
Abstract
Background
Ruxolitinib cream is a topical formulation of ruxolitinib, an inhibitor of Janus kinase 1 and Janus kinase 2.
Objective
We aimed to determine the safety, tolerability, and bioavailability of 1.5% ruxolitinib cream under maximum-use conditions in patients with atopic dermatitis. Efficacy was evaluated as an exploratory objective.
Methods
Eligible patients aged ≥ 12–65 years with atopic dermatitis, an Investigator’s Global Assessment score ≥ 2, and ≥ 25% affected body surface area were enrolled in an open-label, maximum-use phase I study conducted in the USA and Canada. Patients applied 1.5% ruxolitinib cream twice daily to lesions identified at baseline for the first 28 days and continued use only on active lesions for an additional 28 days (extension period). Safety was assessed by frequency, duration, and severity of treatment-emergent adverse events. Plasma concentrations of ruxolitinib and pharmacokinetic parameters were assessed as secondary endpoints.
Results
Overall, 41 patients (median age, 17 years; 51% male) were enrolled and 37 (90.2%) entered the extension period, all of whom completed the study. Treatment-emergent adverse events were reported in 13 patients (31.7%). Treatment-related adverse events were reported in four patients (9.8%). The mean (standard deviation) steady-state plasma concentration was 104 (309) nM during the first 28 days, well below the half-maximal inhibitory concentration of Janus kinase-mediated myelosuppression in the bone marrow (281 nM), and decreased further during the extension period. Higher plasma concentrations were detected in a few patients who were treated for a very high affected body surface area. At day 56, 94.6% of patients achieved ≥ 75% improvement in the Eczema Area and Severity Index.
Conclusions
Under maximum-use conditions, ruxolitinib cream was generally well tolerated, with approximately one-third of patients experiencing treatment-emergent adverse events and few treatment-related adverse events. The mean steady-state plasma concentration of ruxolitinib was well below the level expected to affect bone marrow production of blood cells, with a small number of patients exhibiting higher plasma concentrations. In addition, ruxolitinib cream showed a high level of efficacy in patients with atopic dermatitis involving ≥ 25% affected body surface area.
ClinicalTrials.gov Identifier
NCT03920852.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In this open-label, phase I, maximum-use trial with twice-daily 1.5% ruxolitinib cream in 41 patients with atopic dermatitis, 31.7% experienced a treatment-emergent adverse event, with 9.8% experiencing treatment-related adverse events. |
The mean steady-state plasma concentration of ruxolitinib remained consistently below the half-maximal inhibitory concentration of Janus kinase-mediated myelosuppression. |
Ruxolitinib cream substantially reduced inflammation and itch in patients with atopic dermatitis involving ≥ 25% affected body surface area. |
1 Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disease that substantially impacts patients’ quality of life, in large part because of inadequately controlled chronic itch [1, 2]. Most patients with AD can be effectively managed without systemic therapy [3], and some patients or physicians may prefer topical over systemic treatment, even for patients with large body surface area (BSA) involvement. Commonly used topical treatments include corticosteroids and calcineurin inhibitors, as well as the phosphodiesterase inhibitor, crisaborole ointment [4, 5].
Janus kinases (JAKs) transmit intracellular signals of inflammatory cytokines involved in the pathogenesis of AD and may directly control itch [6, 7]. Ruxolitinib has been demonstrated to be a potent, selective inhibitor of JAK1 and JAK2 in preclinical models [8]. A topical formulation was developed to maximize its clinical effect on the skin and minimize the likelihood of systemic absorption. In studies in adults and adolescents with AD (affected BSA of 3–20% and Investigator’s Global Assessment [IGA] score of 2 or 3), 1.5% ruxolitinib cream twice daily (BID) demonstrated substantial anti-inflammatory activity with antipruritic action vs vehicle and was well tolerated [9,10,11]. Ruxolitinib cream is approved by the US Food and Drug Administration for treating patients 12 years of age or older with mild-to-moderate AD with application to affected areas of up to 20% BSA [12].
The purpose of a maximum-use trial (MUsT) is to evaluate the safety of a topical product when used in exaggerated conditions (e.g., large surface areas for an extended period of time) [13]. Such studies also allow insight into possible efficacy of a topical product in patients with more severe disease who would otherwise be candidates for systemic therapy. The objective of this MUsT was to determine the bioavailability, safety, and tolerability of 1.5% ruxolitinib cream applied BID for 4 weeks in patients with extensive AD lesions. Efficacy was evaluated as an exploratory objective.
