised state of pluripotency/germline enhancers in EpiLC is transient, since these regulatory sequences become progressively silenced and heterochromatinized upon transition to primed pluripotency/EpiSC and subsequent somatic differentiation [19•, 25•, 30]. Like the poised state of pluripotency/germline enhancers, germline competence is also transient and short-lived as it is a unique property of the pre-gastrulating post-implantation epiblast in vivo and of EpiLC in vitro [3, 6]. Hence, it is tempting to speculate that germline competence might be at least partially dependent on the poised hypomethylated state of pluripotency/germline enhancers.
Similarly to the transition from naïve to primed pluripotency, much of what we know about enhancer dynamics during PGC specification has been described using murine in vitro models. However, the Surani and Saitou labs have recently reported two similar protocols whereby hESC can be efficiently differentiated into PGCLC [43•, 44••]. These in vitro models have been already used to reveal the major similarities (e.g., BLIMP1, PRDM14, TFAP2C, NANOG) but also the notable differences (e.g., SOX17) among the TFs involved in human PGC specification with respect to the mouse [43•, 44••]. We anticipate that these in vitro models will be soon used to uncover how enhancer repertoires change during specification of the human germline.
Enhancer Remodeling During Somatic Reprogramming
Ectopic expression of a limited set of transcription factors (OCT4, SOX2, KLF4, C-MYC (OSKM)) is sufficient to reprogram somatic cells towards pluripotency [45, 46]. Both mouse and human induced pluripotent stem cells (iPSC) display remarkable transcriptional, epigenetic, and functional similarities to ESCs, which, not surprisingly, includes highly similar enhancer repertoires [47, 48]. In addition to its obvious and far-reaching clinical applications, the generation of iPSC offers a tractable system to investigate the transcriptional and epigenetic mechanisms controlling developmental transitions [49, 50]. Unfortunately, somatic reprogramming is typically inefficient and great efforts have been devoted to uncovering the major facilitators and roadblocks of this process [30, 51•, 52–55, 56••, 57]. Somatic cells are frequently arrested in a partially reprogrammed state (i.e., pre-iPSC), in which the re-activation of core pluripotency regulators is a major rate-limiting step towards the acquisition of full pluripotency [30, 49, 51•, 52, 54]. Remarkably, the set of genes that are especially refractory to re-activation during reprogramming includes not only pluripotency regulators but also early germline genes, overall coinciding with genes that dynamically change their expression during peri-implantation transitions, as described in previous sections [30, 52, 54]. We will now review evidence suggesting that, similarly to peri-implantation transitions, the re-activation of these pluripotency/germline genes during reprogramming depends on the epigenetic status and accessibility of their associated enhancers (Fig. 1).
Seminal work from the Zaret lab demonstrated that exogenous OSKM TFs were able to bind to a large set of distal sequences already at the initial steps of somatic reprogramming [30]. Importantly, these sequences could be broadly divided into two groups: (i) sites with accessible chromatin in somatic cells, as represented by DNAseI hypersensitivity and presence of H3K4me1/2 [57] and (ii) sites with closed chromatin in which OSK acted as pioneer TFs and facilitated C-MYC binding. Importantly, the second group included important enhancers that in mESC are active and occupied by endogenous pluripotency TFs. The binding of OSKM TFs to these enhancers leads to their subsequent activation, which, interestingly, precedes the induction of their target genes [30, 58]. These results suggest that, despite the major epigenetic barriers that are believed to exist between different cellular states and despite the refractory effect of silent chromatin on TF binding, ectopic OSKM TFs can readily access a remarkable fraction of their endogenous binding sites in ESCs. Importantly though, the reprogramming TFs initially fail to bind a core set of enhancers associated with relevant pluripotency/germline genes, which remain silent and inaccessible, thus impeding the re-activation of their critical target genes [30]. Overall, these observations illustrate the importance of pluripotency/germline enhancers for the acquisition of pluripotency and also raise major questions regarding the inaccessibility of these sequences and the chromatin features that make them so resistant to TF binding. Epigenomic analysis in MEFs and pre-iPSC indicates that these enhancers are hypermethylated and enriched in H3K9me2/3, creating a heterochromatic environment particularly impenetrable for TFs [30, 51•, 52, 58] (Fig. 1). In full agreement with this, enzymes that methylate (e.g., DNMT1, DNMT3A) or demethylate (TET1, TET2, AID) 5mC have been shown to act as inhibitors and facilitators of