
Abstract
We have proposed an optimized homogeneous precipitation method for the synthesis of spherical and well-dispersed pure Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) nanoparticles (NPs) using synthesized lanthanide sulfates and urea as starting materials. Ln2O2SO4 NPs can be easily transformed to Ln2O2S by reduction under H2 flow at 650 °C for Gd, Dy and Ho whereas the reduction with Lu gave only Lu2O3. Particle size was between 500 nm and 3.5 µm. This reduction method allows to obtain Ln2O2S without using H2S which is highly toxic and uncomfortable gas. Ln2O2SO4/Ln2O2S synthetized can be used for release and oxygen storage or doped with luminescent centers for multimodal medical imaging applications.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During this last decade, lanthanide oxysulfate and oxysulfide compounds (Ln = Gd, Ho, Dy and Lu) have been widely used due to their applications in phosphorescent material design, X-ray-computerized tomography, oxygen storage, and radiation detection [1,2,3]. The other remarkable features of lanthanide oxysulfide/oxysulfate are their large accurate sensing capability over a wide range of temperatures, as well as their ability to withstand a harsh environment [4]. Moreover, the crystal structure of Ln2O2SO4 is usually the alternative stacking of a [Ln2O2]2+ layer and a layer of sulfate [SO4]2− and that of Ln2O2S is the alternative stacking of a [Ln2O2]2+ layer and a layer of sulfide (S2−) [5]. Thus, Ln2O2SO4 is an intermediate phase for the synthesis of lanthanide oxysulfide Ln2O2S and is usually used as a precursor for the synthesis of Ln2O2S in reduction atmosphere.
The Ln2O2SO4 materials for oxygen storage and release reported so far have been prepared by several methods, such as calcination of Ln(SO4)3·nH2O [6,7,8], solid-state reaction method using lanthanide oxide and diammonium hydrogen sulfate as the starting materials [9, 10], utilization of precursors of layered Ln-dodecyl sulfate mesophases’ [11], Ln-precipitation [12, 13], thermal decomposition of nanodroplets formed by Ln-acetylacetonates (Ln(acac)3) [14] and hydrothermal decomposition of hydroxyl sulfates of Ln2(OH)4SO4 (Ln = Eu-Lu and Y) [15] .
All of these methods result to bulk materials with an irregular morphology. The synthesis of NPs intended for applications in imaging and medical therapy imposes the control of many physicochemical characteristics such as the crystal structure and the morphology of the particles. The soft chemistry processes for preserving texture, morphology and structure are, therefore, well adapted for the development of these NPs [16]. The homogeneity of the particles obtained, the control of the various steps of germination and growth of the crystallites originating from the liquid are the main advantages of these synthesis processes. Homogeneous precipitation synthesis is one of the most promising techniques because of its advantages such as the relatively simple synthetic route, low cost, ease of mass production.
Lian et al. [17]. have synthesized (Gd1 − x ,Eu x )2O2SO4 sub-microphosphors by homogeneous precipitation method from commercially available Gd2O3, Eu2O3,H2SO4 and (NH2)2CO (urea) starting materials. After precipitation reaction, the precursor which is mostly composed of gadolinium hydroxyl, carbonate and sulfate groups with some crystal water; can be transformed into pure Gd2O2SO4 phase after heating at 900 °C for 2 h in air. They reported that it is only when the molar ratio of urea to Gd2(SO4)3 is equal to 5, there is formation of pure Gd2O2SO4 phase. The particles were quasi-spherical with a mean size of about 500 nm. The same authors have also reported [18] the photoluminescence of (Gd1 − x ,Dy x )2O2SO4 using the same method. In that work, they use molar ratio of urea to Gd2(SO4)3 equal to 400, to form pure Gd2O2SO4 phase. Thus, there is no clearly in literature the optimal conditions about molar ratio of urea to Ln2(SO4)3 that can be used to synthesize Ln2O2SO4 NPs by homogeneous precipitation method.
This paper suggested an optimized homogeneous precipitation method to prepare spherical and well-dispersed pure Ln2O2SO4 and Ln2O2S (Ln = Gd, Ho, Dy and Lu) NPs using synthesized lanthanide (Ln = Gd, Dy, Ho and Lu) sulfates and urea as starting materials. Then, we studied the reduction reaction of Ln2O2SO4 under H2 to form Ln2O2S to avoid the use of dangerous sulfur-based gases.