2 Methods
2.1 Study Design and Patients
An open-label MUsT of adolescents and adults with AD was conducted at one site in Canada and six sites in the USA from 20 June to 26 December, 2019. Eligible patients were aged ≥ 12–65 years with AD for ≥ 2 years, an IGA score ≥ 2, and BSA involvement ≥ 25%. Patients with an unstable course of AD (as determined by the investigator), other types of eczema, immunocompromised status, or any serious illness or medical condition that could interfere with study conduct, interpretation of data, or patients’ well-being were excluded from the study. Use of AD drugs during the washout period before baseline (biologics, five half-lives or 12 weeks; systemic immunomodulating agents, 4 weeks; topicals, 1 week) and during the study was prohibited.
Enrolled patients continuously applied 1.5% ruxolitinib cream BID for 28 days on all AD lesions identified at baseline (except lesions on the scalp), regardless of the reduction in lesion size. Tubes were counted and weighed at each study visit to assess adherence. At day 28, patients with no safety concerns could continue to apply 1.5% ruxolitinib cream BID, but only to active lesions, for an additional 28 days (optional). All patients had follow-up assessments 30 days after the last application of ruxolitinib cream. If patients used moisturizers before enrollment, they could continue throughout the study.
This study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. Written informed consent/assent was provided by all patients before enrollment. The protocols were approved by the relevant institutional review board or ethics committee at each study site.
2.2 Assessments
The primary objective was to assess the safety and tolerability of ruxolitinib cream. Safety was evaluated by the frequency, duration, and severity of adverse events (AEs). Changes from baseline in physical examinations, vital signs, and laboratory data for hematology, serum chemistry, and urinalysis were also analyzed. Secondary endpoints included the assessment of plasma concentrations of ruxolitinib and pharmacokinetic (PK) parameters, including maximum concentration (Cmax), time to Cmax, and area under the curve from 0 to 12 h. Blood samples for PK assessments were collected on days 1 (pre-application, 1, 2, 4, and 12 h post-application), 15 (pre-application and 1 h post-application), and 28 (pre-application, 1, 2, 4, and 12 h post-application). Exploratory endpoints included the proportion of patients who achieved IGA treatment success (IGA score of 0/1 with a ≥ 2-grade improvement from baseline), proportion of patients with a ≥ 75% improvement in the Eczema Area and Severity Index (EASI-75), proportion of patients with a ≥ 4-point improvement in the itch Numerical Rating Scale (NRS4) score, total affected BSA, and EASI score. Efficacy endpoints were measured on days 15, 28, and 56. Mean change from baseline in daily itch NRS score through day 56 was also assessed.
2.3 Statistical Analyses
The sample size was based on other PK studies with ruxolitinib cream and was not based on statistical power calculations. Enrollment of approximately 40 patients was to ensure ≥ 32 evaluable patients, ≥ 12 of whom were aged 12–17 years (with ≥ 6 aged 12–15 years). The sample size was based on feasibility, a common practice for this type of study, and was considered adequate to achieve the objectives of the study, including characterization of PK parameters after a single application and at steady state [13,14,15]. Patient demographics, baseline characteristics, patient disposition, efficacy, and all safety analyses were conducted using the full analysis set (all patients enrolled in the study with one or more applications of ruxolitinib cream). Data were analyzed by descriptive statistics using SAS® software (SAS Institute Inc., Cary, NC, USA; version 9.4). Sample size, frequency, and percentages were used for categorical measurements; sample size, mean, median, standard deviation, standard error of the mean, range, and interquartile range (IQR) were calculated for continuous measurements. Data for continuous measurements are reported at baseline and at each visit (measured values, change from baseline, and percentage change from baseline, if applicable). The PK-evaluable population consisted of all patients with one or more applications of ruxolitinib cream and one or more post-application plasma PK samples with measurable concentrations of ruxolitinib. Pharmacokinetic concentration data were analyzed using descriptive statistics. Patients were also stratified based on the total affected BSA at baseline (25% to < 40% and ≥ 40%) and/or patient age (12–15, 16–17, and ≥ 18 years). Pharmacokinetic parameters were calculated from serial PK concentration data on days 1 and 28 by a noncompartmental analysis (Phoenix® WinNonlin® version 8.2; Certara, Princeton, NJ, USA). The steady-state plasma concentration (Css) of ruxolitinib was calculated as the average of all plasma concentrations across visits on days 15 and 28.
