
Abstract
Loss of the light-sensing photoreceptors results in vision impairment and blindness, representing one of the main causes for disability in industrialized societies. Although the pathophysiology of diseases such as age-related macular degeneration, retinitis pigmentosa, or cone-rod dystrophies is highly heterogeneous, the final endpoint of such conditions is the degeneration of photoreceptors with no established therapies currently available for their replacement. However, recent preclinical studies demonstrated the feasibility of cell transplantation into the adult mammalian retina. Indeed, young post-mitotic photoreceptors represent a suitable cell type for such replacement approach as they showed proper integration and maturation following grafting. Importantly, donor photoreceptors connected to endogenous neurons allowing some functional rescue in mouse models of night blindness. Furthermore, methods for the generation of transplantable photoreceptors from pluripotent stem cells in vitro have been established. Here, we will review recent developments in the field of photoreceptor transplantation and will discuss still existing roadblocks toward clinical application.
Similar content being viewed by others

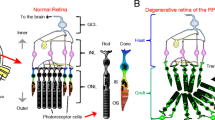

Introduction
In industrialized countries, vision impairment and blindness are primarily caused by degenerative processes within the retina, the light-sensing tissue inside the eye. Age-related macular degeneration (AMD), retinitis pigmentosa (RP), or cone-rod dystrophies are examples of retinal degenerations that eventually lead to loss of vision. A wide range of genetic defects and several environmental factors have been identified as a cause or effectors for such retinal degenerations that show a variety of heterogeneous pathophysiologies. However, in all mentioned conditions, eventually the light-detecting photoreceptors (rods and/or cones) are lost either by direct photoreceptor damage or by dysfunction/loss of supporting cells. The leading cause for blindness in elderly people over 50 years in industrialized countries is AMD (http://www.nei.nih.gov/health/maculardegen/armd_facts.asp). AMD is a multifactorial disease in which age, smoking, and genetic predisposition(s) were identified as major risk factors. Here, the central cone photoreceptors die and cause symptoms including color blindness (achromatopsia), reduced visual acuity, difficulties in light adaptation, and visual scotomas [1]. There are two forms of AMD: non-exudative AMD [also called dry-AMD or geographic atrophy (GA)] and exudative AMD (also called wet-AMD). In dry-AMD, cellular and protein debris, called drusen, accumulate between the retinal pigment epithelium (RPE) and Bruch’s membrane, causing an induction of chronic inflammation in RPE cells that results in RPE and retinal atrophy together with the release of pro-inflammatory cytokines [1]. In wet-AMD, additionally choroidal neovascularization appears, followed by higher permeability, thinning of vessels and disruption of the Bruch´s membrane integrity. Leaking vessels cause subretinal hemorrhage, fluid exudation, retinal detachment, and degeneration of the RPE [1]. Subsequently, mainly cone photoreceptors in the central macula area die. In GA patients, the degeneration can progress over years, while in cases of wet-AMD, sudden vision loss within days/weeks can occur [1].
In contrast to AMD, RP is seen to be an inherited genetic disease with a prevalence of 1:4,000 [2]. Up to now, more than 100 RP-causing mutations were identified and many of them were found in more than 45 photoreceptor—as well as RPE-related genes, leading to RPE and/or photoreceptor malfunction or loss [2, 3]. Although displaying variable pathologies, in most cases initially rod photoreceptors degenerate, resulting in night blindness and reduced peripheral vision (termed tunnel vision). This is followed by secondary degeneration of cones, leading also to loss of central vision and eventually blindness [4]. Depending on the disease-causing mutation, its occurance in RP patients ranges from early (childhood) to late onset and either can result in blindness within a few years or might develop slowly, that patients never get blind within their life time [4]. Although several therapeutic approaches were reported to slow down the progression of retinal degeneration, until now no established treatments exist for these conditions. Among others, surgical interventions including removal of neovascularization [5] or translocation of the macula toward a less affected region [6] were applied in patients. Controversially discussed as well is the nutritional supplementation of RP and AMD patients with antioxidants such as vitamins A, C, E, carotenoids or zinc. Although this treatment is suggested to slow down degeneration, it also carries certain risks, especially regarding the toxicity of high doses of vitamin A when taken over extended time periods or the varying absorption characteristics of vitamin A, depending on formulation or the amount of dietary fat intake of patients [7–11]. However, approaches such as the intravitreal injection of anti-angiogenic factors to reduce neovascularization in wet-AMD by blocking vascular endothelial growth factor A (VEGF-A), such as ranibizumab (Lucentis, Genentech) or bevacizumab (Avastin, Genentech), revealed reduced degeneration and increased visual acuity in clinical trials [12, 13]. Additionally, promising results were achieved while treating retinal dystrophy patients by gene therapy. Mutations in the gene RPE65 lead to early onset of severe retinal dystrophy (Leber’s congenital amaurosis; LCA) with development of poor vision during childhood and loss of vision later on. Replacement of the defective gene via adeno-associated virus (AAV)-mediated subretinal delivery of a vector encoding for RPE65 complementary deoxyribonucleic acid (cDNA) under the control of a human RPE65 [14] or ubiquitous actin promoter [15] showed improvements in recent clinical trials. Patients revealed enhancement in visual function as shown by microperimetry and dark-adapted perimetery [14] beside improvements in subjective and objective measurements of vision (dark adaptometry, pupillometry, electroretinography, nystagmus, and ambulatory behavior) [15]. Although patients’ vision improved significantly over short- and long-term periods, their retina was not protected from an ongoing process of degeneration, indicating the need of an earlier administration or combinational therapy that rescues function and, at the same time, slows down retinal degeneration [16]. Furthermore, such gene therapeutic approach is highly specific for single, known mutations and can only be successfully implemented when applied at early stages of the disease, when most target cells are still present.
