
Abstract
Gelatin hydrolysates from autolysed non-swollen and swollen unicorn leatherjacket skin prepared using partially purified glycyl endopeptidase (GE) from papaya latex were examined for their antioxidative activities. Autolysed swollen skin was more hydrolysed by GE as indicated by higher α-amino group content, compared with autolysed non-swollen skin. ABTS radical scavenging activity and ferric reducing antioxidant power (FRAP) of hydrolysates were increased with increasing levels of GE used. Antioxidative gelatin hydrolysate from autolysed skin, both non-swollen and swollen skins, using 8 % GE termed ‘NS-8GE’ and ‘SS-8GE’ exhibited different modes of action. When both hydrolysates were tested in lecithin liposome system, the efficiency in retardation of lipid oxidation was in a dose-dependent manner. Antioxidative activity of hydrolysates at 5.0 g/L was comparable to that of 0.1 g/L Trolox, in which the oxidation was almost completely inhibited. NS-8GE and SS-8GE also showed their antioxidative activities in gastrointestinal tract model system (GIMs). ABTS radical scavenging activity of both hydrolysates increased, but FRAP decreased in a duodenal condition. Based on size exclusion chromatography, major antioxidative peptides in NS-8GE and SS-8GE had molecular weight of 1,170 and 750 Da, respectively. Therefore, GE could enhance antioxidative activity of autolysed skin. Additionally, swelling process directly determined the modes of actions of resulting gelatin hydrolysates. NS-8GE had high ABTS radical scavenging activity, whereas SS-8GE showed high FRAP.
Similar content being viewed by others


Introduction
Lipid oxidation has been known as a serious cause of food deterioration as well as various human diseases, such as cardiovascular diseases and cancers (Lobo et al. 2010). The reactive oxygen species (ROS) such as hydroxyl radical (•OH), superoxide radical (O •−2 ), hydrogen peroxide (H2O2), hydroperoxyl radical (HO •−2 ), lipid peroxyl radical (LOO•), alkoxyl radical (LO•), and singlet oxygen (1O2) are the main initiators of lipid oxidation through free radical chain reaction (Min and Ahn 2005). Antioxidants are substances that can inhibit the generation of free radicals in a system and prevent or delay the lipid oxidation reaction. Generally, the most common synthetic antioxidants including butylated hydroxytoluene (BHT), butylated hydroxyanisole (BHA), tert-butylhydroquinone (TBHQ) and propyl gallate (PG) have been used to enhance the oxidative stability and inhibit lipid oxidation in lipid containing foods (Min and Ahn 2005; Xu 2008).
Nowadays, natural additives involving antioxidants have gained attention from consumers. Protein hydrolysates, especially from marine processing byproducts, have attracted interest due to their safety as well as bioactivities (Kim and Wijesekara 2010; Gómez-Guillén et al. 2011). Enzymatic hydrolysis has been intensively employed to produce antioxidative gelatin hydrolysates (Gómez-Guillén et al. 2011). However, the high cost of production is mainly associated with enzyme, energy consumed and production process. Recently, autolysis mediated by indigenous protease was used as the aid for production of gelatin hydrolysates with antioxidative activity from unicorn leatherjacket skin that could reduce the amount of commercial proteases and energy used (Karnjanapratum and Benjakul 2014a). The selection of proper enzyme to substrate was crucial for production of antioxidative gelatin hydrolysate from Alaska pollack skin (Kim et al. 2001). Kittiphattanabawon et al. (2012) found that glycyl endopeptidase, the major cysteine protease in papaya latex, showed the effectiveness in cleaving peptide bonds with Gly at P1 of fish gelatin from blacktip shark skin, yielding antioxidative gelatin hydrolysate with high degree of hydrolysis. Additionally, the active fraction of glycyl endopeptidase from papaya latex was prepared using aqueous two-phase system to remove offensive odour compounds (Karnjanapratum and Benjakul 2014b).
To reduce the amount of enzyme and energy used, the autolysis-assisted process mediated by indigenous protease in unicorn leatherjacket skin could be employed prior to further hydrolysis by glycyl endopeptidase from papaya latex. Additionally, pretreatment of skin via swelling process might affect the hydrolysis by protease used. As a consequence, gelatin hydrolysate with different antioxidative activities could be obtained. The objective of this study was to investigate the impact of autolysis-assisted process of non-swollen and swollen skin of unicorn leatherjacket and effect of partially purified glycyl endopeptidase at different levels on antioxidative activities of resulting gelatin hydrolysates.
Materials and methods
Chemicals
Polyethylene glycol (PEG) 6000 was obtained from Fluka (Buchs, Switzerland). 2,4,6-trinitrobenzenesulphonic acid (TNBS), 2,2′-azinobis (3- thylbenzothiazoline-6-sulfonic acid) (ABTS), 6-hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid (Trolox), 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-4′,4″-disulfonic acid sodium salt (ferrozine), pepsin from porcine gastric mucosa (EC 3.4.23.1), pancreatin from porcine pancreas, trypsin from bovine pancreas (EC 3.4.21.4) and bile extract porcine were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Trichloroacetic acid (TCA), ferrous chloride and iron standard solution were obtained from Merck (Darmstadt, Germany). 2-thiobarbituric acid and cumene hydroperoxide were procured from Fluka (Buchs, Switzerland). Ammonium thiocyanate was purchased from Lab-Scan (Bangkok, Thailand). All chemicals were of analytical grade.
