
Abstract
Alcoholic liver disease (ALD) is major cause of chronic liver injury which results in liver fibrosis and cirrhosis. According to the surveillance report published by the National Institute on Alcohol Abuse and Alcoholism, liver cirrhosis is the 12th leading cause of death in the United States with 48 % of these deaths being attributed to excessive alcohol consumption. ALD includes a spectrum of disorders from simple steatosis to steatohepatitis, fibrosis, and hepatocellular carcinoma. Several mechanisms play a critical role in the pathogenesis of ALD. These include ethanol–induced oxidative stress and depletion of glutathione, pathological methionine metabolism, increased gut permeability and release of endotoxins into the portal blood, recruitment and activation of inflammatory cells including bone marrow-derived and liver resident macrophages (Kupffer cells). Chronic alcohol consumption results in liver damage and activation of hepatic stellate cells (HSCs) and myofibroblasts, leading to liver fibrosis. Here we discuss the current view on factors that are specific for different stages of ALD and those that regulate its progression, including cytokines and chemokines, alcohol-responsive intracellular signaling pathways, and transcriptional factors. We also review recent studies demonstrating that alcohol-mediated changes can be regulated on an epigenetic level, including microRNAs. Finally, we discuss the reversibility of liver fibrosis and inactivation of HSCs as a potential strategy for treating alcohol-induced liver damage.
Similar content being viewed by others

Introduction
Alcoholic Liver Disease (ALD)
Chronic alcohol use is a major cause of cirrhosis and liver failure and is the 12th leading cause of death in adult patients in the United States [1••]. ALD progresses from a healthy liver, to steatosis, alcoholic steatohepatitis, fibrosis, and finally cirrhosis, and hepatocellular carcinoma (HCC) [2]. Histologically it is manifested by hepatocyte steatosis, ballooning and apoptosis, lobular inflammation, deposition of extracellular matrix (ECM) and cirrhosis with formation of regenerative nodules [2, 3]. Development of alcohol-induced liver cirrhosis is associated with high levels of proinflammatory and profibrogenic cytokines, increased portal hypertension [2–4], hepatocyte dysplasia, HCC, and hepatic failure. However, only 35 % of alcoholics develop advanced ALD, suggesting that other co-factors are also important for ALD progression. These risk factors include sex, obesity, drinking patterns, genetic and metabolic factors, and cigarette smoking [1••]. Females are more susceptible to alcohol-induced liver damage. Obesity is an important risk factor that synergistically facilitates alcohol-induced damage of hepatocytes, modulating the response of the endoplasmic reticulum and mitochondria to injury and stress, promoting activation of pro-inflammatory macrophages, and causing resistance to insulin and adiponectin [1••, 5••, 6]. Recently identified genetic factors include genetic polymorphism of patatin-like phospholipase domain-containing protein 3 (PNPLA3), whose expression affects the development of alcoholic cirrhosis in patients with ALD [1••, 7–9]. This PNPLA3 polymorphism is also a risk for NAFLD, suggesting a connection between NAFLD and ALD. Finally, environment and dietary habits can also affect ALD progression in alcoholics. Several injury-triggered events play a critical role in the pathogenesis of ALD, which are discussed below.
Stages of ALD
Alcoholic Fatty Liver
Alcoholic fatty liver is an early response to alcohol consumption, develops in more than 90 % of heavy drinkers. Early-mild steatosis occurs in zone 3 (perivenular) hepatocytes; it can also affect zone 2 and even zone 1 (periportal) hepatocytes during ALD progression [1••]. Fatty liver is characterized by the accumulation of fat droplets (mainly triglycerides and phospholipids) in hepatocytes. Alcoholic fatty liver results from a direct (via acetaldehyde) or indirect (via up-regulation of cytochrome P450 2E1) effect of ethanol on up-regulation of SREBP-1c and down-regulation of PPAR-α expression, leading to the induction of fatty acid synthesis and inhibition of β-oxidation [1••, 10]. Alcohol consumption could directly increase transcription of SREBP-1c gene via its metabolite acetaldehyde [11] or indirectly up-regulate SREBP-1c expression and processing, by activating processes and factors that stimulate SREBP-1c expression, such as the endoplasmic reticulum response to cell stress [12, 13], adenosine [14], endocannabinoids [15, 16], LPS signaling via Toll-like receptor (TLR) 4, and its downstream mediators, including IRF-3, Egr-1, or tumor necrosis factor (TNF)-α [17–19, 20••] (reviewed in [1••]). Alcohol also down-regulates factors that reduce SREBP-1c expression, such as AMP-activated protein kinase (AMPK) and Sirtuin1 [21, 22], adiponectin [23, 24], and signal transducer and activator of transcription 3 (STAT3) [20••, 25••, 26, 27] Consistent with this notion, SREBP-1c knockout mice are protected from alcohol-induced fatty liver [27, 28]. In addition several other genes have been identified to play a critical role in regulation of fatty liver disease; and the following knockout mice: HIF-1−/− [29], C3−/− [30], C1qa−/− [31], PKCε−/− [32], and iNOS,−/− [33] develop less steatosis, demonstrating the contribution of these molecules to the pathogenesis of alcoholic fatty liver [1••]. Finally, autophagy has an important role in regulation of steatosis in hepatocytes, and is inhibited during chronic alcohol consumption, resulting in inability to remove lipid droplets from damaged hepatocytes, but is activated during acute alcohol consumption, which may play a compensatory role in preventing acute alcohol-induced liver damage [1••, 20••, 34–36].
Pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharides (LPS) represent the products of intestinal bacteria and activate inflammasome signaling pathways that promote migration of innate immune and vascular cells to sites of injury. In contrast, the process by which injured cells release endogenous danger-associated molecular patterns (DAMPs) that recruit inflammatory cells in absence of infection has been referred to as “sterile inflammation” [37–40]. Recent studies have outlined the synergistic role of sterile inflammation in angiogenesis and HSC activation. High-mobility group box 1 (HMGB1) [41] is a pro-inflammatory mediator released from the nucleus of ethanol-damaged liver parenchymal cells that stimulates recruitment of HSC and liver endothelial cells (LEC) to the site of injury. HMGB1 is an intracellular DNA-binding protein expressed by all mammalian cells that in response to ethanol exposure is translocated from the nucleus of injured hepatocytes into the extracellular space. It interacts with various receptors including TLR2, TLR4, TLR9, and RAGE (receptor for advanced glycation end products) [40], but mediates its pro-fibrogenic signals via TLR4. HSCs and LEC are immediate responders to hepatocyte apoptosis that play a critical role in initiation a cascade of events leading to liver fibrosis in association with angiogenesis [42].
