
Abstract
Phytochemical studies on MeOH extract of stems of Euphorbia griffithii led to the isolation of one new hydrolyzable tannin dimer, corilagiffithiin (1) and one new galloyl-glucoside (2), alongside six known ones (3–8). Their structures and absolute configurations were determined by in depth spectroscopic analyses and comparison of their 1D NMR and MS data with literature reported values. Configurations of sugar moieties were determined by acidic hydrolysis and subsequent GC analysis of their corresponding trimethylsilylated l-cysteine adduct. At a concentration of 50 μM, compounds 1–3 showed no anti-inflammatory activities.
Graphic Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Among the flowering plants, the genus Euphorbia (spurges, Euphorbiaceae) is the third largest with almost 2000 species spread throughout the world [1,2,3]. They inhabit a wide range of habitats and display immense diversity in growth patterns. In their environment, the species vary from small ephemerals to diversified forms of herbaceous annuals, cushion-forming subshrubs, large shrubs, small trees and cactus-like succulents [2]. Spurges often produce toxic white latex when cut [1]. Different communities utilize these plants to treat various kinds of diseases. In India, many species are applied in the management of asthma and respiratory tract inflammations. In Angola, some species are used against skin ailments, gonorrhea, diarrhea, dysentery, asthma, tumors and coughs. In Nigeria, the exudates of the plant are employed as ear drops, in the treatment of boils, sores and enhancement of wound healing [1]. Spurges are also used as ornamental and household plants [1]. Phytochemical investigations have revealed that the Euphorbia genus contains mainly, triterpenoids, diterpenoids, flavonoids, tannins and polyphenols [1, 3]. Pharmacologically, the diterpenoids of Euphorbia have exhibited cytotoxic activities, while the tripernoids and flavonoids have demonstrated possession of anti-inflammation and inhibition of virus replication effects [1, 3, 4].
E. griffithii is a robust rhizomatous perennial whose vertical stems dresses in small, red-tinged leaves and showy orange-red flowers in early summer; it is native to Himalayas and western Asia [5, 6]. It has red stems and dark green leaves, which crop up in spring with a reddish tincture [7]. Exhaustive literature search did not show any evidence of previous phytochemical studies on E. griffithii. That notwithstanding, our study on the stems of E. griffithii disclosed the presence of one new hydrolyzable tannin dimer (1) and one new galloyl-glucoside (2), together with six known ones (3–8). Compounds 1–3 were evaluated for their anti-inflammatory effects. Herein, we present the isolation, structural elucidation of the isolates, and bioassay tests of compounds 1–3.
2 Results, Discussion and Conclusion
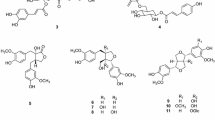
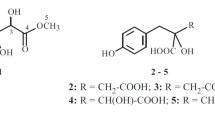
Phytochemical investigation of the stems of E. griffithii, by using various column chromatography, afforded two new compounds (1–2) (Fig. 1). In addition, six known compounds (3–8) were isolated and determined, on the basis of comparison of their NMR spectra and MS data with those reported in the literature, as methyl-O-(3′-O-galloyl)-α-d (3) [8], 3-O-galloyl-d-glucose (4) [9], β-d-glucogallin (5) [10], helioscopinin B (6) [11], methyl gallate (7) [12], and rubusin A (8) [13] (Fig. 1), respectively.