Alternatively, epi- or subretinal implantation of artificial devices, aiming to replace parts of the degenerated retina, was applied in clinical studies to patients suffering from retinal degeneration and demonstrated first promising results. Following implantation of the retinal prosthesis, visual impaired patients were able to perform motion detection tasks which they were not able to do with their native vision [17]. Additionally, identification, localization, and discrimination of objects improved significantly [18], allowing some patients even to identify letters and words following implantation [19].
Cell transplantation into the mammalian retina
The replacement of degenerated photoreceptors in AMD or RP patients by cell transplantation represents another promising treatment alternative. Since the late 1980s, researchers started exploring the possibility of cell therapy in the eye [20–24]. From then on, a wide variety of approaches were evaluated using different cell types (e.g., embryonic or postnatal full retinal [25, 26] or photoreceptor sheets [26, 27], neural stem cells [28], and retinal progenitor cells [29]), which were grafted into different model organisms resulting in some improvements in visual function [30–32]. Starting with pioneering work of Peter Gouras and colleagues in the late 1980s, first pre-clinical evidence for the potential use of postnatal primary retinal cell transplantation for the replacement of photoreceptors was provided [33–37]. Transplanted cells were able to form rudimentary outer segments and synaptic terminals [30, 35]. Further studies by MacLaren et al. [38] and Bartsch et al. [39] identified the most appropriate cell type and correct developmental stage at which donor cells should be transplanted. Using the mouse as a model system, these studies provided evidence that young post-mitotic photoreceptors isolated at postnatal day 4 or 5 (P4/5) rather than proliferating multipotent retinal progenitor/stem cells displayed the potential to correctly integrate into the host tissue after grafting. Following subretinal transplantation, such photoreceptor precursors integrated into the host outer nuclear layer (ONL) and acquired a mature photoreceptor morphology forming synaptic terminals and both inner and outer segments [38, 39]. Interestingly, donor cells isolated from other developmental stages, including the adult retina, were also able to integrate, but in significantly lower numbers [40]. However, following transplantation, the overall integration rates of photoreceptors were very low compared to the total amount of injected donor cells and several strategies were recently evaluated to address this limitation including purification of cells suitable for integration and modulation of the host tissue.
Enrichment of photoreceptors prior transplantation yields increased integration rates
The promising results of cell transplantation studies might open the field of photoreceptor replacement therapies, but the low integration rate of donor photoreceptors demand improvements for such approach. Hence, using transgenic mice expressing fluorescent reporter proteins such as green fluorecent protein (GFP) exclusively in photoreceptors (e.g., Nrl-GFP, Crx-GFP transgenic mice), cells of the photoreceptor lineage were enriched by flow cytometry prior transplantation resulting in significantly increased integration rates [41∙∙]. This approach was brought a step closer to clinical application with the identification of ecto 5′-nucleotidase (Nt5e or CD73) as a cell surface antigen for rod photoreceptors [42∙, 43, 44], The use of cell surface markers allows the enrichment of donor cells by magnetic-activated cell sorting (MACS) [42∙], a technique already routinely used in other clinical settings, e.g., for the enrichment of hematopoietic stem cells, and it further avoids the need for genetic engineering of donor cells for the expression of a reporter protein. Other cell surface antigens specific for young photoreceptors were identified, but the current lack of reliable antibodies limits their application [45]. The use of cell surface markers might be also a reliable method for enrichment of stem cell-derived photoreceptors (see below). However, combination of several markers and/or positive/negative sorting procedures might be necessary to receive highly enriched donor cell fractions as CD73 has been shown to be expressed also on other cell types including T- and B cell subsets [46] or undifferentiated embryonic stem cells (ESCs) [47∙∙]. However, despite increasing integration rates by enrichment procedures, the majority of donor photoreceptors still remain in the subretinal space of the host. As genetically marked (e.g. Nrl-GFP) or CD73-expressing photoreceptors still represent a heterogeneous photoreceptor population, including cells approximately born in a time range from embryonic day (E) 12 to P4, the identification of the most suitable developmental stage for integration and distinct sorting procedures might be advantageous to further improve transplantation efficiencies.
Influence of the host environment on donor photoreceptor integration
Other approaches were investigated to increase the efficiency of photoreceptor transplantation focusing on the host retina. Manipulation of the host environment for regenerative approaches has been performed within other regions of the central nervous system (CNS), e.g., reduction of reactive gliosis, digestion of extracellular matrix (ECM) components, or use of inhibitory antibodies in spinal cord injuries [48, 49]. Indeed, several lines of evidence suggested the influence of the host tissue on donor cell integration also within the retina. Transplantation of in vitro expanded retinal stem/progenitor cells into the embryonic-like retina of the newborn opossum allowed significant increase in the number of integrated donor cells when compared to more mature stages [50]. Furthermore, deficiencies of reactive gliosis indicators such as glial fibrillary acidic protein (GFAP) or vimentin were suggested to have positive effects on retinal cell integration into the adult mouse retina [51]. Conversely, significantly increased numbers of integrated neural stem cells within the adult rodent retina were observed following vitreal transplantation when the host retina was injured during the injection process [52, 53], which was associated with increased GFAP expression in Müller glia cells. First studies evaluated the influence of modulations to the host retina for the integration of transplanted photoreceptor precursor cells. The ectopic expression of growth factors that showed higher expression in the developing retina such as IGF-1, FGF-2, and CNTF had only minor influence on donor photoreceptor integration into the adult mouse retina, with IGF-1 slightly promoting and CNTF reducing cell integration, while FGF-2 showed no significant effect [54]. Furthermore, pharmacologic or genetic disruption of the outer limiting membrane (OLM), a barrier formed by Müller glia endfeed and photoreceptors at the level of inner segments that seals the ONL from the subretinal space, has been shown to significantly increase photoreceptor integration [55, 56]. Another approach that allowed increased donor photoreceptor integration into the host retina was the digestion of ECM components such as chondroitin proteoglycans by chondroitinase ABC [57, 58∙∙]. Recently, Barber et al. [58∙∙] provided evidence that the different integration levels of transplanted rod photoreceptors might also depend on the degeneration mouse model used and the stage of disease progression [58∙∙].