Preparation of skin and autolysed skin
Preparation of skins
The skins of unicorn leatherjacket (Aluterus monoceros) were obtained from a dock, Songkhla, Thailand. The fish with the size of 1.0–1.5 kg/fish (30–35 cm of body length) were caught from the Gulf of Thailand, stored in ice and offloaded approximately 2–3 days after capture. Three different lots of skins were collected. For each lot, skins were pooled and used as the composite sample. Skins were stored in ice with a skin/ice ratio of 1:2 (w/w) and transported to the Department of Food Technology, Prince of Songkla University, Hat Yai within 1 h. Upon arrival, the skins were washed with iced tap water (0–2 °C) and cut into small pieces (0.5 × 0.5 cm2), placed in polyethylene bags and stored at −20 °C until use. The storage time was less than 2 weeks.
Pretreatment of skins
The skins were pretreated to remove non-collagenous proteins following the method of Kaewruang et al. (2013). Fish skins (0.5 × 0.5 cm2) were soaked in 0.05 M NaOH with a skin/alkaline solution ratio of 1:10 (w/v) to remove non-collagenous proteins. The mixture was stirred continuously at room temperature using an overhead stirrer equipped with a propeller (RW 20.n, IKA Labortechnik, Staufen, Germany) at a speed of 150 rpm. The alkaline solution was changed after 2 h and total pretreatment time was 4 h. Pretreated skins were washed with tap water until neutral or faintly basic pH of wash water was obtained.
Preparation of autolysed skin
Use of non-swollen skin
Autolysis was conducted using pretreated skin (non-swollen skin) following the method of Karnjanapratum and Benjakul (2014a). The pretreated skins were mixed with deionised water at a ratio of 1:5 (w/v). The autolysis was performed by incubating the mixture in a water bath (Model W350, Memmert, Schwabach, Germany) at 55 °C for 12 h and terminated by heating at 90 °C for 15 min. The mixture was centrifuged at 5,000×g at 4 °C using a refrigerated centrifuge model Avanti J-E (Beckman Coulter, Inc., Palo Alto, CA, USA) for 10 min to remove the debris. Autolysed skin was collected and referred to as ‘NS’.
Use of swollen skin
To prepare swollen-skin, the pretreated skin was soaked in 0.1 M phosphoric acid with a skin/solution ratio of 1:10 (w/v) for 6 h with a gentle stirring at room temperature as per the method of Kaewruang et al. (2013). The acidic solution was changed every 3 h. Acid-treated skin was washed thoroughly with tap water until wash water became neutral or faintly acidic. To prepare autolysed skin, the swollen skin was mixed with deionised water at a ratio of 1:5 (w/v) and subjected to autolysis as previously described. Autolysed skin obtained was referred to as ‘SS’.
Production of gelatin hydrolysate from autolysed skin using partially purified glycyl endopeptidase (GE)
Preparation of crude extract from papaya (C. papaya) latex
Fresh papaya latex was collected from green papaya fruit cultivated in Songkhla, Thailand. Four to six longitudinal incisions were made on the green papaya fruit using a stainless steel knife. The exuded latex was collected using a receiving container. The latex was then transferred to a beaker and stored below 10 °C and used within 3 h.
The crude extract was prepared using the method of Kittiphattanabawon et al. (2012). The latex was mixed with cold distilled water (≤4 °C) with a latex-to-water ratio of 1:3 (w/v). The mixture was gently stirred at 4 °C for 1 h. Then, the mixture was centrifuged at 9,000×g at 4 °C for 20 min using a refrigerated centrifuge. The supernatant was filtered using a Whatman No. 1 filter paper (Whatman International, Maidstone, Kent, UK), followed by lyophilisation (Scanvac Model Coolsafe 55 freeze dryer, Coolsafe, Lynge, Denmark). The powder obtained was referred to as ‘crude extract’.
Fractionation of glycyl endopeptidase using aqueous two-phase system (ATPS) in combination with ammonium sulphate precipitation
Crude extract powder (1 g) was dissolved in 8 mL of distilled water. The pH of solution was adjusted to 6.0 using 6 M HCl and the volume was made up to 10 mL by distilled water to obtain a concentration of 100 mg/mL prior to fractionation using ATPS.
The glycyl endopeptidase was fractionated using the method of Karnjanapratum and Benjakul (2014b). ATPS with 10 % PEG 6000 and 10 % ammonium sulphate (NH4)2SO4 was used for fractionation of glycyl endopeptidase. The salt-rich bottom phase was collected and further precipitated using ammonium sulphate at 60 % saturation. After centrifugation at 9,000×g at 4 °C for 20 min, the pellet was re-dissolved in distilled water and dialysed against 20 volumes of distilled water, six times. After lyophilisation, the powder referred to as ‘partially purified glycyl endopeptidase, GE’ was stored at −40 °C until use.
Production of gelatin hydrolysates using GE
Autolysed fish skins, both NS and SS, were used as substrates. The solutions (3 %, w/v) were then added with GE at different concentrations (2, 4, 6 and 8 %, based on solid matter). The reaction proceeded at 40 °C for 60 min. After enzyme inactivation by heating at 90 °C for 15 min, the resulting gelatin hydrolysate was centrifuged at 9,000×g at 4 °C for 20 min. The supernatant was collected and lyophilised. The gelatin hydrolysate powders were placed in polyethylene bag and stored at −40 °C. Hydrolysate powders were subjected to analyses.
Analyses
Determination of α–amino group content
The α–amino group content was determined according to the method of Benjakul and Morrissey (1997). To diluted samples (125 μL), 2.0 mL of 0.2 M phosphate buffer, pH 8.2 and 1.0 mL of 0.01 % TNBS solution was added. The solution was mixed thoroughly and placed in a temperature-controlled water bath at 50 °C for 30 min in the dark. The reaction was terminated by adding 2.0 mL of 0.1 M sodium sulphite. The mixture was cooled at room temperature for 15 min. l-leucine standard solutions with concentrations ranging from 0.5 to 5.0 mM were used. The absorbance was read at 420 nm and α–amino group content was expressed in terms of l-leucine.