Chemical structures of 1–8 from E. griffithii
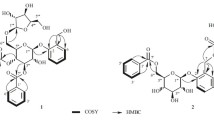
Corilagiffithiin (1) was isolated as a dark brown solid. In HRFAB+, a quasi-molecular ion peak appeared at m/z 1291.1508 [M + Na]+ (calcd for 1291.1510 [M + Na]+), indicating a molecular formula of C54H44O36 for 1. The 1H NMR spectrum displayed signals corresponding to three galloyl moieties (δH 7.14, 7.10, 7.09, each 2H, s), and one hexahydroxydiphenoyl (HHDP) group (δH 6.91, 6.25, each 1H, s). Moreover, 1H NMR spectrum showed a proton signal at δH 7.10 (1H, s), suggestive of the existence of an additional aromatic group. Another characteristic feature of the 1H NMR spectrum was the presence of two glucosyl proton signals at δH 5.81 (1H, d, J = 9.0 Hz,), as well as at δH 6.36 (1H, d, J = 2.0 Hz), which, on the basis of acidic hydrolysis and subsequent GC analysis of their corresponding trimethylsilylated l-cysteine adduct, were determined to be d-glucosyl units. The 13C NMR spectrum showed a total of 54 signals, some of which, on grounds of HMBC experiment, confirmed the presence of three galloyl [δC 120.9, 110.8 × 2, 146.0 × 2, 140.0, 165.26 (ring A, C-1′-7′, resp.); δC 120.2, 110.8 × 2, 146.1 × 2, 139.4, 165.33 (ring E, C-1″-7″, resp.); δC 121.6, 110.4 × 2, 146.2 × 2, 139.0, 166.8 (ring F, C-1″′-7″′, resp.)], one HHDP [δC 125.2, 117.0, 144.8, 137.4, 144.9, 110.0, 167.4 (Unit a, C-1″-7″, resp.); δC 125.3, 117.7, 145.6, 136.9, 147.5, 104.8, 168.5 (Unit a, C-1″′-7″′, resp.)]; and one aromatic [δC 115.3, 143.6, 140.6, 135.8, 139.8, 110.8, 165.7 (Unit b, C-1′-7′, resp.)] groups. Following the guidance of 1H-1H COSY experiment, six carbon signals (δC 94.5, 69.4, 70.9, 62.4, 75.6 and 64.4) were assigned to the 1C4 glucopyranose core II, while six other signals (δC 93.9, 73.7, 75.7, 71.3, 75.2 and 63.9) were allocated to the 4C1 glucopyranose core I [14]. In the HMBC experiment of 1, correlations of δH 6.36 (H-1) and δH 7.10 (H-2′,6′) with δC 165.26 (C-7′) in Unit a; and δH 5.18 (H-2) and δH 7.09 (H-2″,6″) with δC 165.33 (C-7″), and δH 4.53 (H-6) and δH 7.14 (C-2″′,6″′) with δC 166.8 (C-7″′) in Unit b were observed, indicating that the hydroxy groups at C-1 of Unit a and C-2, C-6 of Unit b were acylated by galloyl (A, E and F) groups, resp. (Fig. 2). Location of the HHDP group at O-3/O-6 of the glucose moiety of unit a was revealed by the down-field shifted proton signals of glucosyl H-3 (δH 4.89) and H2-6 [δH 4.71, 4.15] and established by HMBC correlations [H-3 (δH 4.89) and H-6″ (δH 6.91) correlated with δC 167.4 (C-7″); H-6a (δH 4.71), H-6b (δH 4.15) and H-6″′ (δH 6.25) correlated with C-7″′ (δC 168.5)]. On Unit b, HMBC showed correlations from δH 5.81 (H-1) and δH 7.10 (H-6′) with δC 165.7 (C-7′), and from H-6′ (δH 7.10) to C-1′ (δC 115.3), C-2′ (δC 143.6), C-4′ (δC 135.8), and C-5′ (δC 139.8) of the aromatic moiety (ring D) (Fig. 2). Eventually, the planar structures of Units a and b were determined (Fig. 1). Unit a was found matched perfectly corilagin [15] while the structure of unit b was quite similar to 1,2,6-tri-O-galloyl-β-d-glucopyranose [16], by comparisons of their spectral data.

Key1-1H COSY and HMBC correlations of 1–2
The linkage position between the two units, a and b, was determined by the 13C NMR chemical shift values and HMBC correlations. Firstly, HMBC correlations of H-6 and H-6″′ with C-7″′ in unit a, and H-1 and H-6′ with C-7′ in unit b confirmed the locations of aromatic rings C and D on O-6 of Glc-II and O-1 of Glc-I, respectively. The presence of five quaternary carbons [δC 115.3, 143.6, 140.6, 135.8, and 139.8 (C-1′-5′, resp.) in aromatic ring D suggested that it was a galloyl group that had undergone oxidative coupling at C-2′, which was significantly downfield shifted to δC 143.6, hence taking part in an ether linkage. Moreover, it was observed that the 13C NMR resonance of C-5″′ of the HHDP moiety (ring C) was shifted further downfield (δC 147.5), lower than the usual corresponding C-5″′ signal (δC 145.6) of the corilagin [17, 18]. This observation was attributed to formation of an ether linkage at the p-hydroxyl group of the galloyl unit [17]. The HMBC spectrum also showed correlations of H-6″′ (δH 6.25) with C-5″′ (δC 147.5) of the HHDP ring C. Eventually, the structure of compound 1, namely corilagiffithiin was determined as shown in Fig. 1.