Thus, each degeneration mouse model presents itself with its own specific features: different time onset of degeneration, varying levels of gliosis, and OLM integrities [58∙∙] which result in different integration rates of transplanted rod photoreceptors. Such findings highlight the need to tailor therapeutic approaches based on a specific degeneration and the development of distinct strategies to improve the efficiency of cell transplantation into the dystrophic retina. In addition to the manipulation of the host environment, improvements in the surgical procedure such a scleral puncture, retinal pre-detachment, and double injections resulted in a significant increase of integrated photoreceptors [59∙∙]. Such improved methodology resulted in functional rescue of rod-mediated (scotopic) vision in a slow degenerating night blindness mouse model [58∙∙]. Restoration of scotopic vision was assessed by different behavioral (optokinetic head tracking and two-choice water maze) and functional tests (patch-clamping, calcium imaging, and optic intrinsic imaging), confirming functional integration of donor photoreceptors into the host neural circuitry [59∙∙, 60]. Similarly, first indications for the restoration of scotopic vision were also provided following rod photoreceptor transplantation in a late-stage RP mouse model [61∙].
The integration process of transplanted photoreceptors
The transplantation of young post-mitotic photoreceptors into the subretinal space of adult recipients represents an artificial condition for photoreceptor migration and maturation—a situation not appearing during normal life. First investigations into this phenomenon revealed that transplanted donor photoreceptors predominantly integrate as single cells in a defined sequence of morphological changes that include movement of the cell body through the OLM and further migration into the ONL, thereby generating first an apical process that remains in the apical interphotoreceptor matrix and then a basal process growing toward the outer plexiform layer (OPL) forming a synaptic terminal [62∙]. The integration process is almost completed within the first week after transplantation with donor photoreceptors generating synaptic contacts to endogenous second-order neurons and the formation of outer segments [62∙]. Interestingly, the expression of late photoreceptor markers such as α-transducin was delayed compared to normal development. Additionally, some donor photoreceptors integrated as small cell groups (5–20 cells) in clusters or columns or even showed en mass entry (up to 100 cells) into the host ONL, possibly due to injury of the recipient retina following subretinal injection [62∙]. However, the specific cellular and molecular mechanisms that allow young photoreceptors to successfully integrate into the mature mammalian retina are still not well studied. The observation of a predominant radial direction of integration suggests the involvement of Müller glia processes as they vertically span the entire retina from the OLM to the inner limiting membrane (ILM). Indeed preliminary results from our laboratory showed direct contacts between integrating donor photoreceptors with endogenous Müller glia processes (Santos-Ferreira, T and Ader, M; unpublished observation).
Long-term survival of donor photoreceptors
Despite successful integration of donor photoreceptors, long-term survival is critical for a cell replacement approach. However, a significant decrease of integrated donor photoreceptors was observed from four months on after transplantation when compared to earlier time points [63]. Although the retina is seen as an immune-privileged organ [64], the injury and long-term existence of donor cells might induce immune responses. Indeed, activation of resident microglia and infiltration of macrophages and T cells have been observed following photoreceptor transplantation, and long-term survival of grafted photoreceptors has been shown to slightly improve following systemic immune suppression by cyclosporine A treatment [63]. However, the underlying specific mechanisms for long-term graft rejection have not been fully identified. Such knowledge might be essential for the development of distinct immune modulatory regimes as an important prerequisite for future clinical applications to avoid the need for systemic immune suppression with its deleterious side effects.
Transplantation of cone photoreceptors
Contrary to the nocturnal rodent, human vision mainly relies on cone photoreceptors. Thus, it is important to develop strategies for cone photoreceptor replacement. However, to date very little is known about cone photoreceptor transplantation [41∙∙]. Injection of Crx-positive cells isolated at E 15.5, a time point that corresponds with the peak of cone photoreceptor generation in mice, resulted in correct integration of cone photoreceptors, however, also at this ontogenetic stage the majority of integrated cells resembled rod photoreceptors [41∙∙]. Such outcome might reflect the plasticity of the donor cells at the time of isolation and the influence of the host tissue to commit donor cells to a specific cell fate. Despite these advances, the majority of studies focusing on photoreceptor replacement therapies were performed in rodents using rods. Recently, Wang et al. [65] transplanted dissociated retinal tissue isolated from the pig eye at different developmental stages into the adult pig retina. Depending on the age of the donor cells, they observed the generation of more cones when isolated at earlier stages (at E50 or E57) or rod photoreceptors when isolated at later stages (at E65) within the recipient retina [65], in line with previous studies in mice [41∙∙]. However, the lack of an abundant source and enrichment methods for cone photoreceptors currently limits the investigation of cone photoreceptor transplantation for the restoration of cone-mediated visual function.
