
Abstract
Background
Capecitabine monotherapy is a treatment option for selected patients with metastatic colorectal cancer (mCRC) and is administered to up to 17% of patients. Data are limited with regard to adverse events and dosing practices associated with capecitabine monotherapy in real-world situations.
Objectives
The aim of this study was to provide real-world data on adverse event rates and dose adjustments/discontinuations associated with capecitabine monotherapy in patients with mCRC.
Methods
This retrospective study analyzed data from CRC patients scheduled to receive up to eight planned cycles of capecitabine monotherapy between 2009 and 2013 at a single large community hospital in The Netherlands. Data on adverse events (hand-foot syndrome [HFS], gastrointestinal (GI) events, hematological adverse events, and cardiotoxicity), as well as relative dose intensities (RDIs), dose reductions, and discontinuations, were evaluated.
Results
Data from 86 patients (45 females; mean age at the start of treatment, 69 years) were included. A total of 46.5% of patients experienced HFS and 44.2% experienced a GI event at some time during treatment. Hematological events and cardiotoxicity were rare. Most patients (77%) started at below the recommended dose, and patients at the lowest dose also had the lowest median RDIs. Dose reductions and discontinuations occurred in 15–25% of patients who experienced HFS or GI event over the course of eight cycles.
Conclusions
HFS and GI events were very common in patients treated with capecitabine monotherapy in a real-world clinical setting. Most patients started treatment at below the recommended dose, and 15–25% of patients who had HFS or a GI event had a dose reduction or discontinuation.
Similar content being viewed by others

This study provides real-world data on adverse events and dosing practice associated with capecitabine monotherapy in patients with metastatic colorectal cancer. |
Hand-foot syndrome and gastrointestinal (GI) events were seen in almost half of the patients treated with capecitabine monotherapy. |
Dose reductions and discontinuations occurred in 15–25% of patients who experienced hand-foot syndrome or GI events over the course of eight cycles of therapy. |
1 Introduction
Fluoropyrimidine monotherapy is a recommended chemotherapeutic treatment option for patients with metastatic colorectal cancer (mCRC) who are frail or may not tolerate more aggressive therapy [1–3]. Oral capecitabine provides a convenient alternative to the standard intravenous fluoropyrimidine, 5-fluorouracil. In clinical trials, oral capecitabine monotherapy has been shown to be as effective as intravenous 5-fluorouracil as first-line treatment for mCRC, and is generally associated with an improved safety profile with lower rates of stomatitis, alopecia, diarrhea, nausea, and grade 3/4 neutropenia [4–7]; however, reported rates of hand-foot syndrome (HFS) are higher with capecitabine. HFS is characterized by erythema, dysesthesia and/or paresthesia of the palms of the hands or soles of the feet. In more advanced stages, desquamation, ulceration, and blistering can occur. HFS occurs in approximately 54% of patients (17% grade 3/4) who receive capecitabine treatment [4, 6–8]. Grade 3/4 hyperbilirubinemia is also higher with capecitabine and occurs in approximately 23% of patients [4, 6, 7]. The approved regimen for capecitabine is 1250 mg/m2 twice daily for 14 days, followed by 7 days off [9]. In phase III trials, 34% of patients received a reduced dose due to the occurrence of adverse events [4].
In phase III trials including older patients (>70 years of age), who represent a key group in which capecitabine monotherapy may be indicated, grade 3/4 adverse events occurred in 12–22% of patients, including grade 3/4 HFS, diarrhea, venous thromboembolism, neutropenia, thrombocytopenia, and hemorrhage [8, 10]. Eighteen percent of elderly patients experience dose delays due to adverse events while receiving capecitabine monotherapy and 15% discontinue treatment due to adverse events [8, 10].
Most real-world studies of physician prescribing patterns in mCRC have focused on the impact of effective biologic and combination treatments that have extended survival in mCRC in recent years [11–13]. These analyses of retrospective data of treatment patterns have reported that 9–17% of patients receive capecitabine monotherapy as first-line treatment, 5–9% as second-line treatment, and as many as 17% receive this regimen as third-line treatment [11–13]. An observational study of capecitabine-based therapy in routine first-line treatment of mCRC reported that 56% of patients received capecitabine-based treatment—54% of these as combination therapy and 46% of these as monotherapy. Of patients who received monotherapy, 65% were older than 75 years of age [14]. Rates of grade 3/4 adverse events associated with capecitabine monotherapy were highest for HFS, bilirubin elevation, anemia, and neuropathy, which all occurred in 4% of patients [14].