Experimental section
Materials and method
Gadolinium, dysprosium, holmium and lutetium sulfate were synthesized at our laboratory. Urea (99%) was purchased from Reaktiv OAO (Saint-Petersburg, Russia).
Precursors were prepared by coprecipitation method as previously described [1, 2, 19] with some modifications using lanthanide (Ln = Gd, Dy, Ho and Lu) sulfates in deionized water and ethanol, using a protocol based on the decomposition of urea at temperatures around 85 °C. The optimum concentration employed in this study was [Ln3+] = 5.6 × 10−3 M. The concentration of urea was varied (0.125, 0.25 and 0.5 M). So molar ratio of urea to Ln3+ was equal to r = 22.3, 44.6 and 89.3. Solvent employed was deionized water–ethanol with 20 vol% of ethanol. Gd, Dy, Ho and Lu sulfates and urea were dissolved in the solvent, and the solution was placed in a round-bottom flask at reflux for aging at 85 °C in an oil bath, during 120 min for a total volume of 2L under vigorous stirring. The suspension was then centrifuged for 10 min at 6000 rpm. The supernatant of the solution is removed and the solid phase is suspended in deionized water for washing. The solid is dried in an oven at 80 °C overnight.
The precursors were heated at 800 °C during 2 h (5 °C/min) in argon to form Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) NPs.
In our previous work [1, 20, 21], lanthanide oxysulfides were prepared by sulfuration using Ar-H2S gas or H2-S mixture. Unfortunately, H2S is highly toxic and uncomfortable to work with. Therefore, for preparation of oxysulfides we adopted reduction of lanthanide oxysulfates in H2 flow. Ln2O2S (Ln = Gd, Ho, Dy and Lu) NPs were synthesized by heating Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) NPs at 650 °C during 5 h in H2 to form Ln2O2S.
Characterization techniques
X-ray diffractometry (XRD) measurements were performed on a DRON3 X-ray diffractometer using Co-Kα radiation (1.79021 Ȧ). The data were collected with 2θ value from 5° to 80°.
The determination of the different chemical bonds was made by infrared spectroscopy, using a PerkinElmer 100 Series spectrometer. The pellets were prepared by mixing the powders with potassium bromide (1/100 by weight).
Thermal analysis, i.e., thermogravimetry (TG) and differential thermal analysis (DTA), was performed using a Netzsch STA 449 F 3 Jupiter system. DTA and TG with 10 °C/min heating rate in the argon were done on the sample of 50 mg at the same time.
The particle morphology of the synthesized products was observed by a JEOL JSM-6510LV scanning electron microscope (SEM).
Results and discussion
XRD and SEM characterization of NPs
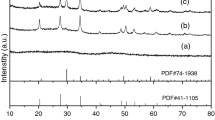
The XRD pattern of precursor (not shown here) did not reveal diffraction peaks. This observation is specific to amorphous or disordered compounds. Figure 1 shows the diffractograms of the precursors with various molar ratio of urea to Ln3+ after annealing in argon at 800 °C. When the molar ratio r is equal to 89.3 [(urea) = 0.5 M], the final products obtained are mainly composed of Ln2O2SO4 and a small amount of Ln2O3. With decreasing of r values to 22.3 [(urea) = 0.125 M], the disappearance of Ln2O3 diffraction peaks can be observed, indicating that more OH- or CO3 2− anions are substituted by SO4 2− anions as a result of excess of Ln2(SO4)3 dosage during homogeneous precipitation. Thus, the concentration of urea (r = 22.3) is chosen as the best condition in the rest of our work.

XRD patterns of product obtained from Dy precursor after annealing at 800 °C during 2 h (5 °C/min) in argon
The XRD patterns (Fig. 2) of the final products of all lanthanides (Ln = Gd, Ho, Dy and Lu) exhibit well-developed peaks, characteristic of lanthanide oxysulfate phases without precipitation of impurities.

XRD patterns of Ln2O2SO4 NPs obtained after annealing of precursor in Ar at 800 °C (2 h)
The lattice parameters were calculated and could be indexed to the orthorhombic phase of Ln2O2SO4 matched with the standard of the literature values [22] and presented in Table 1.
Figure 3 shows XRD patterns of reduction of Ln2O2SO4 (Ln = Gd, Dy and Ho) particles in H2 at 650 °C during 5 h. The patterns show well-developed peaks, characteristic of hexagonal lanthanide oxysulfide phases for Gd, Dy and Ho. All peaks are indexed according the ICDD data in the hexagonal phase, of Gd2O2S(File card No 020-1422), Dy2O2S(File card No 026-0592) and of Ho2O2S(File card No 025-1143), respectively, for Gd, Dy and Ho. No additional peak from impurities can be detected.