reprogramming, respectively [52, 53, 59, 60]. Likewise, H3K9 methyltransferases (HMTs) (e.g., SUV39H1, SUV39H2, SEDB1, EHMT2) act as reprogramming barriers, while H3K9me2/3 demethylases (e.g., JMJD1A, JMJD1B, JMJD2B, JMJD2C) increase reprogramming efficiency [30, 51•, 55]. DNA methylation has been traditionally investigated in the context of promoter regions and repetitive elements, but a major role in controlling enhancer activity, mostly by interfering with TF binding, has been recently appreciated [40, 61, 62•]. On the other hand, reprogramming experiments suggest that H3K9me3/me2 might have an equally important yet largely unappreciated role in enhancer silencing [30, 51•, 55]. The importance of DNA methylation and H3K9me2/3 as epigenetic reprogramming barriers that prevent the re-activation of pluripotency/germline enhancers is in perfect agreement with the epigenetic changes displayed by these regulatory elements during embryogenesis [17•, 18••, 19•, 22••, 25•, 26, 27, 37••, 63, 64] (Fig. 1). In addition to DNA hypermethylation and gain of H3K9me2/3, the chromatin changes observed at pluripotency/early germline enhancers during peri-implantation transitions suggest that complete loss of H3K27ac and H3K4me1/2 can also contribute to the full silencing of these regulatory elements [17•, 18••, 19•, 22••, 25•, 37••]. Accordingly, enzymes and protein complexes mediating histone deacetylation (e.g., HDAC1, MBD3, NuRD) and H3K4 demethylation (e.g., LSD1) at enhancer elements can act as somatic reprogramming barriers [56••, 65, 66].
The re-activation of pluripotency/germline enhancers and their associated genes seems to be an important and early step during PGC specification [5•, 22••, 37••]. Interestingly, enhancer re-activation seems to occur effortlessly in PGCs, which is in stark contrast to the refractory nature of this process during somatic reprogramming. As mentioned earlier, germline competence might require a silent (loss of enhancer RNAs (eRNAs) and H3K27ac) but poised chromatin state (i.e., H3K4me1, bound by TFs such as FOXD3) at pluripotency/early germline enhancers that protects them from DNA hypermethylation [22••, 26–28, 39] (Fig. 1). Like germline competence itself, this poised chromatin state is also transient, progressively transitioning to a fully silent and heterochromatic state as gastrulation and somatic differentiation begins [19•, 25•, 63, 64]. So, why do these regulatory elements demand such a resilient and impenetrable chromatin environment in the soma? From a functional standpoint, spurious re-activation of these enhancers could interfere with proper execution of somatic differentiation programs and/or the maintenance of somatic identity, potentially leading to tumorigenesis [49]. However, mechanistically, it remains unclear how this unique set of regulatory elements are specifically targeted by DNMTs and H3K9 HMTs. One possibility is that this is a rather passive process, in which the absence of positive regulatory inputs (e.g., TFs, co-activators) and transcriptional activity allows the spreading of a default repressive chromatin environment in a rather unspecific and progressive manner. In agreement with this model, most gene promoters are contained within CpG islands that protect them from DNA methylation through the establishment of antagonistic chromatin signatures (e.g., H3K4me2/3, H3K27me3) [28, 67, 68]. In contrast, enhancers typically display low CpG contents that could in principle render them susceptible to DNMTs activity. Nonetheless, this fails to explain why many of the active enhancers in ESC, once silent in somatic cells, do not gain H3K9me2/3 and/or become hypermethylated and are thus easily bound and re-activated by OSKM TFs upon reprogramming [30]. In fact, many enhancers active during embryogenesis remain hypomethylated once silenced in adult somatic tissues in a so-called vestigial state [69]. Alternatively, more direct and targeted regulatory mechanisms might be in place to ensure an efficient silencing of pluripotency/germline enhancers. For example, enhancers associated with core pluripotency genes (e.g., Sox2, Oct4) become silenced and hypermethylated in a targeted manner upon differentiation [63, 64]. In the case of Oct4, the transcriptional repressor GCNF might be involved in this process through the recruitment of DNMT3A/B [70]. Although a general targeting mechanism directing DNA methylation and H3K9me2/3 to pluripotency/germline enhancers has not been described, such a mechanism seems to exit for the promoter regions of genes involved in late germline development and gametogenesis. In this case, the transcription factor MAX and the E2F6 complex recruit H3K9 HMTs (e.g., G9a, GLP) to these promoters in order to ensure stable silencing of the associated genes in ESC and somatic tissues [71•, 72]. Future studies should aim at elucidating the mechanisms involved in the silencing of pluripotency/germline enhancers during embryogenesis, as this might illuminate novel strategies to improve the efficiency of somatic reprogramming.
