
Abstract
Aims
This bioequivalence study aimed to compare rate and extent of absorption of a generic medicinal product of ibandronic acid 150-mg film-coated tablet versus Bonviva®.
Methods
This was a single-centre, open-label, randomized, three-way, three-sequence, reference-replicated, crossover bioequivalence study, under fasting conditions. A single oral dose of ibandronic acid as one 150-mg film-coated tablet was administered in each study period. Each washout period lasted 14 days. Blood samples were collected according to a predefined sampling schedule and up to 48.0 hours after administraton in each period. Plasma concentrations of ibandronic acid were measured using a liquid chromatograph–mass spectrometry/mass spectrometry method. Bioequivalence between generic and reference medicinal products is acceptable if the 90 % confidence intervals (CI) of ratio of least-squares means between the test and the reference product of ln-transformed area under the serum concentration–time curve from time zero to time of last measurable concentration (AUC0–t ) is within the 80.00–125.00 % interval. Prospectively, a scaled average bioequivalence approach for maximum serum concentration (C max) was established.
Results
153 healthy volunteers were enrolled and randomized. After the test formulation (T) and first and second Bonviva® (R) dosing, the C max was 96.71 ± 90.19 ng/mL, 92.67 ± 91.48 ng/mL and 87.94 ± 60.20 ng/mL and the AUC0–t was 390.83 ± 287.27 ng·h/mL, 388.54 ± 356.76 ng·h/mL and 383.53 ± 246.72, respectively. Ratios of T/R and 90 % CI were 100.92 % (94.35–107.94) for AUC0–t , 100.90 % (94.37–107.88) for AUC0–inf and 102.56 % (95.05–110.67) for C max.
Conclusions
Test formulation of ibandronic acid is bioequivalent in rate and extent of absorption to Bonviva® following a 150-mg dose, under fasting conditions.
Similar content being viewed by others


1 Introduction
Ibandronic acid 1-hydroxy-3-[methyl(pentyl)amino]propane-1,1-diyl}bis(phosphonic acid) is a nitrogen-containing bisphosphonate (ATC M05BA06; CAS 114084-78-5) acting as an inhibitor of osteoclast-mediated bone resorption. Ibandronic acid is effective for the treatment and prevention of osteoporosis in postmenopausal women with increased risk of fractures, and a reduction in the risk of vertebral fractures has been demonstrated [1].
The absorption of ibandronic acid in the upper gastrointestinal tract is rapid after oral administration. In fasted state, the maximum observed plasma concentration (C max) is reached within 0.5–2 hours (median 1 hour). The oral bioavailability after oral administration is low (~0.6 %) and highly variable. Bioavailability is reduced by 90 % in the presence of a standard breakfast and by approximately 75 and 30 % when is administered 2 hours after a standard meal and 30 minutes before a meal, respectively. There is no meaningful reduction in bioavailability provided ibandronic acid is taken 60 minutes before a meal [1, 2].
There is no evidence of dose-dependent pharmacokinetics in the range of 2.5–50 mg oral dosage. The exposure following administration of 50, 100 or 150 mg was not dose proportional, with area under the serum concentration–time curve (AUC) and C max presenting greater increase in exposure with increasing dose. The reason for these dose-dependent pharmacokinetics is not fully elucidated [1, 2].
In humans, the apparent terminal volume of distribution is high (~90 L), which is most likely related to substantial distribution within skeletal tissue: the amount of dose removed from the circulation via the bone is estimated to be 40–50 % and the remainder is eliminated unchanged by the kidney. The unabsorbed fraction of ibandronic acid is eliminated unchanged in the faeces. Protein binding in human plasma is approximately 87 % at therapeutic concentrations, and drug–drug interaction due to displacement is unlikely. There is no evidence that ibandronic acid is metabolized in animals or humans. The observed apparent elimination half-life (T ½ el) for ibandronic acid is generally in the range of 10–72 hours. Total clearance of ibandronic acid is low with average values in the range of 84–160 mL/min. Renal clearance (about 60 mL/min in healthy postmenopausal females) accounts for 50–60 % of total clearance and is related to creatinine clearance. The difference between the apparent total and renal clearances is considered to reflect the uptake by bone [1, 2].