XRD patterns of: Ln2O2S particles obtained after reduction in H2 at 650 °C during 5 h (5 °C/min)
For Lu, reduction in H2 gave only lutetium oxide: Lu2O3 (XRD patterns not shown here) even though the reduction temperature was changed to 650, 690, and 800 °C.
Transformation of Ln2O2SO4 to Ln2O2S is commonly classified as a redox reaction and can be described by the equation:
For Lu, the presence of only lutetium oxide: Lu2O3 can be described by the equation:
These results are consistent with those reported by Andreev et al. [20, 21] on the transformation of Ln2O2SO4 bulk materials obtained by solid-state reaction to Ln2O2S under H2 flow.
SEM images corresponding to each Ln2O2SO4 before and after reduction are showed in Fig. 4. Before reduction, NPs are spherical and monodispersed in size with a mean diameter of 500 nm, 3.5 µm, 1.7 µm and 1.2 µm respectively for Gd, Dy, Ho and Lu. After reduction, the morphology of particles is conserved with a reduction in size with a mean diameter of 310 nm, 2.8 µm, 830 nm and 1 µm respectively for Gd, Dy, Ho and Lu. The difference in morphology observed could be explained by a difference in the reactivity of these lanthanides during the precipitation reaction and the effect of the famous “contraction of the lanthanides”. In fact, the particle size is significantly affected by a decrease in the effective ionic radius of the lanthanide ions. Reducing the radius with a simultaneous increase in the nuclear charge leads to an increase in the tendency of the particles to agglomerate. In our SEM pictures observation, two types of particles were identified. The particles sizes differ from nanoscale and monodisperse (Gd) to microscale (Ho, Dy, Lu). This division is not accidental. The synthesized compounds of gadolinium enter into the region of crystal-chemical instability and in their properties are similar to compounds of light rare-earth elements. While the elements from terbium to lutetium are heavy. Within the series of heavy rare earth elements, two families are distinguished: similar terbium (Tb-Tm, Y) and similar ytterbium (Yb-Lu). Thus, the differences in particle morphology are caused by the fact that compounds are formed by rare earth elements being representatives of different families.

SEM images of Ln2O2SO4: before (left) and after (right) reduction in H2 at 650 °C (5 h)
FT-IR spectroscopy characterization of NPs
Figure 5 showed FT-IR analysis spectra of precursor and its annealed product for each lanthanide.

FT-IR spectra of: a precusor and b Ln2O2SO4 particles
The FT-IR spectrum of the precursor for each lanthanide (Fig. 5a) shows bands attributed to the absorption peaks of physically absorbed water, crystal water, hydroxyl groups [23] (near 3500 and 1610 cm−1), the CO3 2− (near 1450 cm−1) and the SO4 2− anions (near 1125 and 680 cm−1), which shows that the precursor is consisted of lanthanide, hydroxyl, carbonate and sulfate groups with some crystal water. After calcination under argon at 800 °C (Fig. 5b), spectrum showed that the peak centered at 3500 cm−1 becomes weak, indicating that the removal of crystal water from the precursor. The SO4 2− absorption bands (near 1125 and 625 cm−1) split into some narrow sharp peaks, the absorption bands become weaker, and a new small absorption peak centered about 500 cm−1 appears, which corresponds to the characteristic vibration peaks of Ln–O bond in the prepared product. These observations are very similar to the ones already discussed in the previous work [1, 17, 18]. We, thus, conclude that the formulation of the NPs elaborated in a water–EtOH (80–20 vol%) mixture is Ln2(OH)2CO3SO4·xH2O (Ln = Gd, Dy, Ho and Lu), the lanthanide hydroxycarbonate sulfate. This precursor has been well transformed into Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) after annealing in argon at 800 °C.