Determination of in vitro antioxidative activities
ABTS radical scavenging activity
ABTS radical scavenging activity of gelatin hydrolysates was determined as described by Binsan et al. (2008). The stock solutions included 7.4 mM ABTS solution and 2.6 mM potassium persulfate solution. The working solution was prepared by mixing the two stock solutions in equal quantities. The mixture was allowed to react for 12 h at room temperature in the dark. The solution obtained (1 mL) was then diluted with 50 mL distilled water in order to obtain an absorbance of 1.1 ± 0.02 units at 734 nm using a UV-1601 spectrophotometer (Shimadzu, Kyoto, Japan). Fresh ABTS solution was prepared for each assay. Sample (150 μL) was mixed with 2,850 μL of ABTS solution and the mixture was allowed to stand at room temperature for 2 h in the dark. The absorbance was then measured at 734 nm using a spectrophotometer. The blank was prepared in the same manner, except that distilled water was used instead of the sample. A standard curve of Trolox ranging from 50 to 600 μM was prepared. The activity was expressed as μmol Trolox equivalent (TE)/g sample.
Hydrogen peroxide scavenging activity
Hydrogen peroxide scavenging activity was assayed according to the method of Wettasinghe and Shahidi (2000) with a slight modification. Briefly, the sample (1 mL) was mixed with 83 µl of 100 mM hydrogen peroxide (prepared in 0.1 M phosphate buffer, pH 7.4). The mixture was allowed to react for 40 min at room temperature. The absorbance at 230 nm of the reaction mixture was read. The blank, in which 0.1 M phosphate buffer was used instead of hydrogen peroxide, was prepared. Trolox (0–10 mM) was used as the standard. The hydrogen peroxide scavenging activity was expressed as mmol Trolox equivalent (TE)/g sample.
Ferric reducing antioxidant power (FRAP)
FRAP was determined according to the method of Benzie and Strain (1996). FRAP reagent was prepared by mixing acetate buffer (30 mM, pH 3.6), 10 mM TPTZ solution in 40 mM HCl and 20 mM iron (III) chloride solution in proportions of 10:1:1 (v/v). The sample solution (100 µL) was mixed with 3 mL of working FRAP reagent and incubated in dark condition at room temperature for 30 min. The absorbance of the reaction mixture was read at 593 nm using a spectrophotometer. The standard curve was prepared using Trolox ranging from 0 to 500 µM. The activity was expressed as µmol Trolox equivalent (TE)/g sample.
Ferrous chelating activity
Chelating activity of gelatin hydrolysates towards ferrous ion (Fe2+) was measured by the method of Thiansilakul et al. (2007) with a slight modification. Sample (200 μL) was mixed with 800 μL of distilled water. Thereafter, 0.1 mL of 2.0 mM FeCl2 and 0.2 mL of 5 mM ferrozine were added. The mixture was allowed to react for 20 min at room temperature. The absorbance was then read at 562 nm. The standard curve of EDTA (0–1.0 mM) was prepared. The control was prepared in the same manner except that distilled water was used instead of the sample. Ferrous chelating activity was expressed as μmol EDTA equivalents (EE)/g sample.
Antioxidative activity of gelatin hydrolysate in lecithin liposome model system
Gelatin hydrolysates prepared using 8 % GE rendering the highest in vitro antioxidative activity were tested in a lecithin liposome model system according to the method of Thiansilakul et al. (2007). Both gelatin hydrolysates were named ‘NS-8GE’ and ‘SS-8GE’ where NS and SS were used as substrates, respectively.
Lecithin liposome system was prepared by suspending lecithin in deionised water at a concentration of 8 mg/mL. The mixture was stirred with a glass rod, followed by sonification for 30 min in a sonicating bath (Elma Model S30H, Singen, Germany). Gelatin hydrolysate (3 mL) was added to the lecithin liposome system (15 mL) to obtain the final concentrations of 2.5 and 5.0 g/L. The mixture was sonicated for 2 min. To initiate the reaction, 40 µL of 0.15 M cupric acetate was added. The mixture was shaken in the dark at 120 rpm using a shaker (Unimax 1010, Heidolph Model, Schwabach, Germany) at 37 °C. The system containing 0.10 g/L Trolox was also prepared. The control was prepared in the same manner, except that distilled water was used instead of gelatin hydrolysates or Trolox. Oxidation in lecithin liposome systems was monitored during 24 h of incubation by determining PV and TBARS values.
Determination of peroxide value (PV)
PV was measured according to the method of Wu et al. (2003). Liposome sample (0.1 mL) was mixed with 4.7 mL of ethanol/distilled water (3:1), 0.1 mL of 30 % ammonium thiocyanate, and 0.1 mL of 20 mM ferrous chloride solution in 3.5 % HCl. After incubation at 40 °C for 3 min, the absorbance was read at 500 nm using a spectrophotometer. PV was calculated from a standard curve of cumene hydroperoxide (0–100 mg/L) and expressed as mg cumene hydroperoxide/L lecithin liposome.