Despite these few studies, real-world data are limited with regard to the adverse events and dosing practice associated with oral capecitabine monotherapy in mCRC in the oncology clinic. While realizing its inherent limitations, this study sought to provide real-world data on the occurrence of adverse events in patients treated with capecitabine monotherapy for mCRC at a single large community hospital.
2 Methods
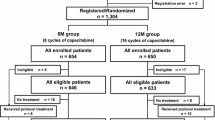
This was a single-center, retrospective study of patients treated at a large community hospital in Zwolle, The Netherlands, for mCRC. Data were collected for toxicity in relation to dose and exposure time for patients diagnosed with adenocarcinoma. We limited our period of analysis to the planned eight cycles (±6 months) of capecitabine monotherapy, as recommended in the Dutch pharmacotherapeutic guidelines for capecitabine monotherapy in mCRC. In these guidelines, capecitabine monotherapy is considered a good option when no immediate response is needed (for instance in case of relatively limited tumor load), or in patients who are deemed too frail to start with combination therapy.
Institutional Review Board approval was obtained for this retrospective analysis, and key data that were collected included capecitabine dose by cycle; adverse event data for hematological events (neutropenia, leukopenia, thrombocytopenia, anemia), cardiac events (angina pectoris, heart failure, myocardial infarction, arrhythmia/conduction disorder, myocarditis, ECG changes), hand-foot syndrome, and gastrointestinal (GI) events (diarrhea, nausea, vomiting, constipation, mucositis, abdominal pain, stomatitis, loss of appetite); and dose reductions and discontinuations. Patient data were excluded if the patient received anticancer therapy other than capecitabine, and only the first eight cycles of therapy were retained for patients who received more than eight cycles of therapy or an additional eight cycles at a later start date.
For the adverse event analyses, patients were counted if they had the adverse event concerned and if they had a dose reduction or discontinuation of treatment during that cycle. The cause of dose reduction or discontinuation was not explicitly stated to be the adverse event in question but was tracked for the patients who had that adverse event in that cycle. Discontinuation in cycle 8 could be due to adverse events, progression, or simply the end of planned treatment. This analysis has not looked beyond eight cycles, but some patients were treated for much longer than eight cycles.
Relative dose intensity (RDI) was calculated for each patient to determine the dose received relative to the planned schedule to dose over eight cycles. Receipt of the starting dose for eight cycles represented 100%. Reduced doses were scored based on their relative proportion of the starting dose. For example, if 1250 mg/m2 twice daily was the starting dose, then a reduction to 1000 mg/m2 was scored as 80% of the dose for that cycle, and a reduction from 1000 to 750 mg/m2 was scored as 75% of the dose for that cycle. RDI was calculated as the number of cycles at the starting dose plus the number of cycles at a reduced dose (e.g. four cycles × 1.0 + four cycles × 0.8) divided by eight total cycles.
3 Results
Data for 86 patients (45 female, 41 male; mean age at start of treatment, 69 years [range 45–83]; 57% ≥70 years of age) treated with capecitabine monotherapy for mCRC between 2009 and 2013 were analyzed for side effects occurring during eight planned cycles of treatment. A total of 355 patients started palliative systemic therapy for mCRC at our center during this time period. Twelve patients started cycle 1 with a dose of 750 mg/m2 twice daily (mean age 64.4 years), 54 patients started at a dose of 1000 mg/m2 twice daily (mean age 71.5 years), and 20 patients started at a dose of 1250 mg/m2 twice daily (mean age 67 years). In total, 35 patients were still taking capecitabine in cycle 8 (Table 1), of whom 49% were on the lowest dose. A total of 41% of patients completed at least four cycles of therapy at the starting dose, and 21% completed eight cycles of therapy at the starting dose. The numbers of patients on the lowest dose stayed relatively constant or increased as patients moved from higher doses in the later cycles of therapy.
3.1 Relative Dose Intensity
A box plot of the RDIs for all 86 patients included in the study is shown in Fig. 1. The median RDIs for patients who started at the 750, 1000, and 1250 mg/m2 twice-daily doses were 37.5, 67.2, and 68.75%, respectively. Twenty-five percent of patients at the 750 mg/m2 twice-daily dose received 100% of the planned dose compared with 18.5% of patients at the 1000 mg/m2 twice-daily dose, and 30% of patients at the 1250 mg/m2 dose.