Epigenetic Changes During Aging and Rejuvenation
Aging and age-associated cellular processes, such as senescence, represent major impediments for somatic reprogramming [73–75••]. Nevertheless, hiPSC have been successfully established from geriatric donors as well as from samples of progeria patients displaying accelerated aging, thus demonstrating that age-related barriers can be overcome and that cells can be rejuvenated [76–78]. As a result, there is growing interest in using somatic reprogramming as a model to molecularly and mechanistically understand age-associated barriers, as this can provide valuable insights into future rejuvenation strategies [79]. Although evidence is still limited, preliminary observations indicate that age-associated epigenetic alterations might represent major reprogramming roadblocks. A work from the Lopez-Otin laboratory has demonstrated that aging-associated NF-KB activation lead to a strong induction of DOT1L and consequently increased levels of H3K79me2 [75••]. DOT1L inhibition greatly facilitated somatic reprogramming and corrected age-associated alterations, thus representing a promising anti-aging target [75••]. Mechanistically, DOT1L increased expression lead to higher H3K79me2 levels at fibroblast-specific genes linked to epithelial-to-mesenchymal transition (EMT), presumably reinforcing their active state and preventing their silencing, which is a critical and early step during fibroblast reprogramming [53, 75••]. Based on this data, it would be interesting to evaluate if the rejuvenating effects of DOT1L inhibition are limited to mesenchymal cell types or instead, more general and effective in epithelial tissues as well. On the other hand, it has been conclusively demonstrated that aging results in hypermethylation of polycomb target genes, an epigenetic phenomenon that has also been observed in cancer [80–83]. Presumably, this hypermethylation locks already silent genes in an even more stable repressive chromatin state. A recent integrative analysis of human reprogramming has revealed an orchestrated re-activation of broad developmental regulators that recapitulate, in an inverse manner, the events occurring during normal embryogenesis [56••]. Since many of these developmental regulators are bona-fide polycomb targets, we speculate that their age-associated hypermethylation might block their re-activation and thus could represent a major reprogramming barrier. These and other examples emphasize that epigenetic alterations are a hallmark of aging, which, chiefly, can be pharmacologically corrected [75••, 84].
A systematic evaluation of how enhancer landscapes and their associated chromatin features are affected by aging and age-associated processes has not been performed yet. However, since aging leads to a loss in cellular homeostasis and considering the major role of enhancers in conferring cellular identity, it is likely that epigenetic alteration of these regulatory sequences will be involved in the aging process [84, 85]. Hence, it would be interesting to evaluate how the chromatin environment of pluripotency/germline enhancers evolves upon aging, as the re-activation of these regulatory elements represents a milestone during somatic reprogramming (Fig. 1). It is conceivable that the levels of silencing epigenetic marks (DNA methylation, H3K9me2/3) will further increase at these enhancers or even extend to other pluripotency regulatory sequences that normally remain hypomethylated in somatic tissues. On the other hand, it has been recently shown that both oncogene-induced senescence and accelerated aging (as represented by progeroid syndromes) dramatically alter the three-dimensional architecture of the human genome, preferentially affecting lamina-associated heterochromatin, also known as lamina-associated domains (LADs) [86–88]. Interestingly, the association of these heterochromatin domains with the nuclear lamina is linked to transcriptional repression and G9a-dependent deposition of H3K9me2 [89, 90]. Since, as stated above, pluripotency/germline enhancers and their associated loci gain H3K9me2/3 and become silenced upon somatic differentiation [25•, 30, 63, 64], it is likely that they will frequently reside within LADs in somatic cells. Moreover, it has been recently shown that pluripotency enhancers frequently contact each other, forming spatially organized genomic clusters that have been proposed to confer robustness to the pluripotent state [91]. We postulate that during aging, pluripotency/germline loci might not only retain heterochromatic marks but also lose their endogenous chromatin topology, which might further complicate the three-dimensional re-organization of the pluripotency network upon reprogramming. Thus, it is tempting to speculate that topological disruption of LADs during senescence and/or aging might exacerbate the re-activation of pluripotency loci during reprogramming.