Alcoholic Steatohepatitis (ASH)
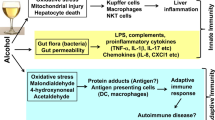
ASH is a syndrome characterized by infiltration of inflammatory cells and by hepatocellular injury. ASH, which develops in 10–35 % of alcoholic patients with steatosis, is usually associated with progressive fibrosis, and is manifested by steatosis, centrilobular ballooning of hepatocytes, neutrophilic infiltration, and Mallory–Denk hyaline inclusions [1••, 43]. In response to chronic alcohol consumption, lipid peroxidation and mitochondrial damage cause apoptosis of hepatocytes, which triggers recruitment of inflammatory cells to the fatty liver, activation of bone marrow-derived and liver resident macrophages (Kupffer cells) and release of pro-inflammatory cytokines (IL-6, IL-1β, TNF-α). Infiltrating neutrophils kill sensitized hepatocytes and further exacerbate alcohol-induced liver injury, which is a prominent feature of ASH. Up-regulation of IL-8, CXCL1 (Gro-α), and IL-17 in the liver contributes to neutrophil infiltration and correlates with the severity of ASH [43, 44]. IL-17 not only acts as a neutrophil chemoattractant, but also stimulates HSCs to produce IL-8 and CXCL1 to facilitate neutrophil recruitment [45, 46]. A rodent model of ASH has demonstrated a pivotal role of neutrophils in pathogenesis of ALD [47, 48••]. Besides neutrophils, T and B lymphocytes are also recruited to the damaged liver, suggesting that the adaptive immune response contributes to alcohol-induced liver damage and activation of myofibroblasts. In addition to a direct hepatotoxic effect, alcohol induces changes in intestinal microflora (dysbiosis), which results in increased intestinal permeability and translocation of bacterial products (LPS) into blood, facilitating activation of myofibroblasts [49••, 50].
Intestinal bacterial overgrowth is common in patients with ALD. Sequencing of bacterial 16S rRNA genes revealed a relative abundance of Bacteroidetes and Verrucomicrobia bacteria in mice fed alcohol compared with a relative predominance of Firmicutes bacteria in control mice. This was associated with downregulation of host gene and protein expression of bactericidal c-type lectins Reg3b and Reg3g in the small intestines [49••]. Alcohol not only causes enteric dysbiosis and bacterial over-growth, but also increases gut permeability and the translocation of bacteria-derived LPS from the gut to the liver [51]. Consistently, increased levels of LPS are observed in patients with ALD [50]. One mechanism by which LPS exacerbates fibrosis is by activation of the innate immune system in the liver. In Kupffer cells, LPS binds to TLR4 to activate the MyD88-dependent and independent (TRIF/IRF-3) signaling pathway, leading to production of oxidative stress and proinflammatory cytokines, including TNF-α, that cause liver inflammation and hepatocellular damage [5••, 52–54]. TLR4 and complement factors also cause Kupffer cells/recruited macrophage to produce cytokines such as interleukin (IL)-6 and IL-10 [2]. Other TLRs expressed by innate immune cells can recognize distinct PAMPs, including bacterial peptidoglycan, double-stranded RNA, and unmethylated DNA, and may also play a role in promoting liver inflammation in ALD.
Alcoholic Liver Fibrosis/Cirrhosis
Alcoholic liver fibrosis/cirrhosis is characterized by excessive accumulation of collagen and other ECM proteins, steatosis, and fibrosis, and is accompanied by release of the major pro-fibrogenic cytokines TGF-β1, mostly produced by bone marrow-derived macrophages and resident Kupffer cells [2, 55–58]. HSCs are the major source of myofibroblasts in hepatotoxic liver injury [59••]. Under physiological conditions, HSCs store Vitamin A and exhibit a quiescent phenotype (qHSCs), but in response to alcohol-induced injury, α-smooth muscle actin (α-SMA) and collagen Type I are upregulated [2]. LPS not only stimulates Kupffer cells/macrophages to produce ROS and TGF-β1, but also promotes HSC activation via direct binding to TLR4 [60]. Acetaldehyde is produced mainly by hepatocytes and acts on HSCs by directly increasing expression of collagen I [61, 62]. Activated natural killer cells inhibit liver fibrosis by causing apoptosis of activated HSCs [63] or producing interferon (IFN)-γ, which induces HSC growth arrest [64].
Regression of Liver Fibrosis
When the cause of liver injury is removed (e.g., alcohol cessation), hepatic fibrosis is reversible in both patients and rodents. Regression of fibrosis is associated with decreased cytokine and ECM production, increased collagenase activity, disappearance of the myofibroblast population and dissolution of the fibrous scar [2, 65]. Only recently has the fate of myofibroblasts during regression been revealed. The previous concept was that myofibroblasts undergo apoptosis on the basis of documented senescence and apoptosis of some aHSCs during reversal of fibrosis. We [66••] and subsequently others [67] have used genetic marking to demonstrate an alternative pathway in which myofibroblasts revert to a quiescent-like phenotype in CCl4-induced liver injury. Of more relevance, we also demonstrated that experimental ALD regresses with the discontinuation of alcohol and that the aHSCs/myofibroblasts revert to an inactivated phenotype [66••].
Inactivation of HSCs (iHSCs) into a Quiescent-Like State During Regression of Liver Fibrosis
The Cre-loxP-system [68] provides a critical tool to study the fate of HSCs and their progeny in mice during fibrosis progression and/or regression. Genetic labeling of aHSCs/myofibroblasts results from crossing mice expressing recombinant Cre under control of collagen-α1(I) promoter (Colα1(I)Cre mice) with reporter mice (Rosa26f/f-YFP mice, ubiquitously expressing a yfp gene in which transcription is blocked by a floxed Stop cassette) [66••]. In the offspring mice (Colα1(I)Cre-YFP mice), genetic Cre-loxP recombination causes excision of the floxP-Stop-floxP sequence from genomic DNA and activation of YFP transcription in cells expressing type I collagen. Following induction of liver injury in these mice, aHSCs and their progeny are permanently labeled by YFP expression [66••]. Phenotypic changes of aHSCs and the mechanism of their inactivation can be now studied during regression of liver fibrosis.
Using this genetic labeling methodology, the fate of HSCs/myofibroblasts during recovery from alcohol-induced liver fibrosis has been elucidated. For the first time, it was demonstrated that half of the myofibroblasts escape apoptosis during regression of liver fibrosis, downregulate fibrogenic genes and acquire a phenotype similar to, but distinct from, quiescent (q)HSCs. Inactivated (i)HSCs more rapidly reactivate into myofibroblasts in response to fibrogenic stimuli and more effectively contribute to liver fibrosis [66••]. Inactivation of HSCs is associated with re-expression of lipogenic genes peroxisome proliferator-activated receptor gamma (PPAR-γ), Insig1 [66••], and CREBP [69]. The role of PPAR-γ in regulation of HSC activation in culture has been proposed. Ectopic over-expression of PPAR-γ results in the phenotypic reversal of activated HSCs to qHSC in culture, and upregulation of adipogenic transcription factors causing morphologic and biochemical reversal of activated HSCs to quiescent-like cells [69–72, 73•]. Findings from mouse models also demonstrate the importance of PPAR-γ for the maintenance of qHSCs, and inactivation of iHSCs during regression of liver fibrosis [66••].