DTA-TG characterization of NPs
The TG/DTA curves (Fig. 6) show the thermal decomposition under argon atmosphere of precursor Dy precursor and offer interesting result to complete the determination of its formula. The TG curve presents a continuous weight loss between 80 and 900 °C with an overall weight loss of approximately 17 wt%. This total weight loss mainly consists of the following steps in the whole temperature range. The weight loss in the temperature range from room temperature to ≈ 300 °C is about 7.5% by mass and represents the progressive removal of physically absorbed water from the precursor. This mass loss corresponds to a weak endothermic peak at around 160 °C in DTA curve. The weight loss between 300 and 500 °C is about 4.1% by mass, which is associated with the complete dehydroxylation of the precursor with the progressive decomposition of CO3 2− anions and emission CO2. This weight loss corresponds to an endothermic peak at 450 °C in the DTA curve. The weight loss in the temperature range from 500 to 693 °C is about 5.4% by mass, which is attributed with the full decomposition of CO3 2− anions and release CO2 of the precursor. Moreover, as shown in Fig. 6, small variations in mass are observed at temperatures higher than 750 °C on the TG curve. This is why we used 800 °C as the optimal heat treatment temperature for the synthesis of Ln2O2SO4 particles. The same thermal decomposition (Curves not shown here) was observed for Gd, Ho and Lu precursor. Thus, the formula of precursor is Ln2(OH)2CO3SO4·xH2O (Ln = Gd, Dy, Ho and Lu). These results are in a good agreement with those reported previously [17, 18] and with those obtained by FTIR analyses and XRD patterns.

TG-DTA curves of Dy precursor in argon
Conclusions
We have proposed an optimized homogeneous precipitation method to synthesize spherical and well-dispersed pure Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) NPs with size between 500 nm and 3.5 µm. If Ln2O2SO4 NPs can be easily transformed to Ln2O2S by reduction under H2 flow at 650 °C for Gd, Dy and Ho, Lu gave only Lu2O3 under H2 flow even though the reduction temperature was changed to 650, 690, and 800 °C. This reduction method allows to obtain Ln2O2S without using H2S which is highly toxic and uncomfortable gas. Ln2O2SO4/Ln2O2S synthesized can be used for release and oxygen storage or doped with luminescent centers for multimodal medical imaging applications.
References
Osseni, S.A., Lechevallier, S., Verelst, M., Dujardin, C., Dexpert, J.G., Neumeyer, D., Leclercq, M., Baaziz, H., Cussac, D., Santran, V., Mauricot, R.: New nanoplatform based on Gd2O2S:Eu3+ core: synthesis, characterization and use for in vitro bio-labelling. J. Mater. Chem. 21, 18365 (2011)
Osseni, S.A., Lechevallier, S., Verelst, M., Perriat, P., Dexpert, J.G., Neumeyer, D., Garcia, R., Mayer, F., Djanashvili, K., Peters, J.A., Magdeleine, E., Gros-Dagnac, H., Celsis, P., Mauricot, R.: Gadolinium oxysulfide nanoparticles as multimodal imaging agents for T-2-weighted MR, X-ray tomography and photoluminescence. Nanoscale. 6, 555–564 (2014)
Hoenderdaal, S., Espinoza, L.T., Marscheider-Weidemann, F., Graus, W.: Can a dysprosium shortage threaten green energy technologies? Energy. 49, 344 (2013)
Haynes, W.M.: Handbook of chemistry and physics. CRC Press/Taylor and Francis, Boca Raton (2014)
Zhukov, S., Yatsenko, A., Chernyshev, V., Trunov, V., Tserko-vnaya, E., Anston, O.: Structural study of lanthanum oxysulfate (LaO)2SO4. Mater. Res. Bull. 32, 43–50 (1997)
Ye, X., Collins, J.E., Kang, Y., Chen, J., Chen, D.T.N., Yodh, A.G., Murray, C.B.: Morphologically controlled synthesis of colloidal upconversion nanophosphors and their shape-directed self-assembly. Proc. Natl. Acad. Sci. 107, 22430–22435 (2010)
Rodrigues, R.V., Marciniak, L., Khan, L.U., Matos, J.R., Brito, H.F., Stręk, W.: Luminescence investigation of Dy2O2S and Dy2O2SO4 obtained by thermal decomposition of sulfate hydrate. J Rare Earths. 34(8), 814 (2016)