Conclusion
The last few years have witnessed a dramatic change in our view of the transcriptional and epigenetic regulatory mechanisms involved in the control of early mammalian embryogenesis. The combination of ESC-based differentiation protocols together with next-generation sequencing approaches have resulted in a more global and mechanistic understanding of how pluripotent states are established, extinguished, or maintained. Recent advances on single-cell sequencing approaches suggest that the possibility of similarly characterizing these regulatory processes in vivo is within reach [89, 92, 93]. We anticipate that, as the comprehension of pluripotency keeps increasing, this will undoubtedly help improving the efficiency of iPSC generation. Last but not least, since somatic reprogramming can be seen as a cellular rejuvenation process, uncovering the major facilitators and barriers of somatic reprogramming can have far-reaching implications for the molecular understanding of aging and for the therapeutical treatment of age-related disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Leitch HG, Smith A. The mammalian germline as a pluripotency cycle. Dev Camb Engl. 2013;140:2495–501.
Kalkan T, Smith A. Mapping the route from naive pluripotency to lineage specification. Philos Trans R Soc Lond B Biol Sci. 2014;369.
Ohinata Y, Ohta H, Shigeta M, Yamanaka K, Wakayama T, Saitou M. A signaling principle for the specification of the germ cell lineage in mice. Cell. 2009;137:571–84.
Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, et al. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–4.
Murakami K, Günesdogan U, Zylicz JJ, Tang WWC, Sengupta R, Kobayashi T, et al. NANOG alone induces germ cells in primed epiblast in vitro by activation of enhancers. Nature. 2016;529:403–7. This study demonstrates the importance of pluripotency TFs and the enhancers they bind for germline induction and specification.
Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 2011;146:519–32.
Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet TIG. 2012;28:276–84.
Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–31.
Hackett JA, Surani MA. Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell. 2014;15:416–30.
Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–9.
Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–23.
Dunn S-J, Martello G, Yordanov B, Emmott S, Smith AG. Defining an essential transcription factor program for naïve pluripotency. Science. 2014;344:1156–60.
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–17.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–83.
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6.
Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, et al. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell. 2013;12:368–82.
Yang S-H, Kalkan T, Morissroe C, Marks H, Stunnenberg H, Smith A, et al. Otx2 and Oct4 drive early enhancer activation during embryonic stem cell transition from naive pluripotency. Cell Rep. 2014;7:1968–81. The importance of this study is described concurrently with reference 18.
Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, et al. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell. 2014;14:838–53. Together with Yang et al., this study provides a global view of the major enhancer landscape remodelling that occurs during the transition from naïve to primed/formative pluripotency. Moreover, these two studies uncovered OTX2 as a major driver of this transition and the associated enhancer reorganization.
Factor DC, Corradin O, Zentner GE, Saiakhova A, Song L, Chenoweth JG, et al. Epigenomic comparison reveals activation of “seed” enhancers during transition from naive to primed pluripotency. Cell Stem Cell. 2014;14:854–63. In this study, the authors profiled the enhancer landscape of EpiSC and uncovered “seed” enhancers as regulatory elements shared between primed pluripotent and somatic cells.
Soucie EL, Weng Z, Geirsdóttir L, Molawi K, Maurizio J, Fenouil R, et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science. 2016. This study demonstrates that differentiated somatic cells and ESC can activate a similar self-renewal gene expression program using distinct sets of enhancers.
Martello G, Sugimoto T, Diamanti E, Joshi A, Hannah R, Ohtsuka S, et al. Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self-renewal. Cell Stem Cell. 2012;11:491–504.
Respuela P, Nikolić M, Tan M, Frommolt P, Zhao Y, Wysocka J, et al. Foxd3 promotes exit from naive pluripotency through enhancer decommissioning and inhibits germline specification. Cell Stem Cell. 2016;18:118–33. This study demonstrates that enhancer decommissioning and the dismantling of the naïve gene expression program are important for the exit from naïve pluripotency and to prevent germline specification.