Generation of photoreceptors from pluripotent stem cells
P4/5 was identified in mice as the optimal developmental stage for isolation of rod photoreceptors and successful transplantation. Translation of this finding to humans would presumably require the isolation of donor cells from fetuses in the second trimester post-conception [66]. Such cell source raises critical ethical concerns beside problems associated with availability and logistics. Thus, an in vitro expandable cell source to provide high amounts of donor cells will be mandatory for the development of cell replacement therapies in the retina. Since the observation that human ESCs (hESCs) are expandable in vitro [67] and that human somatic cells can be reprogrammed to a pluripotent state (human induced pluripotent stem cells, hiPSC) [68], the potential of pluripotent stem cell (PSC)-derived cells for therapeutic approaches are under extensive investigation. Indeed, in recent years several laboratories successfully established protocols that allowed the in vitro generation of retinal cells including RPE and photoreceptors from mouse PSCs. First attempts used 2D culture systems and the inhibition of Wnt/ß-catenin (by, e.g., noggin, Lefty A) and BMP (by, e.g., Dkk1) signaling pathways in combination with growth factors (e.g., IGF-1) to induce the differentiation of PSCs along the retinal lineage [69, 70]. Indeed, expression of early eye field markers including Rx, Pax6, Six3, Six6, and Lhx2 were observed following induction. Further differentiation induced the expression of retinal progenitor cell markers like Chx10 characteristic for the neuroretina and, on the other side, markers specific for developing RPE cells like Mitf [69–71]. Cell specification of PSC-derived retinal progenitors toward more mature retinal phenotypes was achieved by adding several components to the culture medium. For the generation of retinal neurons including photoreceptors, media compositions contained, e.g., RA, taurine, SHH, or FGFs [70], and for the generation of RPE cells, Activin A or Nicotinamide were used [72, 73]. Stem cell-derived photoreceptors expressed characteristic photoreceptor markers including Crx, Nrl, rhodopsin, recoverin, gnat1, red green opsin, and blue opsin. However, the overall amount of photoreceptors generated by these 2D culturing methods was relatively low. On the other hand, stem cell-derived RPE cells showed the formation of cobblestone monolayers and pigmentation besides the expression of ZO1, RPE65, or bestrophin and the capacity of phagoytosis of photoreceptor outer segments [72–74]. Isolation of pigmented cells from differentiating PSC cultures and further passaging was used for the generation of almost pure RPE cell populations and recent improvements of culturing methods allow the complete direct conversion of hESCs toward RPE [73, 75].
Recently, 3D culturing approaches of PSCs showed dramatic improvements in regard to the generation of retinal phenotypes [76∙∙, 77∙∙]. Actually, these protocols demonstrated a remarkable degree of self-organization as PSCs generated optic cup-like structures and stratified retinal tissue in vitro. By culturing PSCs as floating aggregates in medium containing basement-membrane matrix components (matrigel; specifically laminin and entactin) retinal epithelium was generated that spontaneously formed optic vesicle-like structures expressing eye field transcription factors like Rx and Pax6 [76∙∙]. Such evaginated retinal tissue became patterned along the proximal–distal axis with the proximal part differentiating along a RPE phenotype, whereas the distal part invaginated and showed a neural retina marker expression profile reminiscent of an optic cup. Excision of such optic cup-like structures and further cultivation led to spontaneous formation of stratified retina containing three nuclear layers and distinct retinal cell types including photoreceptors, bipolar-, horizontal-, amacrine- and retinal ganglion cells beside Müller glia. Interestingly, inhibition of notch signaling by the chemical compound DAPT further promoted the generation of photoreceptors [76∙∙]. In summary, studies over the recent years provided evidence for successful differentiation of PSCs toward retinal phenotypes. Interestingly, the temporal order of stage-specific expression profiles followed in vivo retinal development and the optimization of the differentiation protocols have led to the generation of stratified retinal tissue that contained high amounts of photoreceptors. Importantly, propagation of PSCs along the retinal and photoreceptor fate was not only achieved with mouse, but also with human PSCs, however, with considerable longer time needed for the efficient generation of specified retinal neurons including photoreceptors [77∙∙, 78].
Transplantation of PSC-derived photoreceptors
The efficient generation of photoreceptors from PSCs raised the question about their potential as donor material for transplantation into the adult mammalian retina. Indeed, evidence for successful integration of mouse ESC-derived photoreceptors was recently provided [47∙∙, 79, 80]. Photoreceptors were generated in these studies using a modified version of the 3D differentiation protocol provided by the Sasai group [76∙∙]. Gonzalez-Cordero et al. [47∙∙] further enriched ESC-derived photoreceptors by flow cytometry due to rod-specific expression of GFP using AAV-mediated transduction whereas Decembrini et al. [80] used a photoreceptor-specific ESC reporter line (Crx-GFP) for this purpose. In vitro-generated photoreceptors correctly integrated into the retina of adult wild-type mice as well as three genetic mouse models of retinal degeneration, similar to primary photoreceptors directly isolated from the developmental retina (see above), and formed cells with the cardinal features of mature photoreceptors, i.e., a nucleus-containing cell body in the ONL, apical located inner and outer segments beside an axonal terminal within the OPL in close proximity to endogenous bipolar cells [47∙∙, 80]. Interestingly, highest integration of mouse ESC-derived photoreceptors was observed when collected for transplantation after 25/26 days of in vitro differentiation, a developmental stage closely resembling primary photoreceptors at P4–P8 in the mouse that showed highest integration rates. However, the absolute number of integrated mouse ESC-derived donor photoreceptors was significantly lower than of photoreceptors directly isolated and enriched from the developmental mouse retina [38, 39, 41∙∙, 42∙, 44, 58∙∙]. Interestingly, whereas photoreceptors derived from 3D-cultured mESCs showed potential for integration into adult hosts, photoreceptors derived from 2D cultures were unable to do so [81]. This might be explained by the observation that photoreceptors from 3D cultures expressed higher levels of postnatal rod genes and might therefore more closely resemble the maturation stage of P4–P8 primary photoreceptors making them more eligible for integration.