3 Results
3.1 Patients
Forty-one patients were treated, with a median (IQR) age of 17 (15–36) years; 51.2% were male, and 68.3% were white (Table 1). The median affected BSA at baseline was 31.2% (IQR, 28.6–44.0%; range, 25.0–90.0%), and the mean (standard deviation [SD]) baseline EASI score was 20.8 (9.2). One patient had a treatment-emergent AE (TEAE) that led to study discontinuation (increased liver function test before the first treatment application, not considered related to treatment). One other patient discontinued, with the recorded reason being “Other.” Two additional patients completed the first treatment period but did not enter the extension period (reasons unknown). Thirty-seven patients (90.2%) continued with therapy in the extension period (days 28–56). Per the protocol, all of these patients were considered to have completed the study, regardless of whether they completed 28 days of treatment during the extension period. The daily amount of ruxolitinib cream applied (mean [SD]) decreased from the first 28-day period (21.8 [13.1] g) to the extension period (9.0 [7.5] g), consistent with the treatment of only active lesions during the extension period.
3.2 Safety
Approximately one-third of patients (31.7%) experienced TEAEs, most of which were mild or moderate in severity (Table 2). One patient had a serious AE not on a treated area (abscess on the lower extremity [investigator determined likely due to a previous tendon surgery in that area approximately 3 months before baseline]), which resolved and was considered unrelated to treatment. No fatal TEAEs were reported. Treatment-emergent adverse events determined by the investigator to be possibly related to treatment were experienced by four patients (9.8%), for a total of six treatment-related TEAEs (all grade 1 [neutropenia, n = 1; aspartate aminotransferase increase, n = 2; alanine aminotransferase increase, n = 1] or grade 2 [dyspnea, n = 1; hemoglobin decrease, n = 1]). The patients with neutropenia and decreased hemoglobin both had plasma concentrations of ruxolitinib well below the ex vivo half-maximal inhibitory concentration (IC50) for inhibition of thrombopoietin (TPO)-stimulated phosphorylation of STAT3 (281 nM) in human whole blood, a surrogate marker to evaluate the potential effects on bone marrow production of blood cells (i.e., platelets) [8]. Both patients also had low neutrophil or hemoglobin levels, respectively, at baseline (Table 2). One patient, noted above, discontinued from the study because of an AE (sponsor and medical monitor’s decision) not considered to be related to treatment. This patient had increased baseline levels in alanine aminotransferase (43 IU/L; grade 1), aspartate aminotransferase (117 IU/L; grade 2), and lactate dehydrogenase (318 IU/L, grade 1). Three patients (as described above) had AEs that resulted in treatment interruptions (grade 1 neutropenia considered by the investigator as related to treatment, grade 2 dyspnea considered related to treatment when applying to lesions on the face [nonsmoker with no history of asthma or hay fever], and grade 3 lower extremity abscess considered not related to treatment). Minimal changes (decreases) in mean hemoglobin within the normal range were observed, as well as transient increases in platelets at day 15 that returned to baseline levels by day 28 (Fig. 1).