The present study aimed to compare the rate and extent of absorption of ibandronate acid (as sodium ibandronate) 150 mg from a test medicinal product (test formulation; Treatment A), manufactured by Tecnimede (Sintra, Portugal) and that of the reference medicinal product (reference formulation; Treatment B; Bonviva®), a surrogate for therapeutic equivalence.
2 Volunteers and Methods
2.1 Study Protocol
The clinical study protocol and related documents were approved by an independent ethics committee (International Review Board Services) and a No Objection Letter (NOL) was obtained from Canadian authorities. The study was conducted in accordance with the most recent version of the Helsinki Declaration and Good Clinical Practice Guideline [3]. Informed consent was obtained from participants prior to initiation of study procedures. The clinical and analytical parts of the study were conducted at Inventive Health’s facility (Québec City, QC, Canada). Pharmacokinetic and statistical analyses were also performed by Inventive Health’s facility (Québec City, QC, Canada).
2.2 Volunteers
The 153 subjects were recruited from the community at large and considered eligible for enrolment as per protocol inclusion and exclusion criteria. Subjects included were males or females of non-childbearing potential, nonsmokers or moderate smokers (no more than nine cigarettes daily), aged 18 years of age and older (≥18 years) and with body mass indices (BMI) greater than 18.5 kg/m2 (>18.5) and less than 30.0 kg/m2 (<30.0). Females of non-childbearing potential included post-menopausal females or surgically sterile females. The screening procedures included collection of anamnesis and demographic data (gender, age, race, body weight [kg], height [cm] and BMI), a physical examination, a resting 12-lead electrocardiogram (ECG), urine illicit drug screen, urine pregnancy test (female subjects) and clinical laboratory tests (haematology, biochemistry, urinalysis, human immunodeficiency virus [HIV], hepatitis C [HCV] antibodies and hepatitis B surface antigen [HBSAg]). The baseline demographic characteristics of the pharmacokinetic population are depicted in Table 1.
A total of 153 healthy subjects were randomly assigned to a treatment in accordance with the computer-generated blocks randomization scheme (block size 6, randomly variable). The randomization scheme was generated using Statistical Analysis System® (SAS®) program version 9.2 (SAS Institute Inc., Cary, NC, USA). This program used the randomized block design to ensure an equal distribution of sequences at multiples of 6 in the list of subject assignment.
Based on results of a previous pilot study, the within-subject coefficients of variance (CVs) should be approximately 39 and 48 % for AUC and C max, respectively. Thus, with these expected CVs and an expected ratio of AUC and C max within 0.90 and 1.11, the study should have a power of at least 90 % to show bioequivalence with 138 subjects. In order to account for possible dropouts, 153 subjects were included in the study.
2.3 Study Design
This study was a single-centre, randomized, single-dose, open-label, three-way, three-sequence, reference formulation-replicated, crossover bioequivalence study to compare the rate and extent of absorption of Tecnimede’s test formulation of ibandronic acid (batch number 15044; expiration date: 04-2013; manufactured by West Pharma, SA, Portugal) with the reference formulation (batch number B1176B01; expiration date: 11-2015; manufactured by Roche Pharma AG, Germany), acquired in the Polish market, under fasting conditions, in healthy volunteers.
After an overnight fast of at least 10 hours, subjects were dosed in the mornings. Subjects were administered the test or the reference formulation, as per the randomization scheme, as a single oral dose of one film-coated tablet containing 150 mg of study medication, with 240 mL of water. Subjects were dosed as specified in the protocol, and subsequently fasted for a period of at least 4 hours. Subjects were served a controlled meal not less than 4 hours post-dose, and at appropriate times thereafter, in each period. Subjects were served standardized post-dose meals similar in composition in each period. With the exception of the volume administered at the time of dosing, fluids were not permitted from 1 hour before dosing to 1 hour after dosing, but, after that period, plain water was permitted ad libitum.
According with a reference formulation-replicated design, the study had three periods (period 1, period 2 and period 3) and the subjects were randomized to three sequences (test-reference-reference [TRR]; reference-reference-test [RRT] and reference-test-reference [RTR]). In each study period, subjects were administered the test formulation (Treatment A) or the reference formulation (Treatment B) as per the randomization scheme. The treatment periods were separated by washout periods of 14 days.