Ethyl-O-(3′-O-galloyl)-α-d-glucopyranoside (2) was obtained as a dark brown solid. Its molecular formula, C15H20O10, was determined based on the HRESIMS quasi molecular ion peak at m/z 359.0987 (calcd for C15H20O10 [M−H]−, 359.0984). The 1H NMR spectrum displayed signals attributable to one methyl at δ 1.25 (1H, t, J = 7.3 Hz, H-2), one α-anomeric proton signal at δ 4.87 (1H, d, J = 3.8 Hz, H-1′), and one set of two methines belonging to a galloyl moiety at δ 7.12 (2H, s, H-2″,6″). The 13C NMR spectrum showed 14 signals signifying the existence of a methyl (δC 15.3), a glucopyranosyl moiety at 99.6, 77.2, 73.3, 73.3, 71.8, 62.3, and a galloyl moiety [ester carbonyl carbon (δC 168.5), a set of two oxygenated quaternary carbons (δC 146.2), and two quaternary carbons (δC 139.6 and 121.7). Moreover, in the 1H–1H COSY spectrum, the proton sequence pattern revealed the presence of a glucopyranose moiety which, owing to the small coupling constant of the anomeric proton [δ 4.87 (1H, d, J = 3.8 Hz, H-1′)] and acidic hydrolysis, was suggested to be α-d-glucopyranosyl moiety [19, 20]. 1H–1H COSY [δH 1.25 (H-2) / δH 3.76 (H-1a), 3.48 (H-1b)], jointly with HMBC correlation [from δH 1.25 (H-2) to δC 64.6 (C-1)] supported the existence of an ethoxyl group in 2. HMBC experiment showed that the protons at δ 5.33 (H-3′ of the glucose moiety) and 7.12 (H-2″ and 6″ of the galloyl group) correlated with δC 168.5 (the carbonyl carbon of the galloyl group), connoting that the hydroxyl group at C-3′ of glucose was acylated by the galloyl group (Fig. 3). A closer look at the planar structure of compound 2 showed that it was constituted of a galloyl [21] and ethyl α-D-glucopyranoside [22]. Thus, compound 2 was identified as ethyl-O-(3′-O-galloyl)-α-d-glucopyranoside.
Compounds 1–3 were evaluated for their anti-inflammatory effects on NO levels in LPS-stimulated RAW 264.7 macrophages. The compounds were inactive under the concentration of 50 μM.
In conclusion, from the methanolic stem extract of E. griffithii, compounds 1–8, comprising two new ones (1–2) and six known ones (3–8), were isolated. Compound 1 is a hydrolysable tannin possessing corilagin linked with 1,2,6-tri-O-galloyl-β-d-glucopyranose, whereas 2 constituted of a galloyl and ethyl-d-glucopyranoside. However, it is important to emphasize that compound 3 was isolated, for the first time, as a natural product. The only mention of compound 3 was by Masayuki et al. [20] and Arapitsas P. et al. [8]. Masayuki et al. [20] obtained compound 3 as a byproduct from acid hydrolysis of castanopsinin G (an ellagitannin containing triterpenoid glycoside core) with 1 N methanolic sulphuric acid [20]. None of the tested compounds 1–3, exhibited anti-inflammatory activities.