Hematologic parameters. Boxes represent the interquartile range, the middle lines represent the median, and the “X” inside the boxes represents the mean. Bars represent data within 1.5× the interquartile range of either quartile 1 or quartile 3. Circles outside the bars represent outliers
3.3 Pharmacokinetic Data
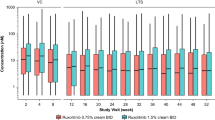
The mean (SD) Css of ruxolitinib was 104 (309) nM with a geometric mean of 26.5 nM (geometric coefficient of variation, 321%) during the first 28-day period in which all lesions at baseline were treated (Table 3). The overall mean plasma concentration peaked at 4 h after topical application on day 1 (Fig. S1a of the ESM) at 241 nM, with lower peaks among patients treated for < 40% BSA (Fig. S1b of the ESM) and adolescents vs adults (Fig. S1c of the ESM). The overall median trough plasma concentration was stable during the first 28 days and was lower on day 56 (Fig. 2a), likely due to the treatment of only active lesions during the second 28-day period, which had decreased substantially since baseline (Fig. 2b); concentrations were lower in patients treated for < 40% BSA (Fig. 2c), also likely due to fewer or a smaller area of treated lesions (Fig. 2d). The mean PK concentration–time profiles on day 28 were relatively flat (Fig. S1d of the ESM), with lower concentrations in patients treated for < 40% BSA (Fig. 1e). Overall, plasma concentrations were lower and the lesional area was smaller in adolescents vs adults (Fig. 2e, Fig. S1f of the ESM), consistent with the smaller treated area in adolescents (and less drug applied; Fig. 2f, Table 3). The peak plasma concentrations at steady state on day 28 were considerably lower than those observed after oral administration of ruxolitinib (15 mg BID) in healthy volunteers on day 10 in a phase I study [16] (Fig. 3). Modest peak/trough ratios were observed at day 28 (mean Cmax/minimum concentration, 2.72; Table 3).

Boxplots of plasma trough concentrations and affected body surface area (BSA) in the (A, B) overall population and by (C, D) baseline BSA and (E, F) age groups. Boxes represent the interquartile range, the middle lines represent the median, and the bars indicate data within 1.5× the interquartile range of either quartile 1 or quartile 3

Steady-state plasma concentrations of ruxolitinib from ruxolitinib cream (1.5% twice daily [BID]) on day 28 vs oral ruxolitinib (15 mg BID) on day 10. Error bars represent standard error of the mean. The data for oral ruxolitinib were included in the phase I publication [16]. AD atopic dermatitis, IC50 half-maximal inhibitory concentration, TPO thrombopoietin
Five patients with the highest mean Css of ruxolitinib were further examined (Table S1 of the ESM). Of these patients, two patients with baseline affected BSA of 45% and 90% (average daily amount of 1.5% ruxolitinib applied during first 28 days, 45.5 g and 75.2 g, respectively; application rate, 2.5 and 2.2 mg/cm2) had Css exceeding the IC50 for TPO-mediated pSTAT3. In these two patients, plasma concentrations were above the IC50 for TPO-stimulated STAT3 phosphorylation on days 1, 15, and 28, and were below this threshold on day 56 (pre-application). No hematologic AEs were observed in these five patients with the highest mean Css of ruxolitinib during the full study period; one patient had a 57% reduction in platelets on day 28, but remained within the normal range throughout the study. Two of these patients reported no AEs during the study; one patient experienced grade 1 alanine aminotransferase (baseline, 18 IU/L; day 15, 94 IU/L) and aspartate aminotransferase elevations (baseline, 30 IU/L; day 15, 67 IU/L) that were considered related to treatment and resolved without dose modification; one patient (described above as a serious AE) had a grade 3 limb abscess considered unrelated to treatment; and one patient had peripheral edema and skin rash (both grade 1) that were considered unrelated to treatment and resolved without dose modification.
Mean (SD) systemic bioavailability was 2.5% (3.6%). There was no increase in Cmax, concentration at 12 h post-application, and area under the curve from 0 to 12 h between day 1 and day 28, suggesting no ruxolitinib accumulation in plasma after repeated applications of ruxolitinib cream BID for 28 days (Table 3). There was no consistent pattern between change in hematologic laboratory parameters and plasma exposure except a transient increase in platelet count on day 15 (first study visit) that was plasma ruxolitinib concentration dependent. The mean platelet counts spontaneously decreased with continuous treatment and returned to (or near) baseline values by day 28 (the next visit).