Determination of thiobarbituric acid reactive substances (TBARS)
TBARS was determined as described by Buege and Aust (1978) with a slight modification. Liposome sample (0.5 mL) was homogenised with 2.5 mL of TBARS solution (0.375 % thiobarbituric acid, 15 % TCA and 0.25 M HCl). The mixture was heated in boiling water for 10 min to develop the pink colour. The mixture was then cooled with running water and centrifuged at 5000xg for 10 min at room temperature using Hettich centrifuge (Hettich Model MIKRO-20, Tuttlingen, Germany). The supernatant was collected and the absorbance at 532 nm was read using a spectrophotometer. TBARS value was calculated from a standard curve of malondialdehyde (MDA) (0–10 mg/L) and expressed as mg MDA/L lecithin liposome.
Changes in antioxidative activities of gelatin hydrolysate in gastrointestinal tract model system
Gastrointestinal tract model system mimicking the conditions of the digestive organs was prepared according to the method of Enari et al. (2008) with slight modifications. Gelatin hydrolysate solution (10 mg/mL in distilled water 100 mL) was mixed with 0.5 ml of 1 M HCl-KCl buffer (pH 1.5), followed by adding 32 U/mL of pepsin solution in 1 M HCl-KCl buffer (pH 1.5) (5 mL) and incubating for 60 min at 37 °C (stomach condition) with a continuous shaking using a Unimax 1010 shaker (Heidolph Model, Schwabach, Germany). Thereafter, the pH of the reaction mixture was raised to 6.8 with 1 M NaHCO3 (1 mL), and the enzyme mixture of bile and pancreatic juice (1 mL) that contained pancreatin (10 mg/mL), trypsin (14,600 U/mL) and bile extract (13.5 mg/mL) in 10 mM phosphate buffer (pH 8.2), was added to the solution, followed by incubation at 37 °C for 3 h (duodenal condition) with a continuous shaking. The digestion was terminated by placing in a boiling water bath for 10 min. During the simulated gastrointestinal digestion, the mixture was randomly taken at 0, 0.5, 1, 2, 3 and 4 h for determination of ABTS radical scavenging activity and FRAP.
Molecular weight distribution
Gelatin hydrolysates (NS-8GE and SS-8GE) were separated by size exclusion chromatography using a Sephadex G-25 gel filtration column (2.5 × 50 cm) (17-0032-01, GE Healthcare Bio-Science AB, Uppsala, Sweden). The experiment was conducted in a walk-in cold room (4 °C) and 2 mL of sample solution (60 mg gelatin/mL) was loaded. After being loaded, the elution was performed using a low pressure chromatography system (BioLogic LP system, Bio-Rad Laboratories Ltd., Hercules, CA, USA) coupled with a fraction collector (Model 2128, Bio-Rad Laboratories Ltd., Hercules, CA, USA). Fifty mM sodium phosphate buffer (pH 7.0) was used as the elution buffer at a flow rate of 0.5 mL/min. The fractions of 3 mL were collected. The absorbance was recorded at 220 and 280 nm. ABTS radical scavenging activity and ferric reducing antioxidant power (FRAP) of each fraction were determined. Blue dextran (2,000,000 Da) was used for void volume measurement. The molecular weight markers included insulin chain B (3,496 Da), vitamin B12 (1,355 Da), glycine-tyrosine (238 Da) and tyrosine (181 Da). MW of fraction with the highest ABTS radical scavenging activity and FRAP was estimated from the plot between available partition coefficient (Kav) and the logarithm of the molecular weight of the protein standards.
Statistical analysis
All experiments were run in triplicate using the three different lots of skin. Data were subjected to analysis of variance (ANOVA) and mean comparisons were carried out using Duncan’s multiple range test (Steel and Torrie 1980). Statistical analysis was performed using the Statistical Package for Social Sciences (SPSS for Windows SPSS Inc., Chicago, IL, USA). The data with P < 0.05 were considered to be statistically significant.
Results and discussion
Effect of GE on hydrolysis of autolysed skin
The hydrolysis of autolysed non-swollen and swollen skin by GE at different levels as monitored by α-amino group content is shown in Fig. 1. Without hydrolysis using GE, NS and SS had the similar α-amino group content (P > 0.05). The α-amino group content of resulting gelatin hydrolysates increased with increasing GE levels for both substrates (NS and SS) used. This result was in accordance with Karnjanapratum and Benjakul (2014a) who reported that the increase in enzyme/substrate (0.5, 1.0 and 2.0 %) resulted in the increase in degree of hydrolysis of gelatin hydrolysate from unicorn leatherjacket skin prepared using papain. When NS was used as substrate, α-amino group contents in resulting hydrolysates were lower than those found in hydrolysate from SS at all enzyme levels used (P < 0.05). The result indicated that peptides in SS were more likely the preferable substrate for GE. Autolysis of gelatin in unicorn leatherjacket skin at 55 °C caused by indigenous protease (Kaewruang et al. 2013) yielded available peptides for further hydrolysis by GE. For swollen skin (SS), α-chains or their cross-link, e.g. β- or γ-chains, could be more repulsed. As a result, those proteins could be cleaved by indigenous protease easily. The resulting peptides might have free exposed glycine along the chain to a higher extent, in comparison with peptides produced from non-swollen skin. When the skin matrix was still compact, cleavage sites might be limited and the resulting peptides were plausibly present as bundles, in which cleavage site, especially glycine, could be hindered. This might result in the lower hydrolysis as indicated by lower α-amino group content.