3 Experimental Section
3.1 General Experimental Procedure
UV spectra were recorded on a UV 210A Shimadzu spectrometer. CD spectra were obtained on a JASCO 810 spectrometer. IR spectra were measured on a Bio-Rad FTS- 135 series spectrometer with KBr pellets. Optical rotations were measured on Rudolph Autopol VI polarimeter (Rudolph Research Analytical, Hacketstown, NJ, USA). One- and two-dimensional (1D and 2D) NMR spectra were recorded in acetone-d6 with Avance 600 spectrometer operating at 600 MHz for 1H and at 150 MHz for 13C. Coupling constants are expressed in hertz, and chemical shifts are given on a δ (parts per million, ppm) scale with tetramethylsilane (TMS) as an internal standard. ESI mass spectra were recorded on a VG Auto Spec-300 spectrometer. High-resolution (HR) ESI mass spectra were recorded on an API QSTAR Pular-1 spectrometer. FAB and HRFAB were measured on Agilent G6230 TOF MS. Anti-inflammatory activity was evaluated on the basis of the ability of the compounds to inhibit nitric oxide (NO) production in LPS-stimulated RAW 264.7 macrophages. Column chromatography was performed on Sephadex LH-20 (25 − 100 μm Pharmacia Fine Chemical Co., Ltd.), MCI gel CHP20P (75 − 100 μm, Mitsubishi Chemical Co., Ltd.) and Toyopearl HW-40F (Tosoh Co., Ltd.). Thin-layer chromatography (TLC) was performed on precoated silica gel plates, 0.20 − 0.25 mm thick (Qingdao Haiyang Chemical Co.), with benzene/ethyl formate/formic acid (1:7:1, 2:7:1, or 3:6:1 v/v/v) or chloroform/methanol/water (7:3:0.5 or 8:2:0.2 v/v/v), and spots were visualized by spraying with 2% ethanolic FeCl3 or 10% H2SO4 in EtOH, followed by heating. Water was purified in a Milli-Q (Millipore, America). Acetonitrile (chromatographic grade) was purchased from Merck (Darmstadt, FR, Germany).
3.2 Plant Material
Stems of E. griffithii were collected in in July 2017 from the vicinity of Baisha township, Yulong county, Lijiang, Yunnan province, China, and identified by Prof. C.R. Yang from Kunming Institute of Botany (KIB), Chinese Academy of Sciences (CAS). A voucher specimen (KIB-Z-2017008) was deposited in the State Key Laboratory of Phytochemistry and Plant Resource in West China of KIB, CAS.
3.3 Extraction and Isolation
The air-dried stems of E. griffithii (10 kg) were extracted with MeOH (three times) under reflux at 60 °C. After removal of the organic solvent, the extract (4.5 kg) was partitioned with H2O and EtOAc to generate H2O extract (1.5 kg) and EtOAc extract (1.2 kg). EtOAc extract was applied to Sephadex LH-20 column, eluting with EtOH to give 11 sub fractions 2.1–2.11. Fr. 2.2 (15.8 g), fr. 2.6 (9.2 g), fr. 2.7 (15.0 g) and fr. 2.8 were separately chromatographed over MCI gel CHP20P [MeOH: H2O (0:1 → 1:0)] to give compounds 3 (39 mg) and 4 (33 mg) from fr. 2.2, 5 (70 mg) from fr. 2.6, and 6 (12 mg) from fr. 2.7, respectively. The water layer was submitted to Sephadex LH-20 column, eluting with MeOH: H2O (0:1 → 1:0), to afford 10 sub fractions, fr. 3.1–3.10. Fr. 3.3 (1.2 g) was applied to Toyopearl HW-40F, eluting with MeOH: H2O (0:1 → 1:0) to yield compounds 7 (502 mg) and 8 (62 mg). Similarly, fr. 3.8 (1.2 g) was submitted to Toyopearl HW-40F, eluting with MeOH: H2O (0:1 → 1:0; acetone) to yield compounds 1 (534 mg) and 2 (360 mg).