The randomization scheme was kept unavailable to the bioanalytical division until completion of the clinical and analytical phases.
2.4 Drug Analysis
A dead-volume intravenous catheter was used for blood collection, which occurred prior to drug administration and 0.167, 0.333, 0.500, 0.750, 1.00, 1.25, 1.50, 1.75, 2.00, 3.00, 4.00, 6.00, 8.00, 12.0, 24.0 and 48.0 hours post-dose in each period. Actual sampling times were used in the statistical analyses. Blood samples were cooled in an ice bath and were centrifuged at 3,000 rpm (corresponding to approximately 1,900 g) for at least 10 minutes at approximately 4 °C (no more than 110 minutes passed between the time of each blood draw and the start of centrifugation). The aliquots were transferred to a −20 °C freezer, pending transfer to the bioanalytical facility.
2.5 Pharmacokinetic Analysis
Pharmacokinetic analyses were performed using Pharsight® Knowledgebase ServerTM (version 4.0.2) and WinNonlin® (version 5.3), which are validated for bioequivalence/bioavailability studies by Inventive Health. Inferential statistical analyses were performed using SAS® (release 9.2) according to the Food and drug Administration (FDA), Health Product and Food Branch of Health Canada and European Medicines Agency (EMA) guidance.
The number of observations (N), mean, standard deviation (SD), CV%, range (minimum and maximum), median and geometric mean were calculated for plasma concentrations of ibandronic acid for each sampling time and treatment. These descriptive statistics were also presented for the AUC from time zero to time of the last non-zero concentration (AUC0–t ), the AUC from time zero to infinity (extrapolated) (AUC0–inf), the C max, the residual area calculated through the equation (1 − AUC0–t /AUC0–inf) × 100 %, time to C max (T max), the T ½ el and the elimination rate constant (K el). The AUC0–t was calculated using the linear trapezoidal rule. AUC0–inf was calculated through the following equation: AUC0–t + (C t /K el), where C t is the fitted last non-zero concentration for that treatment.
2.6 Safety Analysis
Adverse events were listed and coded using Medical Dictionary for Regulatory Activities (MedDRA®), version 15.0. Treatment-emergent adverse events (TEAEs) were summarized descriptively in the safety population, and were tabulated by treatment group, system organ class, preferred term, causality and severity.
2.7 Statistical Analysis
For the purpose of statistical analyses, the safety population included the subjects who received at least one dose of the investigational medicinal product whereas the pharmacokinetic population included the subjects who completed at least two periods including one period with test formulation and other with the reference formulation and for whom the pharmacokinetic profile was characterized. Pharmacokinetic parameters were summarized by treatment. Plasma concentrations were summarized by treatment and timepoint. Individual and mean plasma concentrations, as well as the plots of the plasma levels for all subjects versus time, were graphically displayed for three treatments.
Ln-transformed AUC0–t , AUC0–inf and C max were analysed using general linear model (GLM) procedure in SAS® following the method A recommended by the EMA (CHMP Pharmacokinetics Working Party [PKWP] EMA/618604/2008 Rev. 3). The statistical model included sequence, period, treatment and subject within sequence as fixed factors. The sequence effect was tested using the subject-within-sequence effect as the error term. The treatment and period effects were tested against the residual mean square error. Within-subject coefficient of variation (CVWR) was calculated for the reference product using analysis of variance (ANOVA), on reference data only, with sequence, subject within sequence, and period as fixed effects. The point estimate and the 90 % geometric confidence interval for the test-to-reference geometric mean ratio (T/R) were calculated for AUC0–t , AUC0–inf and C max using the least-squares means statement. K el and T ½ el were also analysed using the GLM Procedure. Wilcoxon’s test was performed on the mean T max for both treatments. All statistical tests were performed at the alpha level of 0.05.