Hepatocellular Carcinoma (HCC)
HCC is a malignant tumor made of cells resembling hepatocytes [74]. It usually arises in a cirrhotic liver [75, 76], and is identified by expression of alpha-fetoprotein (AFP), CD90, CD133, and EpCAM [77]. Several mechanisms have been suggested to contribute to HCC development in patients with alcoholic cirrhosis, including sustained inflammation, immunosuppressive effect of alcohol, impaired hepatocyte proliferation, loss of cell cycle checkpoints, increased tumor cell survival, telomere shortening and chromosomal instability. Accumulating evidence suggests that HCC originates from dedifferentiation and transformation of mature hepatocytes [76, 77]. Progression of HCC is associated with upregulation of IL-6 [78••, 79••], IL-17 [80, 81] and IL-22 [82] and constitutive activation of STAT32 [6, 83••]. Consistent with this, IL-6−/− and IL-22−/− mice are less susceptible to diethylnitrosamine (DEN)-induced HCC than wt mice. In addition to STAT3 [84], the NF-κB, Wnt/β-catenin, and Hedgehog signaling pathways are also implicated in HCC development [80, 85, 86••, 87••, 88]. There are some unique mechanisms that contribute to the development of HCC in patients with ALD [89–92]. These include the formation of acetaldehyde, which is a carcinogen with mutagenic properties, ethanol-stimulated induction of CYP2E1 which metabolizes alcohol, and the immunosuppressive effect of alcohol. LPS and chronic damage to hepatocytes can synergistically promote liver tumorigenesis via up-regulation of cancer stem cells as a mechanism of liver regeneration, indicated by analysis of stem cell markers [93••].
Mouse Models of ALD
A significant attempt to understand the mechanism of ALD development has been made over the past 20 years. However, this work has been hampered by the absence of suitable animal models. First, there are differences in time courses (weeks in mice vs years in human patients). Second, most models of chronic alcohol feeding in mice (such as the Lieber–Decarli diet) do not mirror the stages of ALD in patients. Dr. Gao’s group developed a chronic-plus-binge ethanol feeding model, which induces significant liver inflammation and neutrophil infiltration [48••, 94••] (but no fibrosis), and was highly successful in analyzing the ALD stages of steatosis and steatohepatitis. Serum levels of about 250 IU/L ALT and 420 IU/L AST were reached 9 h post gavage, and correlated with increased expression of hepatic and serum inflammatory cytokines and hepatic oxidative stress. In addition, this model is improved in the intragastric model of ethanol feeding [95••], and achieved a significant level of liver fibrosis in mice after 2 months of injury [66••]. Therefore, the Tsukamoto-French model of alcoholic liver injury can be used to study alcoholic fibrosis [95••]. Using this model, specific components of the gastrointestinal tract and liver were identified as causative in alcohol-induced liver injury. The levels of endotoxin in the blood begin to rise after about two weeks of continuous intragastric administration of ethanol and correlate with histology of alcohol-induced liver injury [96]. Development of alcohol-induced liver injury causes profound changes in gut enteric microflora; and altering the enteric flora with lactobacillus or antibiotics reduces liver injury [96]. Inactivation of Kupffer cells/macrophage in vivo by gadolinium chloride or by a calcium channel blocker markedly reduces inflammatory response in the liver and suppresses the extent of alcoholic liver injury [1••]. Meanwhile, little is known about the effects of ethanol on HCC progression. Only recently, a model of alcohol-induced HCC has been reported in which the addition of ethanol to drinking water increased tumor incidence in DEN-injected male mice [90, 92], suggesting that alcohol consumption promotes hepatic tumorigenesis [90].
Factors Affecting ALD Progression
Hepatoprotective Versus Oncogenic Functions of STAT3
STAT3 [97] plays an important role in pathogenesis of fibrogenic liver injury, ALD and progression to HCC by mediating different functions in T cells, macrophages, hepatocytes, and HSCs. T cell-specific STAT3-deficient mice are resistant to Con A-induced liver inflammation and exhibit reduced IL-17 production [27, 98]. STAT3 also regulates the expression of the RORγt and RORα transcription factors and promotes differentiation of Th17 cells [27]. IL-17A can directly activate STAT3 signaling in macrophages [98, 99••]. Anti-inflammatory cytokine IL-10 also activates STAT3 signaling in macrophages [100, 101], and myeloid-specific STAT3-deficient mice are prone to a higher degree of liver inflammation in liver injury induced by several hepatic toxins [98, 102, 103] including alcohol [25••]. In concordance, the studies from hepatocyte-specific STAT3 knockout mice demonstrate that IL-6, IL-10, and IL-22 produce hepatoprotective and anti-fibrogenic effects via the activation of STAT3 in hepatocytes, and STAT3 in hepatocytes promotes an anti-inflammatory signal to suppress liver inflammation under most conditions [104–107].
For the past decade, the role of STAT3 in the pathogenesis of liver fibrosis was only investigated in inflammatory cells and hepatocytes [27, 47, 103, 108–110]. Only recently it was shown that STAT3 is activated by IL-6 and modulates collagen production in aHSC/myofibroblasts [99••, 111], and mice deficient of STAT3 in HSCs are less susceptible to liver fibrosis, implying that IL-17A, leptin, IL-6, and HGH signaling in HSCs may promote the development of liver fibrosis by activation of STAT3 in HSCs. Interestingly, IL-22 also activates STAT3 in HSCs but induces HSC senescence, thereby inhibiting liver fibrosis [112]. It is unclear why IL-6 and IL-22 have such different effects on HSCs.
At present, the mechanisms underlying the anti-inflammatory functions of STAT3 are not well understood. Even less is known about the tumorigenic effects of STAT3 on progression of ALD to HCC. However, it has been demonstrated that constitutive activation of STAT3 at the end-stage of liver cirrhosis promotes tumor cell survival leading to HCC [27]. Furthermore, deletion of STAT3 in hepatocytes reduced DEN-induced HCC development in mice [27, 113]. Similarly, deletion of STAT3-inhibitory proteins SHP-2 or SOCS3 in hepatocytes resulted in constitutive STAT3 activation, and increased DEN-induced HCC development [83••, 114–116]. In human HCC, increased STAT3 activation is likely due to persistent stimulation from cytokines such as IL-6 and IL-22 [82, 114]. In concordance, overexpression of IL-22 in hepatocytes caused STAT3-dependent activation of a variety of anti-apoptotic genes (Cyclin D1, Bcl-XL, Bcl-2), making these IL-22TG mice more susceptible to DEN-induced liver cancer [114]. Therefore, STAT3 inhibitors may have therapeutic potential for the treatment of HCC.
IL-6 is a cytokine involved in the regulation of several cellular processes including proliferation and differentiation and plays a pivotal role in acute phase response and in the control of the balance between pro-inflammatory and anti-inflammatory pathways. Increased levels of IL-6 are linked to development of HCC, and activation of IL-6 signaling plays a critical role in hepatocyte survival, and activation of pathways responsible for their transformation (outlined in [47, 113, 117, 118]). IL-6 binding to IL-6R and its signal transducing chain, gp130, induces gp130 dimerization and the subsequent activation and dimerization of gp130-associated Janus kinases (JAKs), which leads to JAK phosphorylation, followed by STAT3 activation [97]. Other signaling pathways activated by IL-6 include PI3 kinase, p38 MAP kinase, c-Jun NH2-terminal kinase (JNK), and TORC1-S6K, which ultimately lead to cell proliferation, protection from apoptosis and increased metastatic potential. The human IL-6 gene is located on chromosome 7p21 [119, 120]. A number of studies indicated that the presence of a G/C single nucleotide polymorphism (SNP) at the promoter −174 of the IL-6 gene is related to the IL-6 gene transcription and, that significantly affects production of IL-61 [21]. Two phenotypes for this polymorphism were identified: the high-producer phenotype, including the −174 G/G and −174 G/C genotypes, characterized by higher circulating IL-6 levels; and the low-producer phenotype, including the −174 C/C genotype [120, 121]. The GG IL-6 genotype showed the strongest influence on HCC risk among all the cytokine polymorphisms studied [122] and was associated with high the incidents of HCC in patients with liver cirrhosis [123, 124]. Meanwhile, IL-6 polymorphisms with the low-producer genotype (−174 CC) correlated with the absence of HCC in the same patient population [120, 123, 124].