3.4 Efficacy
The proportion of patients achieving IGA treatment success at day 15 was 20.0% and increased to 35.9% and 56.8% at days 28 and 56, respectively. Most patients had clear or almost clear skin (IGA score of 0/1) by day 28 (61.5%), with a further increase at day 56 (81.1%; Fig. S2 of the ESM); no patient had an IGA score of 0/1 at baseline. The mean (SD) total affected BSA at days 15, 28, and 56 was 15.8% (11.4%), 6.5% (8.2%), and 3.1% (5.4%), respectively; for context, the mean (SD) BSA at baseline was 38.1% (16.3%). The onset of itch relief was rapid, with a mean change from baseline in itch NRS, as measured through patient-reported daily diaries, of − 1.9 at day 1 (approximately 12 h after the first application; Fig. S3 of the ESM). Mean itch NRS continued to decrease; the mean change was − 5.3 at day 20, and reductions were maintained for the remainder of the study. The proportions of patients achieving NRS4 at days 15, 28, and 56 were 58.6%, 82.6%, and 90.5%, respectively. The mean (SD) EASI score at days 15, 28, and 56 was 6.3 (5.2), 2.4 (2.3), and 1.3 (2.2), respectively; for context, the mean (SD) score at baseline was 20.8 (9.2). The proportions of patients achieving EASI-75 at days 15, 28, and 56 were 30.0%, 79.5%, and 94.6%, respectively (Fig. S4 of the ESM). Representative clinical images are shown in Fig. S5 of the ESM.
4 Discussion
This study evaluated the safety, pharmacokinetics, and efficacy of ruxolitinib cream under maximum conditions of use in patients with extensive AD lesions (BSA involvement ≥ 25%). Ruxolitinib cream was generally well tolerated, with most AEs of mild or moderate severity. Transient increases in mean platelet count at day 15 were observed, which were within the normal range and spontaneously returned to baseline levels by day 28. The underlying mechanism is not clear; however, the increased platelet counts did not appear to have clinical implications.
The mean maximal plasma concentration of ruxolitinib at 4 h after topical application on day 1 was 241 nM. At steady state, mean ruxolitinib plasma concentrations were relatively flat over the dosing interval and were lower than the peak on day 1. Over the first 28 days of treatment, during which treated BSA and daily dose of the product were kept constant, plasma concentrations of ruxolitinib showed no significant change (i.e., no sign of accumulation of ruxolitinib in the systemic circulation upon repeated twice-daily applications of the cream). The ruxolitinib plasma concentrations were related to the amount of ruxolitinib cream applied and/or percentage of treated BSA, and the relationship was sublinear, which is consistent with a previous report [17]. The observed decrease in trough plasma concentrations of ruxolitinib beyond day 28 was consistent with the substantial decrease in percentage of affected BSA and the instruction to only treat affected areas after day 28. Plasma concentrations were lower in adolescents than adults, consistent with lower average affected BSA in adolescents.
The mean daily application amount of 1.5% ruxolitinib cream over the first 4 weeks was approximately 3.7-fold higher vs the phase III studies (20.2 vs 5.4 g; mean application rate of 1.5 mg/cm2 for both), which only included patients with BSA ≤ 20% at baseline, and the mean Css was approximately 2.9-fold higher (104 vs 35.7 nM) [17]. However, the overall bioavailability of ruxolitinib in this study was 2.5% and was previously reported to be 7.7% for 0.75% ruxolitinib cream and 6.2% for 1.5% ruxolitinib cream in phase III studies [17]. The lower bioavailability in this MUsT may reflect the previously reported sublinear increase in exposure as the amount of drug product applied is increased (i.e., the power index of the correlation between the amount of drug product applied vs the plasma concentration is < 1) [17]. The small sample size (n = 41) and high interpatient variability (> 100% coefficient of variation) in bioavailability are also possible factors leading to the lower bioavailability observed in this study.
The peak plasma concentrations at steady state reported in this study were considerably lower than those observed after oral administration of ruxolitinib (15 mg BID) in healthy volunteers [16], with the exception of two patients (treated BSA, 45% and 90%). In clinical practice, patients with very high affected BSA are not usually treated with topical therapy alone. For these patients, clinicians should monitor the amount used and ensure that patients stop treating areas that are clear. In the overall study population, the mean Css of ruxolitinib cream over the first 4 weeks of treatment (104 nM) was well below the IC50 for TPO-mediated pSTAT3 (281 nM) [8]. Thrombopoietin-induced pSTAT3 is driven by JAK2 and thus serves as a proxy parameter to assess JAK-related myelosuppression in bone marrow [8]. Two patients had Css above the IC50 for TPO-mediated pSTAT3 but did not experience hematologic AEs, although one patient had a 57% reduction in platelets within the normal range. The current data suggest that a longer duration of treatment with ruxolitinib cream does not result in any further increase in Css over time, especially as affected BSA is reduced because of lesion clearance from treatment. Thus, ruxolitinib plasma concentrations would be expected to progressively diminish in line with the decrease in size of active lesions, as observed during the treatment extension period.