Krishnakumar R, Chen AF, Pantovich MG, Danial M, Parchem RJ, Labosky PA, et al. FOXD3 regulates pluripotent stem cell potential by simultaneously initiating and repressing enhancer activity. Cell Stem Cell. 2016;18:104–17.
Lien W-H, Polak L, Lin M, Lay K, Zheng D, Fuchs E. In vivo transcriptional governance of hair follicle stem cells by canonical Wnt regulators. Nat Cell Biol. 2014;16:179–90.
Zylicz JJ, Dietmann S, Günesdogan U, Hackett JA, Cougot D, Lee C, et al. Chromatin dynamics and the role of G9a in gene regulation and enhancer silencing during early mouse development. eLife. 2015;4. This study shows that the silencing of naïve pluripotency enhancers involves a progressive gain of H3K9me2.
Borgel J, Guibert S, Li Y, Chiba H, Schübeler D, Sasaki H, et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010;42:1093–100.
Osorno R, Tsakiridis A, Wong F, Cambray N, Economou C, Wilkie R, et al. The developmental dismantling of pluripotency is reversed by ectopic Oct4 expression. Dev Camb Engl. 2012;139:2288–98.
Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7.
Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol. 2012;19:1155–60.
Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994–1004.
Theunissen TW, Powell BE, Wang H, Mitalipova M, Faddah DA, Reddy J, et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15:471–87. The importance of this study is described concurrently with reference 32.
Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, et al. Derivation of novel human ground state naive pluripotent stem cells. Nature. 2013;504:282–6. Together with Theunissen et al., this study reports the long-sought conditions to induce naïve pluripotency in hESC.
Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S, et al. 3D Chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18:262–75. Togther with Gafni et al., this study provides a global view of the different enhancer repertoires of naïve and primed human pluripotent cells. Moreover, the overall topological organization of enhancer-promoter contacts in human pluripotent cells.
Saitou M, Yamaji M. Primordial germ cells in mice. Cold Spring Harb Perspect Biol. 2012;4.
Nakaki F, Hayashi K, Ohta H, Kurimoto K, Yabuta Y, Saitou M. Induction of mouse germ-cell fate by transcription factors in vitro. Nature. 2013;501:222–6. This study demonstrates the major roles of BLIMP1, PRDM14, and TFAP2C during PGC induction and specification.
Magnúsdóttir E, Dietmann S, Murakami K, Günesdogan U, Tang F, Bao S, et al. A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat Cell Biol. 2013;15:905–15.
Kurimoto K, Yabuta Y, Hayashi K, Ohta H, Kiyonari H, Mitani T, et al. Quantitative dynamics of chromatin remodeling during germ cell specification from mouse embryonic stem cells. Cell Stem Cell. 2015;16:517–32. This study provides the first global view of the enhancer dynamics during PGC specification.
Aramaki S, Hayashi K, Kurimoto K, Ohta H, Yabuta Y, Iwanari H, et al. A mesodermal factor, T, specifies mouse germ cell fate by directly activating germline determinants. Dev Cell. 2013;27:516–29.
Xu J, Watts JA, Pope SD, Gadue P, Kamps M, Plath K, et al. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23:2824–38.
Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5.
Guibert S, Forné T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012;22:633–41.
Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, et al. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–62.
Sasaki K, Yokobayashi S, Nakamura T, Okamoto I, Yabuta Y, Kurimoto K, et al. Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell. 2015;17:178–94. The importance of this study is described concurrently with reference 44.
Irie N, Weinberger L, Tang WWC, Kobayashi T, Viukov S, Manor YS, et al. SOX17 is a critical specifier of human primordial germ cell fate. Cell. 2015;160:253–68. Together with Sasaki et al., this study reports novel differentiation conditions to induce germ fate in human pluripotent cells.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–52.
Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73.
Apostolou E, Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature. 2013;502:462–71.
Papp B, Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324–43.
Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet. 2013;45:34–42. This study demonstrates that H3K9me2/3 at promoter and enhancer regions of important pluripotency genes represents a major roadblock for somatic reprogramming.
Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55.
Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602.
Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–32.