α-Amino group content of gelatin hydrolysates prepared using partially purified glycyl endopeptidase at different concentrations (2, 4, 6 and 8 % of solid matter). NS autolysed non-swollen skin; SS autolysed swollen skin. Different uppercase and lowercase letters on the bars within the same enzyme concentration and the same substrate, respectively, indicated significant difference (P < 0.05). Bars represent standard deviation (n = 3)
In vitro antioxidative activities of gelatin hydrolysates
Figure 2 shows the antioxidative activities of gelatin hydrolysates prepared using GE at different levels (2–8 % of solid matter) as examined by different assays. ABTS radical scavenging activity of NS (52.72 µmol/g sample) was higher than SS (29.50 µmol/g sample) (P < 0.05). Notably, NS and SS had similar α-amino group content (Fig. 1). Different peptides with various chain length and amino acid sequence might be obtained as influenced by prior swelling. Therefore, the difference in antioxidative activity of autolysed skin (NS and SS) could be observed. The increase in ABTS radical scavenging activity of gelatin hydrolysates was obtained as GE was applied for the second step of hydrolysis. The higher activity was found when GE levels used increased (P < 0.05), regardless of substrate. At the same level of GE, gelatin hydrolysates obtained from SS possessed the lower ABTS radical scavenging activity, compared with those from NS (P < 0.05). It was noted that ABTS radical scavenging activity of resulting gelatin hydrolysates was not in accordance with α-amino group content (Fig. 1). This might reflect the difference in antioxidative peptides in hydrolysate when different substrates were used. Wu et al. (2003) reported that size, level and composition of free amino acids of peptides affected the antioxidative activity. ABTS radical scavenging assay is based on biamperometric measurements using ABTS+/ABTS redox couple. The ability of antioxidants to scavenge the preformed ABTS+ was estimated by the decolourisation (Re et al. 1999). Therefore, hydrolysate prepared from NS using GE more likely showed higher ability of scavenging ABTS radical, compared with those from SS.

ABTS radical scavenging activity (a), hydrogen peroxide scavenging activity (b), ferric reducing antioxidant power (c) and ferrous chelating activity of gelatin hydrolysates prepared using partially purified glycyl endopeptidase at different concentrations (2, 4, 6 and 8 % of solid matter). NS autolysed non-swollen skin; SS autolysed swollen skin. Different uppercase and lowercase letters on the bars within the same enzyme concentration and the same substrate, respectively, indicated significant difference (P < 0.05). Bars represent standard deviation (n = 3)
H2O2 scavenging activity of gelatin hydrolsates prepared using GE at different concentrations (2–8 % of solid matter) is depicted in Fig. 2b. NS (1.17 mmol/g sample) and SS (1.16 mmol/g sample) had a similar H2O2 scavenging activity (P > 0.05). The decrease in H2O2 scavenging activity of gelatin hydrolysates was observed after further hydrolysis by GE, regardless of substrates. However, the resulting gelatin hydrolysate prepared with 4 % GE had a similar H2O2 scavenging activity, compared with the substrates, NS and SS (P > 0.05). These results suggested that the peptides generated after hydrolysis by GE had low ability in scavenging H2O2. Karnjanapratum and Benjakul (2014a) also reported that H2O2 scavenging activity of gelatin hydrolysates from unicorn leatherjacket prepared using papain decreased when the level of papain increased. It was reported that degree of hydrolysis had no relationship with antioxidative activity of protein hydrolysate (Chen et al. 1995). The extensive hydrolysis probably caused the lower antioxidative activity, due to the generation of free amino acids which had lower antioxidative activity, compared to peptides (Hernandez-Ledesma et al. 2005). H2O2, which is a weak oxidising agent, is not directly involved in the initiation of lipid oxidation because its reduction potential is lower than that of unsaturated fatty acid (Choe and Min 2005). However, H2O2 shows indirect contribution to lipid oxidation. H2O2 is the precursor for the generation of hydroxyl radical, which is a strong initiator of lipid oxidation (Choe and Min 2005). Thus, further hydrolysis of autolysed skin by GE slightly lowered H2O2 scavenging activity of resulting hydrolysates.
Ferric reducing antioxidant power (FRAP) of gelatin hydrolsates prepared using GE at different concentrations is shown in Fig. 2c. NS had low FRAP (0.04 µmol/g sample). On the other hand, SS had no FRAP. The activity of gelatin hydrolysates increased when GE levels used increased (P < 0.05), regardless of substrates. Generally, gelatin hydrolysate obtained from SS exhibited the higher FRAP than those from NS, especially when GE at levels higher than 2 % was used. The increases in FRAP were in accordance with increasing α-amino group content of gelatin hydrolysate (Fig. 1). It was suggested that smaller peptides in hydrolysate obtained from SS more likely had the higher ability in reducing TPTZ-FE(III) complex. FRAP is generally used to measure the capacity of a substance in reducing TPTZ-FE(III) complex to TPTZ-Fe(II) complex (Binsan et al. 2008). This was in agreement with Kittiphattanabawon et al. (2012) who reported that FRAP of hydrolysate from blackstip shark skin prepared using papaya latex enzyme increased with increasing degree of hydrolysis. After hydrolysis using GE, the resulting gelatin hydrolysates from both substrates, NS and SS, possibly contained high amounts of peptides which donated electron to free radicals, thereby terminating the chain reaction.
Ferrous chelating activity of gelatin hydrolsates is shown in Fig. 2d. NS (6.79 µmol/g sample) showed the higher ferrous chelating activity than SS (3.46 µmol/g sample) (P < 0.05). After further hydrolysis by GE, the resulting gelatin hydrolysates from NS had the decreases in chelating activity, especially when GE levels increased (P < 0.05). However, the increase in ferrous chelating activity was obtained when 4 % GE was used for hydrolysis. When SS was used as substrate, the ferrous chelating activity of resulting hydrolysates increased as GE levels increased up to 4 % (P < 0.05). Nevertheless, the decrease in chelating activity was observed in hydrolysate prepared using GE level higher than 4 % (P < 0.05). Karnjanapratum and Benjakul (2014a) also reported that ferrous chelating activity of gelatin hydrolysate from unicorn leatherjacket skin decreased with increasing papain levels. The result indicated that different peptides in hydrolysates might have varying ability in sequestering Fe2+. It was noted that gelatin hydrolysates from NS after further hydrolysis using GE showed the higher chelating activity, compared with those from SS. It is well-known that transition metal ions such as iron or copper may catalyse the formation of reactive oxygen species that accelerate lipid oxidation. Thus, gelatin hydrolysates obtained in the present study were able to prevent lipid oxidation via metal chelating ability. Nevertheless, their ability varied, depending upon the level of GE used for hydrolysis.