As is customary for maximum-use studies, this study used an open-label design and therefore lacked a vehicle control. The limited sample size precluded the ability to draw firm conclusions for efficacy outcomes; however, this sample size has conventionally been adequate for establishing PK information. The small number of patients and the short duration of the study did not allow for the capture of potential rare long-term AEs.
Despite the small sample size, efficacy results were similar to improvements observed in phase III studies [11], which only included patients with BSA ≤ 20% at baseline. A majority of patients achieved EASI-75 and itch NRS4 by day 28 in this MUsT, suggesting that patients with more extensive disease may benefit from short-term treatment with ruxolitinib cream. Although less ruxolitinib cream was applied in the extension period, patients continued to improve, with 94.6% of patients achieving EASI-75 at day 56, indicating that patients may be able to maintain response by limited application of ruxolitinib cream to active lesions. The high response rate and maintenance of response when only active lesions were treated suggest that ruxolitinib cream could be considered as an alternative to systemic treatments in some patients with AD and high BSA involvement, although additional studies in this patient population are needed.
5 Conclusions
The results from this MUsT, in combination with previously reported phase III data [11], support ruxolitinib cream as a well-tolerated and effective topical treatment for patients with AD, with low bioavailability and Css well below the IC50 for JAK-mediated myelosuppression for patients with affected BSA < 40%. Higher plasma concentrations of ruxolitinib are likely in patients with very high affected BSA, although no hematologic AEs were observed in this study among patients with the highest mean Css.
References
Wei W, Anderson P, Gadkari A, Blackburn S, Moon R, Piercy J, et al. Extent and consequences of inadequate disease control among adults with a history of moderate to severe atopic dermatitis. J Dermatol. 2018;45(2):150–7.
Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, et al. Patient-burden and quality of life in atopic dermatitis in US adults: a population-based cross-sectional study. Ann Allergy Asthma Immunol. 2018;121(3):340–7.
Simpson EL, Bruin-Weller M, Flohr C, Ardern-Jones MR, Barbarot S, Deleuran M, et al. When does atopic dermatitis warrant systemic therapy? Recommendations from an expert panel of the International Eczema Council. J Am Acad Dermatol. 2017;77(4):623–33.
Eichenfield LF, Tom WL, Berger TG, Krol A, Paller AS, Schwarzenberger K, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116–32.
Wollenberg A, Barbarot S, Bieber T, Christen-Zaech S, Deleuran M, Fink-Wagner A, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657–82.
Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013;2(3):e24137.
Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. 2017;171(1):217–28.
Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–17.
Kim BS, Howell MD, Sun K, Papp K, Nasir A, Kuligowski ME, et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J Allergy Clin Immunol. 2020;145(2):572–82.
Kim BS, Sun K, Papp K, Venturanza M, Nasir A, Kuligowski ME. Effects of ruxolitinib cream on pruritus and quality of life in atopic dermatitis: results from a phase 2, randomized, dose-ranging, vehicle- and active-controlled study. J Am Acad Dermatol. 2020;82(6):1305–13.
Papp K, Szepietowski JC, Kircik L, Toth D, Eichenfield L, Leung DY, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: results from two phase 3, randomized, double-blind studies. J Am Acad Dermatol. 2021;85(4):863–72.
Opzelura™ (ruxolitinib cream). Full prescribing information. Wilmington (DE): Incyte Corporation; 2021.
Bashaw ED, Tran DC, Shukla CG, Liu X. Maximal usage trial: an overview of the design of systemic bioavailability trial for topical dermatological products. Ther Innov Regul Sci. 2015;49(1):108–15.
Jett JE, McLaughlin M, Lee MS, Parish LC, DuBois J, Raoof TJ, et al. Tapinarof cream 1% for extensive plaque psoriasis: a maximal use trial on safety, tolerability, and pharmacokinetics. Am J Clin Dermatol. 2022;23(1):83–91.
Zane LT, Kircik L, Call R, Tschen E, Draelos ZD, Chanda S, et al. Crisaborole topical ointment, 2% in patients ages 2 to 17 years with atopic dermatitis: a phase 1b, open-label, maximal-use systemic exposure study. Pediatr Dermatol. 2016;33(4):380–7.