Relative dose intensities for patient data evaluated over the course of eight planned cycles of oral capecitabine monotherapy. Boxes represent interquartile range (25th–75th quartiles), with median value indicated. Whiskers represent maximum and minimum relative dose intensity values for patients at each starting dose (750 mg/m2 bid, n = 12; 1000 mg/m2 bid, n = 54; 1250 mg/m2 bid, n = 20). bid twice daily
3.2 Rates of Hand-Foot Syndrome
HFS events were common in all cycles and at all dose levels. A total of 46.5% of patients experienced HFS at some time during treatment (Fig. 2a). Newly developing HFS was observed in all cycles, and persistent or recurrent HFS events were responsible for 54.5% of total HFS events (n = 88 events). HFS events appeared to increase over time for patients at all three doses (Fig. 2b), which is most clearly seen at the 1000 mg/m2 dose. After the first cycle (8.1% HFS reported), 15–32% of patients reported HFS in each cycle. Over the course of eight cycles, 22 patients had dose reductions and 15 discontinued treatment during a cycle in which they reported HFS, often within four cycles of treatment (Table 2).

Rates of HFS by cycle: a all patients (n = 86) and b according to dose (twice daily). Percentages were calculated as the number of patients who had HFS, of the number of patients who started treatment at that dose level for that cycle. A total of 27 patients experienced HFS at the 750 mg/m2 twice-daily dose, 45 at the 1000 mg/m2 dose, and 16 at the 1250 mg/m2 dose over the course of treatment. HFS hand-foot syndrome
3.3 Rates of Gastrointestinal Adverse Events
GI events were common in all cycles (Fig. 3a), and most first-time events were in the first three cycles. A total of 44.2% of patients experienced a GI adverse event at some time during treatment. In any given cycle, between 14 and 25% of patients reported GI events. Persistent or recurring GI events accounted for 54.8% of total GI events (n = 84 events). Evaluation of GI events by dose level showed that more patients at the 750 mg/m2 dose level experienced GI events in later cycles, while these events were less common for patients at the 1000 mg/m2 dose level and were not observed for patients at the 1250 mg/m2 dose in cycles 4–8 (Fig. 3b). Over the course of eight cycles, 13 patients had dose reductions and 21 discontinued treatment during a cycle in which they reported a GI event (Table 3). Most of these treatment modifications were performed in the first four cycles of capecitabine therapy. The most common GI events were diarrhea, nausea, vomiting, and abdominal pain (Table 4).

Rates of GI events by cycle: a all patients (n = 86) and b according to dose (twice daily). Percentages were calculated as the number of patients who had a GI event, of the number of patients who started treatment at that dose level for that cycle. A total of 30 patients experienced a GI event at the 750 mg/m2 twice-daily dose, 50 at the 1000 mg/m2 dose, and 4 at the 1250 mg/m2 dose over the course of treatment. GI gastrointestinal
A comparison of the total percentage of patients affected by either HFS or a GI event over eight cycles is shown in Fig. 4.

Rates of GI events and HFS by cycle, all patients (n = 86). GI gastrointestinal, HFS hand-foot syndrome
3.4 Rates of Hematological and Cardiac Adverse Events
Six hematological adverse events occurred in five patients during the first four cycles of therapy. One patient at the 1250 mg/m2 dose had neutropenia in cycle 1 that was treated with a dose interruption and dose reduction to 1000 mg/m2 in cycle 2. The patient experienced neutropenia again in cycle 2 but without dose adjustments. One patient each at the 1000 mg/m2 dose experienced leukopenia and thrombocytopenia in the third cycle. Leukopenia was managed with a dose interruption in that cycle and the thrombocytopenia was managed with a dose reduction to 750 mg/m2. One patient at the 750 mg/m2 dose experienced anemia in cycle 2, and one patient at the 1000 mg/m2 dose experienced anemia in cycle 4. The patient discontinued in this cycle but the recorded data did not explicitly state that anemia was the cause.
Six cardiotoxicity events were reported in five patients (mean age 71 years), i.e. chest pain, unregulated heartbeat, atrial fibrillation with pulmonary embolism, dyspnea on exertion and cough, arrhythmia/conduction disorder. Five of these were at the 1000 mg/m2 dose, and atrial fibrillation recurred in one patient who had been reduced to the 750 mg/m2 dose in a different cycle. There was one dose reduction and one discontinuation among patients who reported a cardiac adverse event for that cycle.