Interleukin 22 (IL-22)
IL-22 is a member of the IL-10 family of cytokines. Expression of IL-22 is restricted to hematopoietic cells. Th17 cells selectively synthesize both IL-17 and IL-22 [125–128]. Expression of IL-22 is also dependent on IL-23, as demonstrated by use of IL-23p19−/− mice [127, 129]. IL-22 can be also produced by subsets of γδT cells, NK and NKT cells [129]. The IL-22 receptor is composed of IL-22R1 and IL-10R2 subunits, and receptor ligation results in STAT3 phosphorylation and activation of the p38 mitogen-activated protein kinase pathway [130, 131]. Expression of IL-22R1 is found only on cells of non-hematopoietic origin, which include hepatocytes and pancreatic acinar cells [129]. IL-22 is implicated in inflammation, immune surveillance and mucosal host defense. In the liver, IL-22 promotes hepatic production of acute phase proteins and plays protective roles against liver injury [129]. IL-22 possesses strong hepatoprotective properties and protects mice from ConA- and alcohol-induced hepatitis [10, 48••, 105]. IL-22−/− mice are more susceptible to CCl4-induced liver fibrosis [99••], while treatment with IL-22 attenuates development of CCl4-induced liver fibrosis in mice [99••]. Therefore, IL-22 might be used to treat patients with ALD because of its antioxidant, antiapoptotic, anti-steatotic, proliferative, and antimicrobial effects [48••, 132]. While the exact action of IL-22 still remains to be resolved, progress has been made using transgenic mice overexpressing IL-22 in hepatocytes [114]. These IL-22TG mice are protected from CCl4-induced liver fibrosis, but are more susceptible to tumorigenesis [114], suggesting that IL-22 may have opposing short-term and long-term effects on the liver. Thus, overexpression of IL-22 drives proliferation and hyperactivation of liver progenitor cells [82, 133]. IL-22−/− mice exhibit reduced tumorigenesis [20••, 84]. It remains unclear if IL-22 promotes survival of tumor cells or facilitates transformation of hepatic progenitors via constitutive activation of STAT3, which drives tumor progression [84].
Interleukin 17 (IL-17)
Interleukin-17 (IL-17)-producing effector CD4+ T (Th17) cells [134, 135] originate from naïve T cells via activation of lineage specific transcription factors [136, 137], regulated by TGF-β1 and IL-6, and other cytokines [138, 139]. IL-17 cytokines are mainly produced by CD4+ Th17 cells, but also by a variety of cells, including γδ T cells, CD8+ T cells, NKT cells, NK cells, innate lymphoid cells, eosinophils, neutrophils, and monocytes [140]. Th17 cells secrete a family of cytokines comprised of IL-17A, IL-17F, IL-17B, IL-17C, and IL-17E [141]. IL-17A homodimers (also known as IL-17) are the most abundant product of Th17 cells, exhibit higher biological activity than other family members, and signal through their cognate receptors IL-17RA and IL-17RC [140]. IL-17RA is ubiquitously expressed, but is strongly induced in hematopoietic cells [142] and fibroblasts [143] in response to stress. IL-17A signaling activates inflammatory signaling in target cells, including STAT3, TRAF6, Act1, JNK, ERK, NF-κB [142, 144]. IL-17 mediates autoimmunity, and the autoimmune inflammatory diseases psoriasis and rheumatoid arthritis respond to anti-IL-17 biological therapies [145]. Most recently, IL-17 has been implicated in liver, lung, and skin fibrosis, and in tumorigenesis [44, 140, 141, 146–151]. Although anti-TNF-α therapy has been ineffective in patients with ALD [1••, 152], anti-IL-17 drugs are a potential novel therapy for ALD.
The role of IL-17 in ALD progression is not understood. However, recent studies implicated IL-17 in regulation of IL-8 production, a chemokine which plays a critical role in neutrophil recruitment into alcohol-damaged liver at the stage of steatohepatitis. In concordance, exposure of hepatic stellate cells to IL-17 in vitro induced production of IL-8 and GRO, the factors that have a strong chemoattractive effect on neutrophils, suggesting that IL-17 plays a role in promoting both liver inflammation and fibrogenesis of ALD [151, 153, 154].
Most of the data implicating IL-17 in the pathogenesis of ALD are based on clinical studies. Elevated levels of serum IL-17 were detected in patients with ALD [154]. The cellular origins of IL-17 in this study were monocytes and T cells in the circulation and infiltrating neutrophils and T cells in the liver [155]. Another study based on immunocolocalization staining with fluorescently-labeled antibodies, demonstrated that liver infiltrating IL-17-expressing cells were mainly composed of neutrophils and T lymphocytes [20••, 151]. The number of hepatic IL-17-producing cells correlated with the severity of ALD-induced fibrosis [20••], suggesting that IL-17 may serve as a marker for ALD progression. Moreover, expression of the IL-17 receptor (IL-17RA) was detected on activated HSCs (aHSCs) in the liver biopsies of ALD patients [151].
Regulation of Th17 Differentiation
TGF-β1, IL-6 and IL-21 drive differentiation of Th17 cells from naïve Th0 cells [134, 138, 139] via activation of retinoid-related orphan receptor γt (ROR γt) [136]. IL-23 is required for Th17 proliferation [156–159]. IL-27 antagonizes expansion of Th17 directly via inhibition of IL-23-producing cells. IL-25 also blocks Th17 responses via release of IL-13 which, in turn, suppresses IL-23, IL-1β1 and IL-6 secretion by dendritic (and other) cells [160, 161]. In response to alcohol-induced liver injury, Th17 cells release IL-17, which causes induction of IL-18, CXC and recruitment of neutrophils into the liver. Neutrophils facilitate hepatocyte injury and activation of BM-derived and liver resident Kupffer cells [148–151, 162, 163]. IL-17 stimulates Kupffer cells to express inflammatory cytokines IL-6, IL-1β, and TNF-α, as well as the major fibrogenic cytokine TGF-β1 via activation of STAT3 and NFκB signaling pathways. Using BM chimeric mice, we determined that deletion of either IL-17A or IL-17RA in inflammatory and Kupffer cells decreases liver fibrosis by 50–55 %, and this effect is mediated via regulation of TGF-β1 production, while deletion of IL-17RA in non-immune liver resident cells decreased liver fibrosis by 25 % [99••]. Furthermore, we demonstrated that IL-17A stimulates activation of HSCs in a STAT3-dependent manner. In turn, murine Th17 cells are believed to produce IL-22, which mediates hepatoprotective functions [105] and facilitates oval cells proliferation, suggesting that murine Th17 may also contribute to tumorigenesis.