According to the regulatory requirements [4] translated into the study protocol, the hypothesis of bioequivalence between a generic medicinal product and a reference medicinal product is accepted if the 90 % geometric confidence intervals of the ratio of least-squares means of the test to reference product of ln-transformed AUC0–t is within the acceptance range of 80.00–125.00 %. For C max, the protocol established a scaled average bioequivalence approach. This approach is based on the CVWR: if the CVWR is inferior or equal to 30 % (≤30 %), the 90 % geometric confidence intervals of the ratio T/R of least-squares means of the ln-transformed C max should be within the acceptable range of 80.00–125.00 % to conclude bioequivalence. On the other hand, if the CVWR for the reference product was superior to 30 % (>30 %) for C max, the bioequivalence acceptance limits for this pharmacokinetic parameter had to be scaled to the within-subject variability of the reference product (to a maximum of 69.84–143.19 %).
For scaled average bioequivalence, the applicant should justify that the calculated CVWR is a reliable estimate and that it is not the result of outliers. Therefore, a box plot analysis using the studentized intra-subject residuals from the ANOVA model including only data for the reference treatment was done using the univariate procedure in SAS®. A box plot was constructed from studentized intra-subject residuals corresponding to the first administration of reference product in each subject. Values that were further away from the box by more than three interquartile ranges were considered outlying observations and these values are indicated by an asterisk in the box plot. The detected outlying observations were excluded, and within subject standard deviation of the log transformed parameter (SWR) was re-estimated and used to widen bioequivalence limits (if eligible) as specified above.
3 Results
3.1 Drug Analysis
3.1.1 Pharmacokinetic Analysis
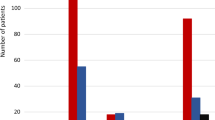
One hundred and fifty-three subjects (47 females and 106 males) were randomized to three sequences of treatment (TRR, RTR and RRT), and received at least one dose of the investigational medicinal products under study. This sample size was considered according to the protocol for safety evaluation (safety population). Nevertheless, as previously stated in the protocol, the subjects used for pharmacokinetic and statistical analysis, the pharmacokinetic population, are those that completed at least two periods including one test and one administration of the reference product and for whom the pharmacokinetic profile was adequately characterized (n = 146). One hundred and forty-two subjects completed all study procedures. The disposition of subjects is presented in Fig. 1.

Disposition of subjects. A (Test) = Tecnimede—Sociedade Técnico—Medicinal S.A., Portugal, ibandronic acid 1 × 150-mg film-coated tablet. B (Reference) = Roche Registration Limited, United Kingdom (Bonviva®), ibandronic acid 1 × 150-mg film-coated tablet
After the test formulation (T) and first and second Bonviva® (R) dosing, the C max was 96.71 ± 90.19 ng/mL, 92.67 ± 91.48 ng/mL and 87.94 ± 60.20 ng/mL and the AUC0–t was 390.83 ± 287.27 ng·h/mL, 388.54 ± 356.76 ng·h/mL and 383.53 ± 246.72 (64.33), respectively (Table 2). No statistically significant difference between treatments was detected using ANOVA for ln-transformed AUC0–t , AUC0–inf and C max. A statistically significant period effect was detected for AUC0–t and AUC0–inf (Table 3). The mean residual area was less than 20 % for the AUCs obtained after administration of the test formulation (3.41 ± 0.84 %) as well as after the first and second administrations of Bonviva® (3.30 ± 0.70 and 3.57 ± 0.95 %, respectively). Mean concentration versus time curves were plotted and are presented in Fig. 2.

Mean plasma ibandronic acid concentrations obtained for the test and reference formulations following a 150-mg dose (log scale). N = 146 for ibandronic acid, N = 146 for Bonviva® (first administration), N = 142 for Bonviva® (second administration), EDTA Ethylene diaminetetraacetic acid
The CVWR for AUC0–t , AUC0–inf and C max were 39.77, 39.45 and 43.23 %, respectively. The limits of the acceptance range based upon the within-subject variability seen in the bioequivalence study using scaled average bioequivalence were 73.01–136.97 %. No statistical outliers were detected for the reference formulation following examination of the distribution of the ln-transformed C max.
The 90 % confidence intervals were 95.05–110.67 for C max, 94.35–107.94 for AUC0–t and 94.37–107.88 for AUC0–inf, which are within the predefined bioequivalence acceptance range of 80.00–125.00 %. For C max, the observed ratio and confidence intervals were also within the limits of acceptance obtained using the scaled average bioequivalence approach. Wilcoxon’s test performed on the t max data showed no statistically significant difference between treatments (p = 0.1382).