Andreev, O.V., Denisenko, Y.G., Sal’nikova, E.I., Khritokhin, N.A., Zyryanova, K.S.: Specifics of reactions of cerium sulfate and europium sulfate with hydrogen. Russ. J. Inorg. Chem. 61(3), 296–301 (2016)
Srivastava, A.M., Setlur, A.A., Comanzo, H.A., Gao, Y., Hannah, M.E., Hughes, J.A., Happek, U.: Optical spectroscopy and thermal quenching of the Ce3+ luminescence in yttrium oxysulfate, Y2O2[SO4]. Opt. Mater. 30, 1499–1503 (2008)
Andreev, P.O., Sal’nikova, E.I., Andreev, O.V., Denisenko, Y.G., Kovenskii, I.M.: Synthesis and upconversion luminescence spectra of (Y1–x–y Yb x Er y )2O2S solid solutions. Inorg. Mater. 53(2), 200–206 (2017)
Zhang, D.J., Eto, M., Ikeue, K., Machida, M.: Low-temperature synthesis of porous praseodymium oxysulfate oxygen storage materials by using a CTA template. J. Ceram. Soc. Jpn. 115, 597–601 (2007)
Shen, W., Naito, S.: Easy precipitation method for preparation of cerium added La2O2SO4 used for oxygen storage. Adv. Mater. Res. 886, 196–199 (2014)
Liu, Y., Tu, D., Zhu, H., Chen, X.: Lanthanide-doped luminescent nanoprobes: controlled synthesis, optical spectroscopy, and bioapplications. Chem. Soc. Rev. 42, 6924–6958 (2013)
Zhang, W., Martinelli, J., Mayer, F., Bonnet, C.S., Szeremeta, F., Djanashvili, K.: Molecular architecture control in synthesis of spherical Ln-containing nanoparticles. RSC Adv. 5, 69861–69869 (2015)
Wang, X., Molokeev, M.S., Zhu, Q., Li, J.G.: Controlled hydrothermal crystallization of anhydrous Ln2(OH)4SO4 as a new family of layered rare-earth hydroxide (Ln = Eu-Lu and Y). Chem. Eur. J. (2017). https://doi.org/10.1002/chem.201703282
Joliver J.P., Henry M., Livabe J.: De la solution à l’oxyde, Savoirs actuels InterEditions/CNRS (1994)
Lian, J., Sun, X., Li, X.: Synthesis, characterization and photoluminescence properties of (Gd 1 − x, Eu x)2 O2SO4 sub-microphosphors by homogeneous precipitation method. Mater. Chem. Phys. 125, 479–484 (2011)
Lian, J., Wang, W., Wang, B.: Photoluminescence of (Gd1-x, Dyx)2O2SO4 phosphors synthesized by homogeneous precipitation method. Adv. Mater. Res. 299, 612–615 (2011)
Lechevallier, S., Lecante, P., Mauricot, R., Dexpert, H., Dexpert-Ghys, J., Kong, H., Kong, H.K., Law, G.L., Wong, K.L.: Gadolinium–europium carbonate particles: controlled precipitation for luminescent biolabeling. Chem. Mater. 22, 6153–6161 (2010)
Andreev, P.O., Sal’nikova, E.I., Kislitsyn, A.A.: Kinetics of the transformation of Ln2O2SO4 into Ln2O2S (Ln = La, Pr, Nd, and Sm) in a hydrogen flow. Russ. J. Phys. Chem. A 87, 9 (2013)
Andreev, P.O., Sal’nikova, E.I., Kovenski, I.M.: Preparation of LnOS (Ln = Gd, Dy, Y, Er, Lu) in flowing hydrogen and hydrogen sulfide. Inorg. Mater. 50(10), 1018–1023 (2014)
Villars, P.: Material phases data system (MPDS), CH-6354 Vitznau, Switzerland SpringerMaterials (2017). http://materials.springer.com/. Accessed 5 May 2017
Liu, G.X., Hong, G.Y., Wang, J.X., Dong, X.T.: Hydrothermal synthesis of spherical and hollow Gd2O3: Eu3+ phosphors. J. Alloy. Comp. 432, 200–204 (2007)
Acknowledgements
This research was supported by Tyumen State University (Russia) by offering PostDoc Grant-2017.
Author information
Authors and Affiliations
Contributions
Sèmiyou. A. OSSENI carried out the synthesis and characterization of nanoparticles, the drafting of the manuscript and interpretation of the data. All authors read and approved the final manuscript. I Sèmiyou. A. OSSENI, the corresponding author of this manuscript, certify that the contributors statements included in this paper are correct and have been approved by all co-authors.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Osseni, S.A., Denisenko, Y.G., Fatombi, J.K. et al. Synthesis and characterization of Ln2O2SO4 (Ln = Gd, Ho, Dy and Lu) nanoparticles obtained by coprecipitation method and study of their reduction reaction under H2 flow. J Nanostruct Chem 7, 337–343 (2017). https://doi.org/10.1007/s40097-017-0243-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-017-0243-4