Epigenetic Regulation
Understanding the mechanisms of HSC inactivation during regression of liver fibrosis is critical for identifying new targets for therapy. Inactivation of HSCs is most likely regulated at an epigenetic level (vs genetic mutations) [164, 165]. Epigenetics are heritable changes in gene function that occur without a change in the DNA sequence [166]. These changes, including nucleosome dynamics and histone modifications, cause structural alterations in the chromatin structure, and regulate gene expression. Post-translational modifications of the core histone subunits of nucleosomes by methylation, acetylation, and phosphorylation [167, 168, 169••] are a fundamental mechanism by which the transcriptional activity of an associated gene locus can be regulated.
Epigenetic Regulation of HSCs, the Role of PPARγ in HSC Activation
Extensive studies have demonstrated that ethanol affects metabolism of methionine and DNA methylation. Methionine metabolism occurs primarily in the liver, where homocysteine is methylated to methionine and then S adenosylmethionine (SAMe) in a transmethylation reaction catalyzed by methionine adenosyltransferase [170]. SAMe is a methyl donor in methylation and has an important role in inducing DNA and histone methylation. Long-term ethanol consumption reduces hepatic levels of SAMe and DNA and histone methylation, increasing expression of genes that regulate stress response and alcoholic liver injury [1••, 12]. PPARγ expression is associated with the adipogenic features of qHSC and must be silenced for the cell to activate into a myofibroblast [69, 171•, 172••, 173]. A multi-step epigenetic network that controls activation of HSCs has been described, and involves activation of MeCP2 which causes alterations at the H3K27 methylation sites and generates transcriptionally repressed chromatin structure in the PPARγ promoter [69, 164]. Consistent with this observation, over-expression of PPARγ in cultured aHSCs results in reversion of HSC activation, and reacquisition of their adipogenic characteristics [173, 174].
Epigenetic Regulation of HSC Activation Via miRNAs
MicroRNAs (miRNAs) are short, noncoding RNAs that are average 22–23 nucleotides long. They control expression of genes involved in cell growth, differentiation, and apoptosis and are believed to be involved in the pathogenesis of liver disease, especially cancer [175]. They regulate gene expression by interacting with the 3′ untranslated region (3′-UTR) of target gene mRNA to repress translation or enhance mRNA cleavage. Several studies have demonstrated the role of miRNAs in ALD [176–178]. Ethanol exposure up-regulates miRNA-155 in macrophages, which increases TNF-α production (via increased mRNA stability) [177]; short-term ethanol exposure up-regulates miRNA-212 in intestinal epithelial cells, which down-regulates zonula occludens-1 protein [179], a factor that maintains intestinal permeability. Expression of liver miRNAs has also been shown to be significantly altered in ethanol-fed mice [180], but the functions of these miRNAs in the pathogenesis of ALD are not clear. Recent study has delineated an important role of miRNA-214 in regulation of CCN2 (CTGF) [181], the second member of the cystein-rich-61/connective tissue growth factor/nephroblastoma-overexpressed protein that plays a critical role in hepatic fibrogenesis due to the ability to directly activate HSCs into myofibroblasts [182]. miRNA-214 inhibits CCN2 expression in HSCs by binding to the CCN2 3′-UTR [183]. Thus, quiescent HSCs expressed high level of miRNA-214, while increased expression of CCN2 mRNA in ethanol-activated HSCs was associated with reciprocal downregulation of miRNA-214 [182, 183]. Furthermore, HSC can produce nano-size exosomes (small vesicles released extracellularly that arise by inward budding from the limiting membranes of multivesicular bodies) which transfer miRNA-214 to neighboring HSC or hepatocytes to inhibit CCN2 3′-UTR activity and suppress CCN2 expression. Exosomes from HSCs were a conduit for uptake of miRNA-214 by HSCs and hepatocytes [183], suggesting that exosomal transfer of miRNA-214 is a paradigm for the regulation of CCN2-dependent fibrogenesis and identifying intercellular regulation of exosomal miRNA as a target for anti-fibrotic therapy [176, 183].
Ethanol Metabolism
Evidence suggests that alcohol metabolites can aggravate liver fibrosis by direct activation of HSCs. During ethanol metabolism, ethanol is converted into acetaldehyde, then to acetate. Acetate rapidly releases into circulation and is eventually metabolized to CO2 in heart, skeletal muscle, and brain cells. Acetaldehyde (the first metabolite of ethanol) upregulates expression of collagen Type I in HSCs. Several mechanisms were identified to mediate acetaldehyde effects in HSCs. Acetaldehyde was shown to induce expression of Col1a2 gene [184] via a de novo protein synthesis-independent, PI3K-dependent mechanism. Acetaldehyde induces phosphorylation and activation of Smad2 and 4, and downregulation of Smad7 [185]. In turn, adenosine is a regulatory nucleoside that is generated in response to cellular stress, damage, tissue hypoxia, inflammation, and alcohol exposure. Hepatic level of adenosine is strongly increased in animal models of alcohol-induced liver injury, and ethanol and its metabolite acetaldehyde were shown to stimulate accumulation of extracellular adenosine through its action on nucleoside transporter [186, 187]. Extracellular adenosine is generated by ecto-5′-nucleotidase (CD73), and mediates its functions via the adenosine A2A receptor (A2AR). A2AR is functionally present on HSCs, and acetaldehyde-induced production and accumulation of extracellular adenosine results in adenosine binding to adenosine A2AR receptor and its subsequent activation [188, 189]. Adenosine-A2AR signaling in HSCs triggers Gs-cAMP-PKA-SRC-ERK1/2-MAPK signaling cascade which mediates collagen Type I production and ECM production in HSC activation and ECM production [190]. These findings explain why antagonists of A2AR signaling, such as caffeine (1, 3, 7-trimethylxanthine), a non-selective adenosine receptor antagonist, could reduce ALD, and provide a novel pathway for anti-fibrotic therapy [191].
Conclusions
Despite extensive studies, the available therapy for ALD remains limited. Recent improvements in mouse models of ALD are providing new insights into the pathogenesis of the different stages of ALD, which may provide new targets for therapy.