An important topic generally discussed concerning the use of PSC-derived cells for regenerative medicine are safety issues, particularly the generation of tumors upon transplantation. Indeed, undifferentiated PSCs are characterized by teratoma formation after transplantation into host tissue including the retina [81, 82] and, thus, in a therapeutic setting the avoidance of contamination by undifferentiated PSCs within the injected cell suspension will be mandatory. Recent studies suggested two general routes to prohibit tumor growth following transplantation into the retina: (i) depletion of remaining pluripotent stem cells and enrichment of target cells and/or (ii) complete differentiation of PSCs prior to transplantation. Indeed, following a 20-day differentiation period and subsequent transplantation of PSC-derived retinal cells into wild-type or retinal degeneration mice tumor formation was regularly observed [79, 81, 82]. Furthermore, mouse iPSCs differentiated in a 2D culture system for 33 days along the retinal lineage still contained up to 30 % of cells expressing the pluripotency marker SSEA-1 [82]. However, by using magnetic bead depletion of cells expressing SSEA-1, tumorigenicity was significantly reduced and completely avoided following double depletion [82]. On the other side, prolonged differentiation times combined with enrichment of photoreceptor precursors by flow cytometry [81] and improved protocols using 3D culture systems [47∙∙, 80] or inhibition of the Wnt signaling pathway [79] allowed the generation of purified PSC-derived photoreceptors that resembled the developmental stage of post-mitotic photoreceptors. Such conditions fully avoided tumor formation after transplantation into the retina [47∙∙, 79, 80].
Importantly, efficient generation of retinal phenotypes or optic cup-like tissue was also observed following differentiation of hESCs or hiPSCs [77∙∙, 78, 83–86] and first studies also showed integration of hESC- and hiPSC-photoreceptors into the adult mouse retina [83, 86] leading to some visual rescue in a mouse model of retinal degeneration [83]. Additionally, methods have been developed for freezing and thawing of hESC-derived optic cup-like tissue that would allow storage and transportation of donor material [77∙∙]. However, the differentiation times for the generation of transplantable photoreceptors from human PSCs remains long, with more than 80 days of differentiation in vitro, but photoreceptor production from hESCs could be accelerated by inhibiting Notch signaling using DAPT [77∙∙]. However, human PSC-derived photoreceptors showed low integration rates following transplantation into the mouse retina [83, 86] and the formation of mature photoreceptors containing proper outer segments and synaptic terminals allowing functional repair remains to be investigated in more detail in future studies. Interestingly, a recent study by Zhong et al. [78] provided first evidence for the generation of rudimentary outer segment-like structures in vitro following prolonged (week 27) cultivation of human iPSC-derived optic cups.
Summary/Conclusion
Proof-of-concept studies in pre-clinical animal models have provided strong evidence for the feasibility of photoreceptor replacement in the adult mammalian retina. Furthermore, recent developments in the differentiation of PSCs along retinal lineages now allow the generation of sufficient amounts of donor material, and PSC-derived photoreceptors might therefore currently represent the most promising source for cell replacement studies in retinal degenerations. Further improvements in regard to integration efficiencies by modulating the recipient tissue and the directed generation of rod or cone photoreceptors beside their specific enrichment will be of utmost importance for developing cell-based strategies toward clinical applications aiming to treat retinal degenerative diseases. Whether other techniques such as direct conversion of somatic cells, as it has been reported for the generation of dopaminergic neurons for the treatment of Parkinson’s disease [87], will also allow the generation and provision of transplantable photoreceptors has still to be investigated but might represent another attractive route for generating sufficient amounts of donor material.
References
Papers of particular interest, published recently, have been highlighted as: ∙ Of importance ∙∙ Of major importance
Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358(24):2606–17.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–809.
Ramsden CM, Powner MB, Carr A-JF, Smart MJK, da Cruz L, Coffey PJ. Stem cells in retinal regeneration: past, present and future. Development. 2013;140(12):2576–85.
Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40.
Hawkins BS, Bressler NM, Miskala PH, Bressler SB, Holekamp NM, Marsh MJ, et al. Surgery for subfoveal choroidal neovascularization in age-related macular degeneration: ophthalmic findings: SST report no. 11. Ophthalmology. 2004;111(11):1967–80.
Mruthyunjaya P, Stinnett SS, Toth CA. Change in visual function after macular translocation with 360 degrees retinectomy for neovascular age-related macular degeneration. Ophthalmology. 2004;111(9):1715–24.
Age-Related Eye Disease Study Research Group. A randomized, placebocontrolled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417–36.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761–72.
Fielder AR. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(11):1463 author reply 1463–1466.
Gaynes BI. AREDS misses on safety. Arch Ophthalmol. 2003;121(3):416–7.