From the above measurements, gelatin hydrolysate prepared with 8 % GE from both NS (NS-8GE) and SS (SS-8GE) showed the highest radical scavenging activity and electron donating activity, which are crucial for termination of chain reaction. Therefore, NS-8GE and SS-8GE were selected for determination of antioxidative activity in lecithin liposome model system in comparison with NS and SS.
Antioxidative activity of selected gelatin hydrolysates in lecithin liposome model system
Oxidation of lecithin liposome system added with gelatin hydrolysates at different concentrations (2.5 and 5.0 g/L) was monitored by determining peroxide value (PV) (Fig. 3a) and TBARS values (Fig. 3b) during incubation of 48 h at 37 °C. For the control (without hydrolysate or trolox), the sharp increase in PV was found within the first 42 h of incubation (P < 0.05), in which the highest value (138.74 ± 3.64 mg cumene hydroperoxide/L lecithin liposome) was found at 42 h of incubation. Thereafter, the sharp decrease in PV was observed up to 48 h. The sharp decrease in PV after 42 h of incubation was plausibly caused by the decomposition of hydroperoxide to the secondary products (Frankel et al. 1997). The system added with NS and SS showed the lower PV than the control throughout 48 h of incubation (P < 0.05). In general, the rate of increase varied with the concentration of gelatin hydrolysate used. On the other hand, the system added with NS-8GE and SS-8GE showed very low increase in PV throughout the incubation of 48 h. Ability in prevention of hydroperoxide formation was comparable to that of 0.1 g/L trolox, especially for NS-8GE and SS-8GE at high concentration tested (5.0 g/L). NS-8GE and SS-8GE possessed the higher antioxidative activity in lecithin liposome system than their substrates (NS and SS). This was in accordance with higher antioxidative activities of hydrolysates, especially ABTS radical scavenging activity and FRAP, as shown in Fig. 2a, c, respectively. NS-8GE and SS-8GE showed both radical scavenging activity and reducing antioxidant power. Gelatin hydrolysates from seabass and bigeye snapper skins displayed antioxidative activity in lecithin liposome systems (Senphan and Benjakul 2014, Phanturat et al. 2010).

The formation of peroxide value (a) and TBARS (b) in lecithin liposome system containing gelatin hydrolysates from unicorn leatherjacket skin. NS autolysed non-swollen skin, NS-8GE autolysed non-swollen skin hydrolysed using 8 % partially purified glycyl endopeptidase, SS autolysed swollen skin. SS-8GE autolysed swollen skin hydrolysed using 8 % partially purified glycyl endopeptidase. Bars represent standard deviation (n = 3)
Changes in TBARS of liposome system during incubation of 48 h are depicted in Fig. 3b. The sharp increase in TBARS was observed after 18 h of incubation in the control and systems added with NS and SS at all levels (P < 0.05), indicating the formation of the secondary lipid oxidation products. The systems containing gelatin hydrolysate at 5.0 g/L had lower TBARS than those added with hydrolysate at a lower level (P < 0.05). There were no marked changes in TBARS of lecithin liposome system added with 1.0 g/L trolox. Slight increases in TBARS were observed in the system containing 5.0 g/L NS-8GE and SS-8GE throughout 48 h of incubation. The result suggested that NS-8GE and SS-8GE, especially at the high concentration (5.0 g/L) could retard the formation of the secondary oxidation products effectively. After 36 h of incubation time, the decreased TBARS was noticeable, suggesting the loss of oxidation products. The secondary products with low molecular weight could be lost easily (Stahnke 1995). Thus, NS-8GE and SS-8GE were able to retard the early stages as well as the advanced stage of oxidation in lecithin liposome system.
Changes in antioxidative activities of gelatin hydrolysate in gastrointestinal tract model system
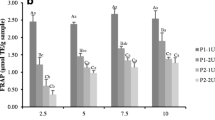
The gastrointestinal tract model system (GIMs) was used to mimic the digestive system containing several proteases. The changes in antioxidative activity of NS-8GE and SS-8GE, as monitored by ABTS radical scavenging activity and FRAP, throughout the system are presented in Fig. 4. ABTS radical scavenging activity decreased after pepsin digestion (P < 0.05) but increased under duodenal condition during 1 and 6 h. After digestion in duodenum, proteases in pancreatin juice might cleave the peptides and release some new peptides with higher antioxidative activities, especially radical scavenging activity as evidenced by the increased ABTS radical scavenging activity. It was found that ABTS radical scavenging activity was higher for SS-8GE than NS-8GE throughout ingestion system. The results suggested that NS-8GE and SS-8GE had different peptides, which could be further hydrolysed at varying degrees. As a result, the generated peptides more likely had different chain length as well as amino acid sequence. This directly determined ABTS radical scavenging activity. Kittiphattanabawon et al. (2012) also found the increased antioxidative activity of gelatin hydrolysate from blacktip shark skin prepared using papaya latex after being ingested in the simulated model system. The gelatin hydrolysate from seabass skin prepared using protease from hepatopancreas of Pacific white shrimp showed the increase of antioxidative activity with further hydrolysis in intestinal simulated system (Senphan and Benjakul 2014). On the other hand, FRAP of both NS-8GE and SS-8GE decreased after ingestion in the GIMs over digestion time of 0–4 h. The extensive hydrolysis during digestion might provide small peptides with low ability on donation of electron to free radicals. The result suggested that radical scavenging activity of NS-8GE and SS-8GE could be increased in the gastrointestinal tract.