Abbreviations
- Col:
-
Collagen α1(I)
- α-SMA:
-
α-Smooth muscle actin
- qHSCs:
-
Quiescent hepatic stellate cells
- aHSCs:
-
Activated hepatic stellate cells
- iHSCs:
-
Inactivated hepatic stellate cells
References
Papers of particular interest, published recently, have been highlighted as: • Of importance, •• Of major importance
•• Gao, B, Bataller, R (2011) Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141:1572–1585. This review described the pathological process of alcoholic liver diseases from steatosis, alcoholic hepatitis, alcoholic fibrosis to the end stage hepatocellular carcinoma
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218
Diehl AM (1997) Alcoholic liver disease: natural history. Liver Transpl Surg 3:206–211
Adachi T et al (2005) NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology 41:1272–1281
•• Gao B (2004) Therapeutic potential of interleukin-6 in preventing obesity- and alcohol-associated fatty liver transplant failure. Alcohol 34:59–65. This review discussed the protective effect of IL-6 administration on alcoholic/steatotic liver isograft, and proposed IL-6 treatment to expand the liver donnor source for liver transplantation
Naveau S et al (1997) Excess weight risk factor for alcoholic liver disease. Hepatology 25:108–111
Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA (2010) Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 42:21–23
Stickel F et al (2011) Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology 53:86–95
Trepo E et al (2011) Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J Hepatol 55:906–912
Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B (2011) Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res 35:787–793
You M, Fischer M, Deeg MA, Crabb DW (2002) Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem 277:29342–29347
Esfandiari F et al (2010) Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology 51:932–941
Ji C, Deng Q, Kaplowitz N (2004) Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology 40:442–451
Peng Z et al (2009) Adenosine signaling contributes to ethanol-induced fatty liver in mice. J Clin Invest 119:582–594
Jeong WI et al (2008) Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab 7:227–235
Osei-Hyiaman D et al (2008) Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest 118:3160–3169
Zhao XJ et al (2008) TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 181:3049–3056
Petrasek J et al (2011) Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 53:649–660
McMullen MR et al (2005) Early growth response-1 transcription factor is essential for ethanol-induced fatty liver injury in mice. Gastroenterology 128:2066–2076
•• Lafdil F, Miller AM, Ki SH, Gao B (2010) Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol 7:250–254. This review described pathological role of Th17 by releasing cytokines, IL-17A and IL-22 during hepatic diseases. IL17A promotes biliary epithelial cells, hepatic stellate cells and kupffer cells secreting proinflammatory cytokines, while IL-22 protects hepatocytes from apoptosis
You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW (2004) The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 127:1798–1808
You M, Liang X, Ajmo JM, Ness GC (2008) Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am J Physiol Gastrointest Liver Physiol 294:G892–G898
Shen Z, Liang X, Rogers CQ, Rideout D, You M (2010) Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 298:G364–G374
You M, Rogers CQ (2009) Adiponectin: a key adipokine in alcoholic fatty liver. Exp Biol Med 234:850–859
•• Horiguchi N et al (2008) Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 134:1148–1158. This report firstly described the dural role of IL-6 intracellular effector, STAT3, during pathogenesis of alcoholic liver diseases. Activation of STAT3 in hepatocytes suppresses expression of SREBP-1 and results in attenuation of steatosis induced by alcohol consumption. Meanwhile, activated STAT3 in hepatocytes promotes inflammation. In the Kupffer cells, activated STAT3 is mainly the effector of IL-10 to reduce pro-inflammatory cytokines secretion
Wang H, Lafdil F, Kong X, Gao B (2011) Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int J Biol Sci 7:536–550
Gao B, Wang H, Lafdil F, Feng D (2012) STAT proteins: key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J Hepatol 57:430–441
Ji C, Chan C, Kaplowitz N (2006) Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol 45:717–724
Nath B et al (2011) Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 53:1526–1537
Pritchard MT et al (2007) Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology 132:1117–1126
Cohen JI, Roychowdhury S, McMullen MR, Stavitsky AB, Nagy LE (2010) Complement and alcoholic liver disease: role of C1q in the pathogenesis of ethanol-induced liver injury in mice. Gastroenterology 139:664–674, 674 e661
Kaiser JP et al (2009) PKCepsilon plays a causal role in acute ethanol-induced steatosis. Arch Biochem Biophys 482:104–111
McKim SE et al (2003) Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology 125:1834–1844
Hernandez-Gea V et al (2012) Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142:938–946
Donohue TM Jr (2009) Autophagy and ethanol-induced liver injury. World J Gastroenterol 15:1178–1185
Wu D, Wang X, Zhou R, Cederbaum A (2010) CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem Biophys Res Commun 402:116–122
Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB (2004) Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 18:1272–1282
Kawai T et al (2001) Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 167:5887–5894
Kawai T, Akira S (2007) TLR signaling. Semin Immunol 19:24–32
Kubes P, Mehal WZ (2012) Sterile inflammation in the liver. Gastroenterology 143:1158–1172
Muller S et al (2001) New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J 20:4337–4340
Thabut D, Shah V (2010) Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol 53:976–980
Wang HJ, Gao B, Zakhari S, Nagy LE (2012) Inflammation in alcoholic liver disease. Annu Rev Nutr 32:343–368
Gaffen SL (2009) Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 9:556–567
Jones CE, Chan K (2002) Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-alpha, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol 26:748–753
Dhanda AD, Lee RW, Collins PL, McCune CA (2012) Molecular targets in the treatment of alcoholic hepatitis. World J Gastroenterol 18:5504–5513
Gao B (2005) Cytokines, STATs and liver disease. Cell Mol Immunol 2:92–100
•• Ki SH et al (2010) Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology 52:1291–1300. This work described a new murine alcoholic liver disease model: chronic plus binge alcohol consumption. By utilizing this model, the authors demonstrated IL-22 treatment ameliorated alcohol induced steatosis and hepatitis via activation of hepatic STAT3 signaling
•• Yan AW et al (2011) Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53:96–105. This paper demonstrated the alcohol assumption could promote intestinal bacterial overgrowth and translocation. Such microflora changes might be induced by suppression of Reg3b and Reg3g expressions within small intestine
Yan AW, Schnabl B (2012) Bacterial translocation and changes in the intestinal microbiome associated with alcoholic liver disease. World J Hepatol 4:110–118
Rao R (2009) Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50:638–644
Fujimoto M et al (2000) Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res 24:48S–54S
Fukui H (2005) Relation of endotoxin, endotoxin binding proteins and macrophages to severe alcoholic liver injury and multiple organ failure. Alcohol Clin Exp Res 29:172S–179S
Seki E, Brenner DA (2008) Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48:322–335