Sridharan R, Gonzales-Cope M, Chronis C, Bonora G, McKee R, Huang C, et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1γ in reprogramming to pluripotency. Nat Cell Biol. 2013;15:872–82.
Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, et al. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell. 2015;162:412–24. This study provides a global, systematic and thorough view of the molecular events accompanying the reprogramming of human somatic cells.
Taberlay PC, Kelly TK, Liu C-C, You JS, De Carvalho DD, Miranda TB, et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147:1283–94.
Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105.
Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–4.
Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, Fujita R, et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–5.
Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–9.
Feldmann A, Ivanek R, Murr R, Gaidatzis D, Burger L, Schübeler D. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013;9:e1003994. Together with previous work from the Schübeler lab, this study demonstrates that TFs can antagonize DNA methylation at regulatory elements such as enhancers.
Athanasiadou R, de Sousa D, Myant K, Merusi C, Stancheva I, Bird A. Targeting of de novo DNA methylation throughout the Oct-4 gene regulatory region in differentiating embryonic stem cells. PLoS One. 2010;5:e9937.
Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol. 2006;8:188–94.
Luo M, Ling T, Xie W, Sun H, Zhou Y, Zhu Q, et al. NuRD blocks reprogramming of mouse somatic cells into pluripotent stem cells. Stem Cells Dayt Ohio. 2013;31:1278–86.
Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70.
Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell. 2014;55:347–60.
Boulard M, Edwards JR, Bestor TH. FBXL10 protects Polycomb-bound genes from hypermethylation. Nat Genet. 2015;47:479–85.
Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, et al. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45:1198–206.
Sato N, Kondo M, Arai K. The orphan nuclear receptor GCNF recruits DNA methyltransferase for Oct-3/4 silencing. Biochem Biophys Res Commun. 2006;344:845–51.
Maeda I, Okamura D, Tokitake Y, Ikeda M, Kawaguchi H, Mise N, et al. Max is a repressor of germ cell-related gene expression in mouse embryonic stem cells. Nat Commun. 2013;4:1754. This study shows that MAX, a TF, is able to mediate the silencing of late germline genes in mESC through the recruitment of H3K9 methyltransferases to promoter regions.
Velasco G, Hubé F, Rollin J, Neuillet D, Philippe C, Bouzinba-Segard H, et al. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc Natl Acad Sci U S A. 2010;107:9281–6.
Wang B, Miyagoe-Suzuki Y, Yada E, Ito N, Nishiyama T, Nakamura M, et al. Reprogramming efficiency and quality of induced Pluripotent Stem Cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages. PLoS Curr. 2011;3:RRN1274.
Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–9.
Soria-Valles C, Osorio FG, Gutiérrez-Fernández A, De Los AA, Bueno C, Menéndez P, et al. NF-kB activation impairs somatic cell reprogramming in ageing. Nat Cell Biol. 2015;17:1004–13. This study shows that DOT1L imposes an epigenetic barrier to somatic reprogramming in a NF-kB and ageing dependent manner. Moreover, this barrier can be overcome pharmacologically, opening new epigenetic strategies towards cellular rejuvenation.
Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45.
Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248–53.
Liu G-H, Barkho BZ, Ruiz S, Diep D, Qu J, Yang S-L, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5.
Liu G-H, Ding Z, Izpisua Belmonte JC. iPSC technology to study human aging and aging-related disorders. Curr Opin Cell Biol. 2012;24:765–74.
Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42.
Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6.
Rakyan VK, Down TA, Maslau S, Andrew T, Yang T-P, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19.
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–51.
McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, et al. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–9.
Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015;10:471–83.
Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;163:134–47.
Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, et al. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–92.
de Wit E, Bouwman BAM, Zhu Y, Klous P, Splinter E, Verstegen MJAM, et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227–31.
Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016.
Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33:1165–72.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Patricia Respuela and Alvaro Rada-Iglesias declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Age-related Stem Cell Modifiers
Rights and permissions
About this article

Cite this article
Respuela, P., Rada-Iglesias, A. Enhancer Remodeling During Early Mammalian Embryogenesis: Lessons for Somatic Reprogramming, Rejuvenation, and Aging. Curr Stem Cell Rep 2, 263–272 (2016). https://doi.org/10.1007/s40778-016-0050-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-016-0050-8