The least-squares means ratios, the 90 % geometric confidence intervals, and the CVWR for the reference product are presented in Table 4.
3.2 Safety Analysis
A total of 279 TEAEs were reported by 91 of the 153 subjects who received at least one dose of the medicinal product (safety population): 96 TEAEs were reported by 28.8 % (42/146) of the subjects when they received the test medicinal product (Treatment A) and 183 TEAEs were reported by 47.7 % (73/153) of the 153 subjects when they received the reference medicinal product (Treatment B). Myalgia was reported by 38 subjects, diarrhoea by 22 subjects and abdominal pain by 16 subjects, corresponding to 24.8, 17.6 and 10.5 % of the safety population (n = 153), respectively. After the causality assessment of the 279 TEAEs, 70 were judged as ‘probable/likely’, 176 as ‘possible’ and 33 as ‘unlikely’.
When comparing the number of subjects for each MedDRA® preferred term, there are no relevant differences between treatments with the exception of the headache and myalgia TEAEs, which were reported by 11 and 19 subjects, respectively, after the administration of Treatment A and by 21 and 29 subjects, respectively, after the administration of Treatment B. The severity of each TEAE was graded as mild (n = 223), moderate (n = 50) or severe (n = 6). No serious adverse event was reported in this study.
4 Discussion and Conclusions
Ibandronic acid is a bisphosphonate compound indicated for the treatment and prevention of osteoporosis in post-menopausal women and the reduction of skeletal complications of malignant disease. The absorption in the upper gastrointestinal tract is rapid after oral administration with an absolute bioavailability of about 0.6 %.
A generic medicinal product is considered to be bioequivalent to a reference medicinal product when the 90 % confidence interval around the estimated ratio of geometric means of AUC and C max is between 0.80 and 1.25 [4]. As per regulatory and scientific requirements, when a generic medicinal product and a reference medicinal product are compared, a single-dose, crossover design is recommended [4].
In studies with crossover design, the amplitude of the confidence interval is proportional to the within-subject SD of the pharmacokinetic parameter and reciprocally proportional to the square-root of the number of subjects [5]. Consequently, the regulatory bioequivalence limits of 0.80 and 1.25 are frequently penetrated when the intra-individual variation is high unless the number of subjects is also large.
Ibandronic acid is a highly variable drug and, although the reference literature confirms acceptance of widening of confidence intervals in Europe, based on non-replicate designs [2], the latest update in the bioequivalence guideline requires that, in order to widen the intervals for C max, a replicate design must be used. Besides the fact of allowing for the widening of the intervals for C max, replicate designs possess the advantage of reducing the sample size of subjects required to demonstrate bioequivalence between the two formulations.
However, one of the main issues with a replicate design is the increased duration of the clinical phase of the study, since a full replicate study with four periods will require three washout intervals and a partial replicate with three periods will require two washout intervals. This represents more than three or two times, respectively, the required amount of time to conduct a simple crossover study. Therefore, by increasing the duration of the study and the number of dosing periods, replicate designs normally exhibit a higher dropout rate, which impacts negatively on the required sample size.
The issue with the higher dropout rates is evident by analysing the bibliographic references, which shows 15.8 and 12.5 % dropout rates for full replicate studies [6, 7], while, according to our experience, we achieved a dropout rate of 7.2 % for this partial replicate study and a 4.2 % dropout rate in a pilot crossover study (data on file).
So, in trying to achieve a compromise between an extended duration of the clinical phase and reducing the sample size without much impact from the dropout rate, we decided to conduct this study as a partial replicate design with three periods, including two administrations of the reference formulation in each sequence. This turned out to be a favourable decision since, according to the guidelines [4], the replicate design allowed for the scaled bioequivalence approach for C max and the duration of the clinical phase was contained and acceptable (37 days as opposed to the required 54 days in a four-period full replicate design), which led to a dropout rate lower than the one observed for full replicate studies. Further to this, the results of the study demonstrated that the within-subject variability for C max of the reference formulation was more than 30 % and this value was not the result of the presence of outliers.