O’Shea RS, Dasarathy S, McCullough AJ, Practice Guideline Committee of the American Association for the Study of Liver, D. & Practice Parameters Committee of the American College of Gastroenterology (2010) Alcoholic liver disease. Hepatology 51:307–328
Lucey MR, Mathurin P, Morgan TR (2009) Alcoholic hepatitis. N Engl J Med 360:2758–2769
Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–1669
Seki E et al (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13:1324–1332
•• Friedman SL, Roll FJ, Boyles J, Bissell DM (1985) Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci USA 82:8681–8685. The first paper indicated that hepatic stellate cells are the major collagen deposition cells during liver fibrosis
Gao B et al (2011) Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 300:G516–G525
Natori S et al (2001) Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol 34:248–253
Mello T, Ceni E, Surrenti C, Galli A (2008) Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects Med 29:17–21
Gao B, Radaeva S, Park O (2009) Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 86:513–528
Radaeva S et al (2006) Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 130:435–452
Iredale JP et al (1998) Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 102:538–549
•• Kisseleva T et al (2012) Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA 109:9448–9453. This paper firstly observed the inactivation of hepatic stellate cells during liver fibrosis regression, rather than apoptosis
Troeger JS et al (2012) Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 143:1073
Gilbertson L (2003) Cre–lox recombination: Cre-ative tools for plant biotechnology. Trends Biotechnol 21:550–555
She H, Xiong S, Hazra S, Tsukamoto H (2005) Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem 280:4959–4967
Wells RG (2008) The role of matrix stiffness in regulating cell behavior. Hepatology 47:1394–1400
Gaca MD et al (2003) Basement membrane-like matrix inhibits proliferation and collagen synthesis by activated rat hepatic stellate cells: evidence for matrix-dependent deactivation of stellate cells. Matrix Biol 22:229–239
Tsukamoto H (2005) Fat paradox in liver disease. Keio J Med 54:190–192
• Tsukamoto H (2005) Adipogenic phenotype of hepatic stellate cells. Alcohol Clin Exp Res 29:132S–133S . This review summarized the transcription factor codes (PPAR-gamma, C/EBPs, SREBP-1c and LX-alpha) that specified the adipocyte fate of hepatic stellate cells which maintained its quiescent status
Sell S (1990) Is there a liver stem cell? Cancer Res 50:3811–3815
Alison MR (2005) Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev 1:253–260
Wu XZ, Chen D (2006) Origin of hepatocellular carcinoma: role of stem cells. J Gastroenterol Hepatol 21:1093–1098
Shen Y, Cao D (2012) Hepatocellular carcinoma stem cells: origins and roles in hepatocarcinogenesis and disease progression. Front Biosci 4:1157–1169
•• Naugler WE et al (2007) Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317:121–124. The paper discovered the differential expression of IL-6 between male and female mice, when they were under diethylnitrosamine (DEN) challenging. It explained the gender disparity in liver cancer
•• He G et al (2013)Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 155:384–396. This report discovered the IL-6 signaling from HCC progenitor is required for its maligance and differentiation into cancer cells
Gu FM et al (2011) IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Molecular cancer 10:150
Li J et al (2011) Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PLoS One 6:e21816
Jiang R et al (2011) Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology 54:900–909
•• He G et al. (2010) Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 17:286–297. This report elucidated the role of IKK-beta in inhibiting JNK or STAT3 to eliminate progression of HCC progenitor differentiate into mature cancer
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Sicklick JK et al (2006) Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis 27:748–757
•• Karin M (2009) NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1:a000141. NF-kappaB is activated by pro-inflammatory cytokines IL-17 and TNFs from activated macrophages and lymphocytes. Downstream genes of NF-kappaB promote cancer cell proliferation and survival
•• Sakurai T et al (2008) Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14:156–165. Carcinogenesis effect of Ikk-beta is mediated by IL-1α releasing and ROS accumulation
White BD, Chien AJ, Dawson DW (2012) Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology 142:219–232
Morgan TR, Mandayam S, Jamal MM (2004) Alcohol and hepatocellular carcinoma. Gastroenterology 127:S87–S96
McKillop IH, Schrum LW (2009) Role of alcohol in liver carcinogenesis. Semin Liver Dis 29:222–232
Brandon-Warner E, Schrum LW, Schmidt CM, McKillop IH (2012) Rodent models of alcoholic liver disease: of mice and men. Alcohol 46:715–725
Brandon-Warner E, Walling TL, Schrum LW, McKillop IH (2012) Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res 36:641–653
•• Machida K et al (2009) Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA 106:1548–1553. The synergistical effect of HCV infection and alcohol consumption is mediated by TLR4 signaling. TLR4 expression is firstly elevated by HCV protein NS5A. Alcohol consumption permealizes gastrointestinal tract to facilitate bacterial translocation into liver and endotoxin released by intestinal bacterial induces TLR4 downstream gene Nanog expression
•• Bertola A, Mathews S, Ki S, Wang H, Gao B (2013) Mouse chronic plus binge ethanol feeding model (the NIAAA model). Nat Protoc, in press. This paper described the method of chronic alcohol consumption and single high dose of alcohol binge, causing more severe steatosis and neutrophil infiltration than chronic alcohol feeding alone
•• Ueno A et al (2012) Mouse intragastric infusion (iG) model. Nat Protoc 7:771–781. This paper reported a novel alcohol feeding protocol by implanting gastrostomy catheter into gastrointestinal tract to create the alcoholic liver disease model. This ALD model is characterized by elevated alanine aminotranferase levels and severe hepatic steatosis
Thurman RG et al (1998) The role of gut-derived bacterial toxins and free radicals in alcohol-induced liver injury. J Gastroenterol Hepatol 13(Suppl):S39–S50
Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW (2002) Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285:1–24
Lafdil F et al (2009) Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology 137(2125–2135):e2121–e2122
•• Meng F et al (2012) Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 143:765–776, e761–763. In response to liver injury (hepatotoxicity and biliary obstruction), IL-17A expression is increased and IL-17A directly activates hepatic stellate cells by STAT3 signaling
Murray PJ (2006) STAT3-mediated anti-inflammatory signalling. Biochem Soc Trans 34:1028–1031
El Kasmi KC et al (2006) General nature of the STAT3-activated anti-inflammatory response. J Immunol 177:7880–7888
Wang H et al (2010) Interplay of hepatic and myeloid signal transducer and activator of transcription 3 in facilitating liver regeneration via tempering innate immunity. Hepatology 51:1354–1362
Horiguchi N et al (2010) Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology 51:1724–1734
Hong F et al (2002) Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest 110:1503–1513
Zenewicz LA et al (2007) Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27:647–659
Kroy DC et al (2010) Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology 51:463–473
Sakamori R et al (2007) Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology 46:1564–1573
Weng H, Li H, Dooley S (2011) Inflammation does not always kill hepatocytes during liver damage. Hepatology 54:366; author reply 367.