ABTS radical scavenging activity (a) and ferric reducing antioxidant power (b) of gelatin hydrolysates from unicorn leatherjacket skin in gastrointestinal tract model system. NS-8GE autolysed non-swollen skin hydrolysed using 8 % partially purified glycyl endopeptidase; SS-8GE autolysed swollen skin hydrolysed using 8 % partially purified glycyl endopeptidase. Bars represent standard deviation (n = 3)
Molecular weight distribution
Elution profiles of NS-8GE and SS-8GE on the Sephadex™-G15 gel filtration chromatography are shown in Figs. 5 and 6, respectively. Based on A 280, gelatin hydrolysates showed three major peaks, indicating the presence of peptides containing aromatic amino acid with varying MW (Figs. 5a and 6a). Different elution profiles between NS-8GE and SS-8GE were generally obtained. SS-8GE showed the higher peak of peptides containing aromatic amino acids with low MW than NS-8GE. Based on A 220 representing peptides, it was noted that NS-8GE showed higher content of larger peptides, in comparison with SS-8GE. The result indicated that prior autolysis and swelling process used for preparing substrate might enhance hydrolysis by GE. Moreover, this result was in accordance with free α-amino group content of SS-8GE which was higher than that of NS-8GE as shown in Fig. 1. ABTS radical scavenging activity and FRAP of fractions of NS-8GE and SS-8GE are depicted in Figs. 5b and 6b, respectively. NS-8GE fraction with MW of 1,170 Da exhibited the highest ABTS radical scavenging activity, followed by the peptide with MW of 3,380 Da (Fig. 5b). Additionally, NS-8GE contained peptides (2,150 Da, 1,010 Da and 220 Da) which possessed the ability for donating electrons to free radicals. For SS-8GE, the fraction with MW of 750 exhibited the highest FRAP, followed by that with MW of 1,590 Da. ABTS radical scavenging activity was also found in fractions with MW of 1,850 and 1,010 Da. Both gelatin hydrolysates showed ABTS radical scavenging activity and FRAP, in which NS-8GE and SS-8GE possessed the ability to retard the oxidation mainly via their radical scavenging and electron donating activities, respectively. The small peptides generally possess high antioxidative activity (Kittiphattanabawon et al. 2012; Phanturat et al. 2010). However, the level and amino acid composition of peptide also affected the antioxidative activity (Wu et al. 2003). Gelatin hydrolysate from skin of unicorn leatherjacket prepared using autolysis-assisted process with MW of 2,200 Da exhibited higher antioxidative activity than those with MW of 870 Da (Karnjanapratum and Benjakul 2014a). Yarnpakdee et al. (2014) found that Nile tilapia protein hydrolysates contained antioxidative peptides with different modes of action. Fraction containing peptides with MW of 513 Da and 1,484 Da showed the strongest ABTS radical scavenging activity and metal chelating activity, respectively (Yarnpakdee et al. 2014). The result suggested that NS-8GE and SS-8GE contained peptides with different sizes and varying modes of action.

Elution profile of gelatin hydrolysate (NS-8GE) from unicorn leatherjacket skin subjected to Sephadex G-25 size exclusion chromatography. Absorbance at 280 nm (filled circle), 220 nm (opened circle) (a) ABTS radical scavenging activity (opened triangle) and ferric reducing antioxidant power (opened box) (b) molecular weight of markers (right arrow) molecular weight of selected fractions (dashed right arrow)

Elution profile of gelatin hydrolysate (SS-8GE) from unicorn leatherjacket skin subjected to Sephadex G-25 size exclusion chromatography. Absorbance at 280 nm (filled circle), 220 nm (opened circle) (a) ABTS radical scavenging activity (opened triangle) and ferric reducing antioxidant power (opened box) (b) molecular weight of markers (right arrow) molecular weight of selected fractions (dashed right arrow)
Conclusion
Antioxidative gelatin hydrolysate could be produced from both autolysed non-swollen and swollen skin of unicorn leatherjacket skin with 8 % partially purified glycyl endopeptidase from papaya latex. Gelatin hydrolysates possessed antioxidative activities and showed the effectiveness in retarding lipid oxidation. However, modes of action of both hydrolysates were different, more likely governed by swelling process. Peptide having MW of 1,170 Da with the dominant radical scavenging activity was found in NS-8GE, whereas that with MW of 750 Da having the profound FRAP was observed in SS-8GE. Therefore, autolysis mediated by indigenous protease in the skin could be applied for production of gelatin hydrolysate with antioxidative activity.