Massof RW, Fishman GA. How strong is the evidence that nutritional supplements slow the progression of retinitis pigmentosa? Arch Ophthalmol. 2010;128(4):493–5.
Rosenfeld PJ, Heier JS, Hantsbarger G, Shams N. Tolerability and efficacy of multiple escalating doses of ranibizumab (Lucentis) for neovascular age-related macular degeneration. Ophthalmology. 2006;113(4):623.e1.
Spaide RF, Laud K, Fine HF, Klancnik JM Jr, Meyerle CB, Yannuzzi LA, et al. Intravitreal bevacizumab treatment of choroidal neovascularization secondary to age-related macular degeneration. Retina. 2006;26(4):383–90.
Bainbridge JWB, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–9.
Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374(9701):1597–605.
Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA. 2013;110(6):E517–25.
Dorn JD, Ahuja AK, Caspi A, da Cruz L, Dagnelie G, Sahel J-A, et al. The detection of motion by blind subjects with the epiretinal 60-electrode (Argus II) retinal prosthesis. JAMA Ophthalmol. 2013;131(2):183–9.
Stingl K, Bartz-Schmidt KU, Besch D, Braun A, Bruckmann A, Gekeler F, et al. Artificial vision with wirelessly powered subretinal electronic implant alpha-IMS. Proc Biol Sci. 2013;280(1757):20130077.
Da Cruz L, Coley BF, Dorn J, Merlini F, Filley E, Christopher P, et al. The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br J Ophthalmol. 2013;97(5):632–6.
Del Cerro M, Notter MF, del Cerro C, Wiegand SJ, Grover DA, Lazar E. Intraretinal transplantation for rod-cell replacement in light-damaged retinas. J Neural Transpl. 1989;1(1):1–10.
Del Cerro M, Notter MF, Grover DA, Olchowka J, Jiang LQ, Wiegand SJ, et al. Retinal transplants for cell replacement in phototoxic retinal degeneration. Prog Clin Biol Res. 1989;314:673–86.
Del Cerro M, Gash DM, Rao GN, Notter MF, Wiegand SJ, Gupta M. Intraocular retinal transplants. Invest Ophthalmol Vis Sci. 1985;26(8):1182–5.
Gouras P, Lopez R. Transplantation of retinal epithelial cells. Invest Ophthalmol Vis Sci. 1989;30(8):1681–3.
Lopez R, Gouras P, Brittis M, Kjeldbye H. Transplantation of cultured rabbit retinal epithelium to rabbit retina using a closed-eye method. Invest Ophthalmol Vis Sci. 1987;28(7):1131–7.
Peng Q, Thomas BB, Aramant RB, Chen Z, Sadda SR, Seiler MJ. Structure and function of embryonic rat retinal sheet transplants. Curr Eye Res. 2007;32(9):781–9.
Schuschereba ST, Silverman MS. Retinal cell and photoreceptor transplantation between adult New Zealand red rabbit retinas. Exp Neurol. 1992;115(1):95–9.
Huang JC, Ishida M, Hersh P, Sugino IK, Zarbin MA. Preparation and transplantation of photoreceptor sheets. Curr Eye Res. 1998;17(6):573–85.
Banin E, Obolensky A, Idelson M, Hemo I, Reinhardtz E, Pikarsky E, et al. Retinal incorporation and differentiation of neural precursors derived from human embryonic stem cells. Stem Cells. 2006;24(2):246–57.
Klassen H. Transplantation of cultured progenitor cells to the mammalian retina. Expert Opin Biol Ther. 2006;6(5):443–51.
Klassen HJ, Ng TF, Kurimoto Y, Kirov I, Shatos M, Coffey P, Young MJ. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest Ophthalmol Vis Sci. 2004;45(11):4167–73.
McGill TJ, Cottam B, Lu B, Wang S, Girman S, Tian C, Huhn SL, Lund RD, Capela A. Transplantation of human central nervous system stem cells: neuroprotection in retinal degeneration. Eur J Neurosci. 2012;35(3):468–77.
Seiler MJ, Aramant RB, Thomas BB, Peng Q, Sadda SR, Keirstead HS. Visual restoration and transplant connectivity in degenerate rats implanted with retinal progenitor sheets. Eur J Neurosci. 2010;31(3):508–20.
Gouras P, Du J, Kjeldbye H, Yamamoto S, Zack DJ. Long-term photoreceptor transplants in dystrophic and normal mouse retina. Invest Ophthalmol Vis Sci. 1994;35(8):3145–53.
Gouras P, Du J, Gelanze M, Kwun R, Kjeldbye H, Lopez R. Transplantation of photoreceptors labeled with tritiated thymidine into RCS rats. Invest Ophthalmol Vis Sci. 1991;32(5):1704–7.
Gouras P, Du J, Gelanze M, Lopez R, Kwun R, Kjeldbye H, et al. Survival and synapse formation of transplanted rat rods. J Neural Transpl Plast. 1991;2(2):91–100.
Gouras P, Du J, Kjeldbye H, Kwun R, Lopez R, Zack DJ. Transplanted photoreceptors identified in dystrophic mouse retina by a transgenic reporter gene. Invest Ophthalmol Vis Sci. 1991;32(13):3167–74.
Gouras P, Du J, Kjeldbye H, Yamamoto S, Zack DJ. Reconstruction of degenerate rd mouse retina by transplantation of transgenic photoreceptors. Invest Ophthalmol Vis Sci. 1992;33(9):2579–86.
MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, et al. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444(7116):203–7.