Shigekawa M et al (2011) Involvement of STAT3-regulated hepatic soluble factors in attenuation of stellate cell activity and liver fibrogenesis in mice. Biochem Biophys Res Commun 406:614–620
Plum W et al (2010) Lack of glycoprotein 130/signal transducer and activator of transcription 3-mediated signaling in hepatocytes enhances chronic liver injury and fibrosis progression in a model of sclerosing cholangitis. Am J Pathol 176:2236–2246
Tan Z et al (2013) IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol 191:1835–1844
Kong X et al (2012) Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 56:1150–1159
Wang H et al (2011) Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am J Pathol 179:714–724
Park O et al (2011) In vivo consequences of liver-specific interleukin-22 expression in mice: implications for human liver disease progression. Hepatology 54:252–261
Riehle KJ et al (2008) Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med 205:91–103
Bard-Chapeau EA et al (2011) Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 19:629–639
Johnson C et al (2012) Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl Gastrointest Cancer 1:58–70
Umemura A et al (2014) Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab 20:133–144
Bowcock AM et al (1988) The human “interferon-beta 2/hepatocyte stimulating factor/interleukin-6” gene: dNA polymorphism studies and localization to chromosome 7p21. Genomics 3:8–16
Giannitrapani L, Soresi M, Balasus D, Licata A, Montalto G (2013) Genetic association of interleukin-6 polymorphism (−174 G/C) with chronic liver diseases and hepatocellular carcinoma. World J Gastroenterol 19:2449–2455
Fishman D et al (1998) The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest 102:1369–1376
Ognjanovic S, Yuan JM, Chaptman AK, Fan Y, Yu MC (2009) Genetic polymorphisms in the cytokine genes and risk of hepatocellular carcinoma in low-risk non-Asians of USA. Carcinogenesis 30:758–762
Falleti E et al (2010) Genetic polymorphisms of interleukin-6 modulate fibrosis progression in mild chronic hepatitis C. Hum Immunol 71:999–1004
Falleti E et al (2009) Interleukin-6 polymorphisms and gender: relationship with the occurrence of hepatocellular carcinoma in patients with end-stage liver disease. Oncology 77:304–313
Cupedo T et al (2009) Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC + CD127 + natural killer-like cells. Nat Immunol 10:66–74
Vivier E, Spits H, Cupedo T (2009) Interleukin-22-producing innate immune cells: new players in mucosal immunity and tissue repair? Nat Rev Immunol 9:229–234
Aujla SJ et al (2008) IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14:275–281
Takatori H et al (2009) Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med 206:35–41
Wolk K et al (2011) Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol 186:1228–1239
Kotenko SV et al (2001) Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10R beta) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J Biol Chem 276:2725–2732
Lejeune D et al (2002) Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem 277:33676–33682
Radaeva S, Sun R, Pan HN, Hong F, Gao B (2004) Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 39:1332–1342
Feng D et al (2012) Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology 143:188–198 e187
Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM (2006) Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 24:677–688
Steinman L (2007) A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med 13:139–145
Ivanov II et al (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell 126:1121–1133
Yang XO et al (2008) T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28:29–39
Bettelli E et al (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238
Mangan PR et al (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234
Kolls JK, Linden A (2004) Interleukin-17 family members and inflammation. Immunity 21:467–476
Iwakura Y, Ishigame H, Saijo S, Nakae S (2011) Functional specialization of interleukin-17 family members. Immunity 34:149–162
Yao Z et al (1995) Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 3:811–821
Andoh A et al (2002) IL-17 selectively down-regulates TNF-alpha-induced RANTES gene expression in human colonic subepithelial myofibroblasts. J Immunol 169:1683–1687
Subramaniam SV, Cooper RS, Adunyah SE (1999) Evidence for the involvement of JAK/STAT pathway in the signaling mechanism of interleukin-17. Biochem Biophys Res Commun 262:14–19
Miossec P, Kolls JK (2012) Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discovery 11:763–776
Ye P et al (2001) Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–527
Shen F, Gaffen SL (2008) Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine 41:92–104
Faust SM et al (2009) Role of T cell TGFbeta signaling and IL-17 in allograft acceptance and fibrosis associated with chronic rejection. J Immunol 183:7297–7306
Wilson MS et al (2010) Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207:535–552
Longhi MS et al (2004) Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol 41:31–37
Lemmers A et al (2009) The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 49:646–657
Affo S et al (2012) Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut 62:452–460
Maltby J, Wright S, Bird G, Sheron N (1996) Chemokine levels in human liver homogenates: associations between GRO alpha and histopathological evidence of alcoholic hepatitis. Hepatology 24:1156–1160
Dominguez M et al (2009) Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 136:1639–1650
Hammerich L, Heymann F, Tacke F (2011) Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol 2011:345803
Langrish CL et al (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Murphy CA et al (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198:1951–1957
Oppmann B et al (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715–725
Cua DJ et al (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421:744–748
Fort MM et al (2001) IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 15:985–995
Hurst SD et al (2002) New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol 169:443–453
Wilson NJ et al (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8:950–957
Ge J et al (2010) Implication of Th17 and Th1 cells in patients with chronic active hepatitis B. J Clin Immunol 30:60–67
Mann J et al (2010) MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138:705–714, 714 e701–704
Tsukamoto H, Zhu NL, Asahina K, Mann DA, Mann J (2011) Epigenetic cell fate regulation of hepatic stellate cells. Hepatol Res 41:675–682
Pepke S, Wold B, Mortazavi A (2009) Computation for ChIP-seq and RNA-seq studies. Nat Methods 6:S22–S32
Park PJ (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10:669–680
Henikoff S (2008) Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet 9:15–26
•• Heinz S, Glass CK (2012) Roles of lineage-determining transcription factors in establishing open chromatin: lessons from high-throughput studies. Curr Top Microbiol Immunol 356:1–15. The methods to study epigenetic changes in different cell types are symmarized in this paper
Mato JM, Martinez-Chantar ML, Lu SC (2008) Methionine metabolism and liver disease. Annu Rev Nutr 28:273–293
• Tsukamoto H, Zhu NL, Wang J, Asahina K, Machida K (2012) Morphogens and hepatic stellate cell fate regulation in chronic liver disease. J Gastroenterol Hepatol 27(Suppl 2):94–98. Important findings on epigenetic regulation of alcohol-activated myofibroblasts are described in this paper
•• Yang MD et al (2012) Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor gamma in hepatic stellate cells for their antifibrotic effect. Hepatology 55:1271–1281. This paper demonstrates novel pathway of regulation of HSCs
Hazra S et al (2004) Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem 279:11392–11401
Jaster R et al (2005) Peroxisome proliferator-activated receptor gamma overexpression inhibits pro-fibrogenic activities of immortalised rat pancreatic stellate cells. J Cell Mol Med 9:670–682
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
McDaniel K et al (2014) The functional role of microRNAs in alcoholic liver injury. J Cell Mol Med 18:197–207
Bala S et al (2011) Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor alpha (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem 286:1436–1444
Bala S, Marcos M, Szabo G (2009) Emerging role of microRNAs in liver diseases. World J Gastroenterol 15:5633–5640
Tang Y et al (2008) Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin Exp Res 32:355–364
Dolganiuc A et al (2009) MicroRNA expression profile in Lieber–DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol Clin Exp Res 33:1704–1710
Brigstock DR et al (2003) Proposal for a unified CCN nomenclature. Mol Pathol 56:127–128
Huang G, Brigstock DR (2012) Regulation of hepatic stellate cells by connective tissue growth factor. Front Biosci (Landmark Ed) 17:2495–2507
Chen L et al (2014) Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology 59:1118–1129
Svegliati-Baroni G et al (2005) Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology 42:343–352
Reyes-Gordillo K et al (2014) Mechanisms of action of acetaldehyde in the up-regulation of the human alpha2(I) collagen gene in hepatic stellate cells: key roles of Ski, SMAD3, SMAD4, and SMAD7. Am J Pathol 184:1458–1467
Perez-Aso M, Fernandez P, Mediero A, Chan ES, Cronstein BN (2014) Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3. FASEB J 28:802–812
Chan ES et al (2013) Adenosine A(2A) receptors promote collagen production by a Fli1- and CTGF-mediated mechanism. Arthritis Res Ther 15:R58
Peng Z et al (2008) Ecto-5′-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. Nucleosides Nucleotides Nucleic Acids 27:821–824
Peng Z et al (2008) Ecto-5′-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J 22:2263–2272
Che J, Chan ES, Cronstein BN (2007) Adenosine A2A receptor occupancy stimulates collagen expression by hepatic stellate cells via pathways involving protein kinase A, Src, and extracellular signal-regulated kinases 1/2 signaling cascade or p38 mitogen-activated protein kinase signaling pathway. Mol Pharmacol 72:1626–1636
Wang H et al (2014) Caffeine inhibits the activation of hepatic stellate cells induced by acetaldehyde via adenosine A2A receptor mediated by the cAMP/PKA/SRC/ERK1/2/P38 MAPK signal pathway. PLoS One 9:e92482
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, J., Liu, X., Gao, B. et al. New Approaches for Studying Alcoholic Liver Disease. Curr Pathobiol Rep 2, 171–183 (2014). https://doi.org/10.1007/s40139-014-0053-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-014-0053-z