4 Discussion
An important and ongoing point of attention influencing treatment outcomes for cancer patients is the tolerability of chemotherapeutic drugs. This is even more important in the palliative setting. The gold standard in clinical research is to investigate these questions in randomized controlled clinical trials but these are expensive and cumbersome trial designs and are rarely suitable for assessing daily practical questions. A good alternative to get more insight into these types of questions is with so-called real-world studies. In this real-world study, a retrospective analysis was performed on data from patients treated for eight planned cycles of therapy with a commonly used chemotherapeutic drug (capecitabine) for mCRC. We chose to analyze only patients receiving capecitabine monotherapy to reduce unwanted interactions and influence by other anticancer drugs in the treatment. We were able to evaluate the rates of adverse events in patients for whom treatment was selected based on each patient’s clinical situation and personal preference in real-world oncology treatment decision-making situations rather than based on selective clinical trial inclusion criteria.
In this study, we have evaluated dosing adjustments and adverse events in patients treated with capecitabine monotherapy for mCRC. We evaluated rates of occurrence and persistence of HFS, GI events, hematological adverse events, and cardiotoxicity over the course of eight scheduled cycles of capecitabine monotherapy and rates of dose reductions and discontinuation. The rates of adverse events reported in this study are similar to those of reported clinical trials of capecitabine monotherapy. The rate of HFS in this study (46.5% overall) is consistent with rates observed in phase III clinical trials of 30–53.5% [4, 8, 10] and with the rate of 42% reported in an observational study that included patients who received capecitabine as monotherapy or in combination treatment [14]. The rate of GI events in this study was 44.2%; previous studies have reported that between 11 and 50% of patients experience one GI event, including diarrhea, vomiting, nausea, or abdominal pain, while receiving capecitabine monotherapy [4, 8, 10]. Our results are consistent with these findings. Neutropenia, observed in only one patient in this study (1.1%) has been reported to occur in 1% of patients in clinical trials [4, 8]. Rates of other hematological adverse events were also low in this study, similar to previous studies [4, 8, 10]. Cardiotoxicity, observed in 5% of patients in this study, was either very rare (approximately 1%) or not reported due to occurring at lower than the 5% threshold for reporting in previous studies [4, 8, 10]. It was not possible to establish if this difference could be explained by the current population being more frail than those described in previous controlled trials.
Most patients in this study (77%) started under the approved dose of 1250 mg/m2 twice daily, 63% started at 1000 mg/m2 twice daily, and 14% started at 750 mg/m2 twice daily. Of note, the reduced starting doses used here are not the recommended reduced starting doses for special populations (75% of starting dose for renal impairment) [9], and phase III trials evaluated a starting dose of 1250 mg/m2 twice daily or used 1000 mg/m2 twice daily in elderly patients ≥70 years of age [4, 8, 10]. Patients in this study who received the 1000 mg/m2 twice-daily dose had a mean age of 71.5 years, consistent with age as an explanation for the use of this reduced dose. However, patients in the study who received 750 mg/m2 twice daily had a mean age of 64.4 years, suggesting that this population was considered frail by their physician. Although this suggests that physicians are reducing the starting dose of capecitabine in anticipation of adverse events, our real-world data did not provide an explicit explanation for these treatment decisions.
Dose reductions and treatment discontinuations were common in this study, occurring in 17–24% of patients who experienced HFS and 15–25% of patients who experienced a GI event. Dose reductions or cessation of treatment most likely due to adverse events occurred predominantly within the first four cycles of therapy. Timely recognition and management of the clinically relevant HFS and GI toxicity is therefore of utmost importance in order to prevent early termination of treatment.
Cassidy et al. reported that 34% of patients starting treatment at 1250 mg/m2 twice daily required a dose reduction for adverse events, while Cunningham et al. reported that 15% of elderly patients who started capecitabine treatment at 1000 mg/m2 twice daily discontinued due to adverse events [4, 8]. In addition, Feliu et al. reported that dose delays occurred in 18% of elderly patients treated with capecitabine 1250 mg/m2 twice daily [9]. In our analysis, the occurrence of HFS and GI events was not related to the dose of capecitabine, which may suggest that lower starting doses and dose reductions do not improve adverse event rates, nor do they prevent them from occurring. In an observational study by Stein et al., the incidence of HFS increased with duration of treatment and was higher in younger patients than in older patients (46 vs. 37%; p = 0.0014) despite similar median daily doses of capecitabine [14].
It is unclear whether dose reductions might negatively impact efficacy outcomes. Cassidy et al. reported a similar risk of disease progression in patients who required dose modification while receiving capecitabine monotherapy compared with those who did not, while patients who required dose modifications while taking 5-fluorouracil/leucovorin had a 12% higher risk of disease progression [4]. Stein et al. reported that patients who experienced HFS had higher response rates, progression-free survival (PFS), and overall survival (OS) than patients without HFS. The authors postulated that a trend in improved PFS and OS in patients who received a capecitabine dose reduction might be related to the occurrence of HFS in this population [14].