However, it is important to point out that a replicate design may not be the solution if high within-subject variability is observed for the AUC parameter, which was not the case for ibandronic acid, since the bioequivalence guideline does not allow for the widening of intervals for that pharmacokinetic parameter [4].
The treatment periods should be separated by a washout period of at least five T ½ el in order to guarantee that the drug concentrations are below the lower limit of quantification at the beginning of each period [4]. In this study, the treatment periods were separated by a washout of 14 days. When reviewing the published data on ibandronic acid pharmacokinetic properties, the authors noticed that the published half-life of ibandronic acid ranges from 10 to 60 hours [1] and, in one study in postmenopausal women that received a single oral dose of ibandronic acid150 mg, a mean T ½ el of 72 hours was observed [8]. In the current study, the T ½ el of ibandronic acid was approximately 10 hours for both formulations, which is in line with published studies but also in the lower limit of the range of values published. This may be explained by the fact that the participants in the study were healthy volunteers since similar half-life values were observed in phase I pharmacokinetic studies with the reference product in healthy subjects.
The mean residual area was less than 20 % for all treatments indicating that a sampling over a period of 48 hours was sufficient.
A statistically significant period effect was detected for AUCs. A statistically significant period effect could be an indication of an equal carryover effect. However, since there was no detectable pre-dose concentration at any of the study periods and there was no sequence effect, there is no indication of carryover effect. As the intra-subject variability was smaller for the AUCs as compared with C max, the power of the study was higher for these parameters. Consequently, small differences between periods could be detected which should not be clinically meaningful.
In this bioequivalence study, all the ratios and 90 % geometric confidence intervals were within the acceptance ranges. The conventional acceptance range of 0.80 and 1.25 was even met for C max (Table 4). Based on these results, it can be concluded that the test formulation of ibandronic acid is bioequivalent to the test reference Bonviva® following a 1 × 150-mg dose under fasting conditions.
The number of subjects reporting TEAE and the number of TEAE reported after intake of reference medicinal product (Treatment B—Bonviva®) is higher than the number of subjects reporting TEAE and the number of TEAE reported following intake of the test medicinal product (Treatment A—test formulation). These differences between treatments can be explained by study design, a reference-replicate crossover study, since all subjects who completed the study received two doses of the reference medicinal product and only one dose of the test medicinal product.
References
Barrett J, Worth E, Bauss F, Epstein S. Ibandronate: a clinical pharmacological and pharmacokinetic update. J Clin Pharmacol. 2004;44(9):951–65.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) European public assessment report (EPAR). Summary of product characteristics for Bonviva (Ibandronic acid). Last Update: 3 April 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000501/WC500052652.pdf.
International Conference on Harmonisation. Guideline for Good Clinical Practice (ICH E6).
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence CPMP/EWP/QWP/1401/98 Rev. 1. 20 January 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf.
Tothfalusi L, Endrenyi L, Arieta AG. Evaluation of bioequivalence for highly variable drugs with scaled average bioequivalence. Clin Pharmacokinet. 2009;48(11):725–43.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) European public assessment report (EPAR) for ibandronic acid Sandoz. Issued: 17 February 2011. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002367/WC500109886.pdf.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) European public assessment report (EPAR) for ibandronic acid Teva. Issued: 17 September 2010 http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001195/WC500097557.pdf.
Reginster JY, Wilson KM, Dumont E, Bonvoisin B, Barrett J. Monthly oral ibandronate is well tolerated and efficacious in postmenopausal women: results from the monthly oral pilot study. J Clin Endocrinol Metab. 2005;90(9):5018–24.
Acknowledgements
Conflict of Interest
Tecnimede is the Sponsor of this study. Augusto Filipe, Pedro Pedroso, Susana Almeida and Rita Neves are employees of the Sponsor of this study. Sylvie Boudreault is an employee of the contract research organization contracted to perform this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Filipe, A., Pedroso, P., Almeida, S. et al. Bioequivalence Study of Two Formulations of Ibandronic Acid 150-mg Film-Coated Tablets in Healthy Volunteers Under Fasting Conditions: A Randomized, Open-Label, Three-Way, Reference-Replicated Crossover Study. Drugs R D 14, 105–112 (2014). https://doi.org/10.1007/s40268-014-0044-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-014-0044-x