Bartsch U, Oriyakhel W, Kenna PF, Linke S, Richard G, Petrowitz B, et al. Retinal cells integrate into the outer nuclear layer and differentiate into mature photoreceptors after subretinal transplantation into adult mice. Exp Eye Res. 2008;86(4):691–700.
Gust J, Reh TA. Adult donor rod photoreceptors integrate into the mature mouse retina. Invest Ophthalmol Vis Sci. 2011;52(8):5266–72.
∙∙ Lakowski J, Baron M, Bainbridge J, Barber AC, Pearson RA, Ali RR, et al. Cone and rod photoreceptor transplantation in models of the childhood retinopathy Leber congenital amaurosis using flow-sorted Crx-positive donor cells. Hum Mol Genet. 2010;19(23):4545–59. First report on cell transplantation resulting in the integration of cone photoreceptors.
∙ Eberle D, Schubert S, Postel K, Corbeil D, Ader M. Increased integration of transplanted CD73-positive photoreceptor precursors into adult mouse retina. Invest Ophthalmol Vis Sci. 2011;52(9):6462–71. The authors took advantage of a cell surface marker (CD73) to isolate and enrich rod photoreceptor using magnetic associated cell sorting (MACS) circumventing the need for the expression of a transgenic reporter protein in photoreceptors. CD73- enrichment resulted in significant increased donor photoreceptor integration.
Koso H, Minami C, Tabata Y, Inoue M, Sasaki E, Satoh S, et al. CD73, a novel cell surface antigen that characterizes retinal photoreceptor precursor cells. Invest Ophthalmol Vis Sci. 2009;50(11):5411–8.
Lakowski J, Han Y-T, Pearson RA, Gonzalez-Cordero A, West EL, Gualdoni S, et al. Effective transplantation of photoreceptor precursor cells selected via cell surface antigen expression. Stem Cells. 2011;29(9):1391–404.
Postel K, Bellmann J, Splith V, Ader M. Analysis of cell surface markers specific for transplantable rod photoreceptors. Mol Vis. 2013;19:2058–67.
Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto- 5′-nucleotidase (CD73). Purinergic Signal. 2006;2(2):351–60.
∙∙ Gonzalez-Cordero A, West EL, Pearson RA, Duran Y, Carvalho LS, Chu CJ, et al. Photoreceptor precursors derived from three-dimensional embryonic stem cell cultures integrate and mature within adult degenerate retina. Nat Biotechnol. 2013; 31(8):741–47. The authors report the selection of GFP-labeled photoreceptors from 3D ESC-derived optic cups and their successful integration into the adult mouse retina following transplantation.
Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416(6881):636–40.
GrandPré T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417(6888):547–51.
Sakaguchi DS, Van Hoffelen SJ, Theusch E, Parker E, Orasky J, Harper MM, et al. Transplantation of neural progenitor cells into the developing retina of the Brazilian opossum: an in vivo system for studying stem/progenitor cell plasticity. Dev Neurosci. 2004;26(5–6):336–45.
Kinouchi R, Takeda M, Yang L, Wilhelmsson U, Lundkvist A, Pekny M, et al. Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat Neurosci. 2003;6(8):863–8.
Nishida A, Takahashi M, Tanihara H, Nakano I, Takahashi JB, Mizoguchi A, et al. Incorporation and differentiation of hippocampus-derived neural stem cells transplanted in injured adult rat retina. Invest Ophthalmol Vis Sci. 2000;41(13):4268–74.
Ader M, Meng J, Schachner M, Bartsch U. Formation of myelin after transplantation of neural precursor cells into the retina of young postnatal mice. Glia. 2000;30(3):301–10.
West EL, Pearson RA, Duran Y, Gonzalez-Cordero A, MacLaren RE, Smith AJ, et al. Manipulation of the recipient retinal environment by ectopic expression of neurotrophic growth factors can improve transplanted photoreceptor integration and survival. Cell Transpl. 2012;21(5):871–87.
West EL, Pearson RA, Tschernutter M, Sowden JC, MacLaren RE, Ali RR. Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp Eye Res. 2008;86(4):601–11.
Pearson RA, Barber AC, West EL, MacLaren RE, Duran Y, Bainbridge JW, et al. Targeted disruption of outer limiting membrane junctional proteins (Crb1 andZO-1) increases integration of transplanted photoreceptor precursors into the adult wild-type and degenerating retina. Cell Transpl. 2010;19(4):487–503.
Suzuki T, Akimoto M, Imai H, Ueda Y, Mandai M, Yoshimura N, et al. Chondroitinase ABC treatment enhances synaptogenesis between transplant and host neurons in model of retinal degeneration. Cell Transpl. 2007;16(5):493–503.
∙∙ Barber AC, Hippert C, Duran Y, West EL, Bainbridge JWB, Warre-Cornish K, et al. Repair of the degenerate retina by photoreceptor transplantation. Proc Natl Acad Sci USA. 2013;110(1):354–9. Barber and colleagues showed that rod photoreceptor replacement therapy has to be tailored to each degeneration type, taking into account its pathophysiology, such as ONL thickness, gliosis and OLM integrity.
∙∙ Pearson RA, Barber AC, Rizzi M, Hippert C, Xue T, West EL, et al. Restoration of vision after transplantation of photoreceptors. Nature. 2012;485(7396):99–103. Here, the authors significantly improved the transplantation method for photoreceptors, resulting in the first functional restoration of scotopic vision in a night blindness mouse model at the cellular and behavioral level.