Shi JG, Chen X, McGee RF, Landman RR, Emm T, Lo Y, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol. 2011;51(12):1644–54.
Gong X, Chen X, Kuligowski ME, Liu X, Liu X, Cimino E, et al. Pharmacokinetics of ruxolitinib in patients with atopic dermatitis treated with ruxolitinib cream: data from phase II and III studies. Am J Clin Dermatol. 2021;22(4):55–66.
Acknowledgments
The authors thank the patients and their families as well as investigators and site personnel. The authors also thank Evan Cimino, Brad Yuska, Xing Liu, and Susan Lockhead for their work in support of the ruxolitinib cream clinical development program, as well as May Venturanza, Michael Kuligowski, Kofi Wagya, Jim Lee, and Jennifer Lofland for their contributions to the article. Writing assistance was provided by Tania Iqbal, PhD, and Joshua Solomon, PhD, of ICON (Blue Bell, PA, USA), and was funded by Incyte Corporation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Incyte Corporation (Wilmington, DE, USA).
Conflicts of interest/Competing interests
RB has served as an advisory board member, consultant, speaker, and/or investigator and received honoraria and/or grants from AbbVie, Arcutis, Arena Pharma, Aristea, Asana BioSciences, Bellus Health, Bluefin Biomedicine, Boehringer Ingelheim, CARA, Dermavant, Eli Lilly, EMD Serono, Evidera, Galderma, GlaxoSmithKline, Incyte, Inmagene Bio, Kiniksa, Kyowa Kirin, LEO Pharma, Novan, Pfizer, Ralexar, RAPT Therapeutics, Regeneron, Respivant, Sanofi-Genzyme, Sienna, Target RWE, and Vyne Therapeutics and is an employee and shareholder of Innovaderm Research. RSC has served as a principal investigator for Incyte Corporation. TR has served as an investigator for AbbVie, Arcutis, AstraZeneca, Bausch Health, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, Dermira, Galderma, GlaxoSmithKline, Eli Lilly, Incyte Corporation, Janssen, LEO Pharma, Pfizer, Regeneron, Sanofi-Genzyme, and UCB Biopharma. ZZ was an employee and shareholder of Incyte Corporation at the time of this study. SY and XG are employees and shareholders of Incyte Corporation. ML is an investigator for AbbVie, Arcutis, AstraZeneca, Bausch Health, Boehringer Ingelheim, Boston Pharmaceuticals, Bristol Myers Squibb, Dermavant, Dermira, Eli Lilly, Incyte Corporation, Janssen, Kiniksa Pharmaceuticals, LEO Pharma, Pfizer, RAPT Therapeutics, Reistone Biopharma, Regeneron, and UCB Biopharma.
Ethics approval
This study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The protocols were approved by the relevant institutional review board or ethics committee at each study site.
Consent to participate
Written informed consent/assent was provided by all patients before enrollment.
Consent for publication
Written informed consent/assent was provided by all patients before enrollment.
Availability of data and material
Incyte Corporation (Wilmington, DE, USA) is committed to data sharing that advances science and medicine while protecting patient privacy. Qualified external scientific researchers may request anonymized datasets owned by Incyte for the purpose of conducting legitimate scientific research. Researchers may request anonymized datasets from any interventional study (except phase I studies) for which the product and indication have been approved on or after 1 January, 2020 in at least one major market (e.g., USA, European Union, Japan). Data will be available for request after the primary publication or 2 years after the study has ended. Information on Incyte’s clinical trial data sharing policy and instructions for submitting clinical trial data requests are available at: https://www.incyte.com/Portals/0/Assets/Compliance%20and%20Transparency/clinical-trial-data-sharing.pdf?ver=2020-05-21-132838-960.
Code availability
Not applicable.
Authors’ contributions
RB contributed to the study design and data collection. RSC and TR contributed to data collection. ZZ and SY contributed to the study design and data analysis. XG contributed to data analysis. ML contributed to the study design. All authors contributed to drafting and critical appraisal of the manuscript and approved the final version for submission.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bissonnette, R., Call, R.S., Raoof, T. et al. A Maximum-Use Trial of Ruxolitinib Cream in Adolescents and Adults with Atopic Dermatitis. Am J Clin Dermatol 23, 355–364 (2022). https://doi.org/10.1007/s40257-022-00690-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-022-00690-3