Abbreviations
- FRAP:
-
Ferric reducing antioxidant power
- PV:
-
Peroxide value
- TBARS:
-
Thiobarbituric acid reactive substances
- GIMs:
-
Gastrointestinal tract model system
References
Benjakul S, Morrissey MT (1997) Protein hydrolysates from Pacific whiting solid wastes. J Agric Food Chem 45:3423–3430
Benzie IFF, Strain JJ (1996) The ferric reducing ability of plasma (FRAP) as a measure of ‘‘antioxidant power’’: the FRAP assay. Anal Biochem 239:70–76
Binsan W, Benjakul S, Visessanguan W, Roytrakul S, Tanaka M, Kishimura H (2008) Antioxidative activity of Mungoong, an extract paste, from the cephalothorax of white shrimp (Litopenaeus vannamei). Food Chem 106:185–193
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Meth enzymol 52:302–310
Chen HM, Muramoto K, Yamauchi F (1995) Structural analysis of antioxidative peptides from soybean β-conglycinin. J Agric Food Chem 43:574–578
Choe E, Min DB (2005) Chemistry and reactions of reactive oxygen species in foods. J Food Sci 70:R142–R159
Enari H, Takahashi Y, Kawarasaki M, Tada M, Tatsuta K (2008) Identification of angiotensin I-converting enzyme inhibitory peptides derived from salmon muscle and their antihypertensive effect. Fisheries Sci 74:911–920
Frankel E, Huang SW, Aeschbach R (1997) Antioxidant activity of green teas in different lipid systems. J Am Oil Chem Soc 74:1309–1315
Gómez-Guillén MC, Giménez B, López-Caballero ME, Montero MP (2011) Functional and bioactive properties of collagen and gelatin from alternative sources: a review. Food Hydrocolloid 25:813–1827
Hernandez-Ledesma B, Davalos A, Bartolome B, Amigo L (2005) Preparation of antioxidant enzymatic hydrolysates from α-lactalbumin and β-lactoglobulin. Identification of active peptides by HPLC-MS/MS. J Agric Food Chem 53:588–593
Kaewruang P, Benjakul S, Prodpran T (2013) Molecular and functional properties of gelatin from the skin of unicorn leatherjacket as affected by extracting temperatures. Food Chem 138:1431–1437
Karnjanapratum S, Benjakul S (2014a) Characteristics and antioxidative activity of gelatin hydrolysates from unicorn leatherjacket skin as affected by autolysis-assisted process. J Food Process Preserv (in press)
Karnjanapratum S, Benjakul S (2014b) Glycyl endopeptidase from papaya latex: partial purification and the use for production of fish gelatin hydrolysate. Food Chem (in press)
Kim SK, Wijesekara I (2010) Development and biological activities of marine-derived bioactive peptides: a review. J Funct Foods 2:1–9
Kim SK, Kim YT, Byun HG, Nam KS, Joo DS, Shahidi F (2001) Isolation and characterization of antioxidative peptides from gelatin hydrolysate of Alaska pollack skin. J Agric Food Chem 49:1984–1989
Kittiphattanabawon P, Benjakul S, Visessanguan W, Shahidi F (2012) Gelatin hydrolysate from blacktip shark skin prepared using papaya latex enzyme: antioxidant activity and its potential in model systems. Food Chem 135:1118–1126
Lobo V, Patil A, Phatak A, Chandra N (2010) Free radicals, antioxidants and functional foods: impact on human health. Pharmacogn Rev 4:118–126
Min B, Ahn DU (2005) Mechanism of lipid peroxidation in meat and meat products-a review. Food Sci Biotechnol 14:152–163
Phanturat P, Benjakul S, Visessanguan W, Roytrakul S (2010) Use of pyloric caeca extract from bigeye snapper (Priacanthus macracanthus) for the production of gelatin hydrolysate with antioxidative activity. LWT: food. Sci Technol 43:86–97
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26:1231–1237
Senphan T, Benjakul S (2014) Antioxidative activities of hydrolysates from seabass skin prepared using protease from hepatopancreas of Pacific white shrimp. J Funct Foods 6:147–156
Stahnke L (1995) Dried sausages fermented with Staphylococcus xylosus at different temperatures and with different ingredient levels-Part II. Volatile components. Meat Sci 41:193–209
Steel RGD, Torrie JH (1980) Principles and procedures of statistics: a biometrical approach, 2nd edn. McGraw-Hill, New York
Thiansilakul Y, Benjakul S, Shahidi F (2007) Antioxidative activity of protein hydrolysate from round scad muscle using alcalase and flavourzyme. J Food Biochem 31:266–287
Wettasinghe M, Shahidi F (2000) Scavenging of reactive-oxygen species and DPPH free radicals by extracts of borage and evening primrose meals. Food Chem 70:17–26
Wu HC, Chen HM, Shiau CY (2003) Free amino acids and peptides as related to antioxidant properties in protein hydrolysates of mackerel (Scomber austriasicus). Food Res Int 36:949–957
Xu Z (2008) Antioxidants in grains, vegetables and fruits. Food Sci Tech Bull Funct Foods 5:61–70
Yarnpakdee S, Benjakul S, Kristinsson HG, Kishimura H (2014) Antioxidant and sensory properties of protein hydrolysate derived from Nile tilapia (Oreochromis niloticus) by one- and two-step hydrolysis. J Food Sci Technol (in press)
Acknowledgments
The authors would like to express their sincere thanks to Thailand Research Fund under the Royal Golden Jubilee Ph.D. Program to Supatra Karnjanapratum (PHD/0019/2554) and the Grant-in-Aid for dissertation from Graduate School, Prince of Songkla University, Thailand for financial support. The TRF Distinguished Research Professor Grant was also acknowledged.
Conflict of interest
The authors declare that they have no competing interests.
Authors’ contributions
SB developed the initial idea and designed the study. SK was responsible for conducting experiments and analysis of data. SB and SK carried out the analytical work. SK wrote the manuscript with assistance from SB. SB read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Karnjanapratum, S., Benjakul, S. Antioxidative gelatin hydrolysate from unicorn leatherjacket skin as affected by prior autolysis. Int Aquat Res 7, 101–114 (2015). https://doi.org/10.1007/s40071-014-0088-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40071-014-0088-0