Homma K, Okamoto S, Mandai M, Gotoh N, Rajasimha HK, Chang Y-S, et al. Developing rods transplanted into the degenerating retina of Crx-knockout mice exhibit neural activity similar to native photoreceptors. Stem Cells. 2013;31(6):1149–59.
∙ Singh MS, Charbel Issa P, Butler R, Martin C, Lipinski DM, Sekaran S, et al. Reversal of end-stage retinal degeneration and restoration of visual function by photoreceptor transplantation. Proc Natl Acad Sci USA. 2013;110(3):1101–6. The MacLaren lab provided evidence for the rescue of night vision in a late stage Retinitis Pigmentosa mouse model following photoreceptor transplantation.
∙ Warre-Cornish K1, Barber AC, Sowden JC, Ali RR, Pearson RA. Migration, integration and maturation of photoreceptor precursors following transplantation in the mouse retina. Stem Cells Dev. 2014;23(9):941–54. This study describes in detail the cellular mechanism of integration following rod photoreceptor transplantation into the adult mouse retina.
West EL, Pearson RA, Barker SE, Luhmann UFO, Maclaren RE, Barber AC, et al. Long-term survival of photoreceptors transplanted into the adult murine neural retina requires immune modulation. Stem Cells. 2010;28(11):1997–2007.
Streilein JW, Ma N, Wenkel H, Ng TF, Zamiri P. Immunobiology and privilege of neuronal retina and pigment epithelium transplants. Vision Res. 2002;42(4):487–95.
Wang W, Zhou L, Lee SJ, Liu Y, Fernandez de Castro J, Emery D, et al. Swine cone and rod precursors arise sequentially and display sequential and transient integration and differentiation potential following transplantation. Invest Ophthalmol Vis Sci. 2014;55(1):301–9.
Martins RAP, Pearson RA. Control of cell proliferation by neurotransmitters in the developing vertebrate retina. Brain Res. 2008;1192:37–60.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Lamba DA, Karl MO, Ware CB, Reh TA. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci USA. 2006;103(34):12769–74.
Osakada F, Ikeda H, Mandai M, Wataya T, Watanabe K, Yoshimura N, et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 2008;26(2):215–24.
Meyer JS, Shearer RL, Capowski EE, Wright LS, Wallace KA, McMillan EL, et al. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc Natl Acad Sci USA. 2009;106(39):16698–703.
Idelson M, Alper R, Obolensky A, Ben-Shushan E, Hemo I, Yachimovich-Cohen N, et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell. 2009;5(4):396–408.
Zhu Y, Carido M, Meinhardt A, Kurth T, Karl MO, Ader M, et al. Threedimensional neuroepithelial culture from human embryonic stem cells and its use for quantitative conversion to retinal pigment epithelium. PLoS One. 2013;8(1):e54552.
Westenskow PD, Moreno SK, Krohne TU, Kurihara T, Zhu S, Zhang Z-N, et al. Using flow cytometry to compare the dynamics of photoreceptor outer segment phagocytosis in iPS-derived RPE cells. Invest Ophthalmol Vis Sci. 2012;53(10):6282–90.
Buchholz DE, Pennington BO, Croze RH, Hinman CR, Coffey PJ, Clegg DO. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl Med. 2013;2(5):384–93.
∙∙ Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, et al. Selforganizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472(7341):51–6. This study showed for the first time the generation of optic cup-like tissue from mouse ESCs in a 3D culture system.
∙∙ Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, et al. Selfformation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10(6):771–85. Generation of optic cup-like tissue from human ESCs and en bloc cryopreservation of human stratified neural retina.
Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. 2014;10(5):4047.
Cui L, Guan Y, Qu Z, Zhang J, Liao B, Ma B, et al. WNT signaling determines tumorigenicity and function of ESC-derived retinal progenitors. J Clin Invest. 2013;123(4):1647–61.
Decembrini S, Koch U, Radtke F Moulin A, Arsenijevic Y. Derivation of traceable and transplantable photoreceptors from mouse embryonic stem cells. Stem Cells Reports. 2014;2(6):853–65.
West EL, Gonzalez-Cordero A, Hippert C, Osakada F, Martinez-Barbera JP, Pearson RA, et al. Defining the integration capacity of embryonic stem cell-derived photoreceptor precursors. Stem Cells. 2012;30(7):1424–35.
Tucker BA, Park I-H, Qi SD, Klassen HJ, Jiang C, Yao J, et al. Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PLoS One. 2011;6(4):e18992.
Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;4(1):73–9.
Meyer JS, Howden SE, Wallace KA, Verhoeven AD, Wright LS, Capowski EE, et al. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells. 2011;29(8):1206–18.
Phillips MJ, Wallace KA, Dickerson SJ, Miller MJ, Verhoeven AD, Martin JM, et al. Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest Ophthalmol Vis Sci. 2012;53(4):2007–19.
Lamba DA, McUsic A, Hirata RK, Wang P-R, Russell D, Reh TA. Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One. 2010;5(1):e8763.
Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature. 2011;476(7359):224–7.
Disclosure
Tiago Santos-Ferreira, Kai Postel, and Marius Ader have not reported any conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tiago Santos-Ferreira and Kai Postel contributed equally to this work.
Rights and permissions
About this article

Cite this article
Santos-Ferreira, T., Postel, K. & Ader, M. Photoreceptor transplantation into the mammalian retina. Curr Ophthalmol Rep 2, 91–99 (2014). https://doi.org/10.1007/s40135-014-0042-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40135-014-0042-8