This study provides some insights into the clinical decisions that were considered necessary in the best interests of the patient and what impact these decisions had on the dosing and schedule of capecitabine. However, there were significant limitations of this study, including its small size, its retrospective nature and lack of control group, and the quality of the real-world data we were able to obtain. The patient record data used in this study often did not include clear reasons for treatment discontinuation or dose reductions, therefore these could not be directly correlated to adverse events. In addition, they did not include consistent information on the grade of adverse events, which would have been informative.
5 Conclusions
This study has provided important information on the rates of adverse events and dosing practices in patients scheduled to be treated with eight cycles of capecitabine monotherapy for mCRC in a real-world setting. The most frequently occurring adverse events were HFS and GI toxicity. These adverse events often led to dose reductions or even termination of treatment, possibly impairing the benefit of fluoropyrimidines in these patients. This information should be of value to practitioners who treat patients with mCRC, particularly older or frail patients.
References
Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii1–9.
National Comprehensive Cancer Network (NCCN) Guidelines. Colon Cancer. v2.2016. Available at: http://www.nccn.org. Accessed 24 Mar 2016.
National Comprehensive Cancer Network (NCCN) Guidelines. Rectal Cancer. v1.2016. Available at: http://www.nccn.org. Accessed 24 Mar 2016.
Cassidy J, Twelves C, Van Cutsem E, Hoff P, Bajetta E, Boyer M, et al. First-line oral capecitabine therapy in metastatic colorectal cancer: a favorable safety profile compared with intravenous 5-fluorouracil/leucovorin. Ann Oncol. 2002;13:566–75.
Van Cutsem E, Hoff PM, Harper P, Bukowski RM, Cunningham D, Dufour P, et al. Oral capecitabine vs intravenous 5-fluorouracil and leucovorin: integrated efficacy data and novel analyses from two large, randomised, phase III trials. Br J Cancer. 2004;90:1190–7.
Van Cutsem E, Twelves C, Cassidy J, Allman D, Bajetta E, Boyer M, et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: results of a large phase III study. J Clin Oncol. 2001;19:4097–106.
Hoff PM, Ansari R, Batist G, Cox J, Kocha W, Kuperminc M, et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first-line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J Clin Oncol. 2001;19:2282–92.
Cunningham D, Lang I, Marcuello E, Lorusso V, Ocvirk J, Shin DB, et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol. 2013;14:1077–85.
Feliu J, Escudero P, Llosa F, Bolaños M, Vicent JM, Yubero A, et al. Capecitabine as first-line treatment for patients older than 70 years with metastatic colorectal cancer: an Oncopaz Cooperative Group study. J Clin Oncol. 2005;23:3104–11.
Xeloda (capecitabine) prescribing information. South San Francisco; Genentech, Inc.; 2015.
Zafar SY, Marcello JE, Wheeler JL, Rowe KL, Morse MA, Herndon JE, et al. Longitudinal patterns of chemotherapy use in metastatic colorectal cancer. J Oncol Pract. 2009;5:228–33.
Hess GP, Wang PF, Quach D, Barber B, Zhao Z. Systemic therapy for metastatic colorectalcancer: patterns of chemotherapy and biologic therapy use in US medical oncology practice. J Oncol Pract. 2010;6:301–7.
Abrams TA, Meyer G, Schrag D, Meyerhardt JA, Moloney J, Fuchs CS. Chemotherapy usage patterns in a US-wide cohort of patients with metastatic colorectal cancer. J Natl Cancer Inst. 2014;106:djt371.
Stein A, Quidde J, Schröeder JK, Göhler T, Tschechne B, Valix AR, et al. Capecitabine in the routine first-line treatment of elderly patients with advanced colorectal cancer-results from a non-interventional observation study. BMC Cancer. 2016;16:82.
Acknowledgements
The authors would like to acknowledge the contributions of Margriet Bisschoff for her meticulous work in collecting the patient data, and Sandy Field, PhD, Medical Writer, for the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jan Willem B. de Groot and Wilko Coers have received consulting fees and/or honorarium for writing support from Nordic Pharma International. Laura W. Leicher, Metin Tascilar, and Jacques C. de Graaf have not received any financial or other support.
Funding
This study was made possible with financial support from Nordic Pharma International.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Leicher, L.W., de Graaf, J.C., Coers, W. et al. Tolerability of Capecitabine Monotherapy in Metastatic Colorectal Cancer: A Real-World Study. Drugs R D 17, 117–124 (2017). https://doi.org/10.1007/s40268-016-0154-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-016-0154-8