3.3.1 Compound 1
Dark brown solid (MeOH); [α] −79.7 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 276 (2.34), 248 (1.54), 219 (4.87) nm; IR (KBr) νmax 3856, 3405, 1717, 1616, 1532, 1512, 1449, 1350, 1212, 1074, 1036, 971, 872, 837, 765, 663, 620; 1H NMR (acetone-d5, 600 MHz) δH Unit a: 7.10 (2H, s, H-2′,6′), 6.91 (1H, s, H-6″′), 6.36 (1H, d, J = 2.0 Hz, H-1), 6.25 (1H, s, H-6″′), 4.89 (1H, br s, H-3), 4.71 (1H, t, J = 11.0 Hz, H-6a), 4.47 (overlapped H-4, 5), 4.15 (1H, dd, J = 11.0, 8.2 Hz, H-6b), 4.10 (1H, br s, H-2), Unit b: 7.14 (2H, s, H-2″′,6″′), 7.10 (1H, s, H-6′), 7.09 (2H, s, H-2″,6″), 5.81 (1H, d, J = 9.0 Hz, H-1), 5.18 (1H, dd, J = 9.6, 8.4 Hz, H-2), 4.53 (1H, dd, J = 12.2, 1.9 Hz, H-6a), 4.47 (1H, dd, J = 12.2, 1.9 Hz, H-6b), 3.89 (1H, m, H-5), 3.67 (1H, t, J = 9.1 Hz, H-4), 3.58 (1H, t, J = 9.4 Hz, H-3); 13C NMR (acetone-d5, 150 MHz) δC Unit a: 168.5 (COO, C-7″′), 167.4 (COO, C-7″), 165.26 (COO, C-7′), 147.5 (C, C-5″′), 146.0 × 2 (C, C-3′, 5′), 145.6 (C, C-3″′), 144.9 (C, C-5″), 144.8 (C, C-3″), 140.0 (C, C-4′), 137.4 (C, C-4″), 136.9 (C, C-4″′), 125.3 (C, C-1″′), 125.2 (C, C-1″), 117.7 (C, C-2″′), 117.0 (C, C-2″), 110.8 × 2 (CH, C-2′,6′), 110.0 (CH, C-6″), 104.8 (CH, C-6″′), 94.5 (CH, C-1), 75.6 (CH, C-5), 70.9 (CH, C-3), 69.4 (CH, C-2), 64.4 (CH2, C-6), 62.4 (CH, C-4), Unit b: 166.8 (COO, C-7″′), 165.7 (COO, C-7′), 165.33 (COO, C-7″), 146.2 × 2 (C, C-3″′,5″′), 146.1 × 2 (C , C-3″,5″), 143.6 (C, C-2′), 140.6 (C, C-3′), 139.8 (C, C-5′), 139.4 (C, C-4″′), 139.0 (C, C-4″′), 135.8 (C, C-4′), 121.6 (C, C-1″′), 120.2 (C, C-1″), 115.3 (C, C-1′), 110.8 × 3 (CH, C-6′, 2″,6″), 110.4 × 2 (CH, C-2″′,6″′), 93.9 (CH, C-1), 75.7 (CH, C-5), 75.2 (CH, C-3), 73.7 (CH, C-2), 71.3 (CH, C-4), 63.9 (CH2, C-6); HRFAB+m/z 1291.1508 [M + Na]+ (calcd for C54H44O36Na [M + Na]+, 1291.1510).
3.3.2 Compound 2
Dark brown solid (MeOH); [α] 74.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 373 (0.01), 371 (0.00), 324 (0.06), 322 (0.06), 275 (1.21), 241 (0.24), 217 (2.96), 200 (1.84) nm; IR (KBr) νmax 3385, 2978, 2934, 1701, 1614, 1534, 1448, 1378, 1336, 1232, 1152, 1090, 1039, 960, 887, 815, 767, 751, 700, 645, 602, 545, 472 cm −1; 1H NMR (CD3OD, 600 MHz) δH 7.12 (1H, s, H-2″,6″), 5.33 (1H, t, J = 9.6 Hz, H-3′), 4.87 (1H, d, J = 3.8 Hz, H-1′), 3.80 (1H, dd, J = 11.9, 2.2 Hz, H-6′a), 3.76 (1H, m, H-1a), 3.72 (1H, m, H-6′b), 3.68 (1H, m, H-2′,5′), 3.64 (1H, dd, J = 3.7 Hz, 9.9 Hz, H-4′), 3.48 (1H, m, H-1b), 1.25 (1H, t, J = 7.3 Hz, H-2); 13C NMR (CD3OD, 150 MHz) δC 168.5 (COO, C-7″), 146.2 (C, C-3″, 5″), 139.5 (C, C-4″), 121.7 (C, C-1″), 110.4 (CH, C-2″, 6″), 99.6 (CH, C-1′), 77.2 (CH, C-3′), 73.3 (CH, C-2′, 5′), 71.8 (CH, C-4′), 64.6 (CH2, C-1), 62.3 (CH2, CH2OH), 15.3 (CH3, C-2); ESIMS m/z 359 [M + H]+; negative ion HRESIMS m/z 359.0987 (calcd for C15H20O10 [M−H]−, 359.0984).
3.3.3 Acid Hydrolysis of Compounds 1–2 and GC Analysis
Compounds, 1–2 (3–5 mg), were separately dissolved in 5% HCl (2 mL) and heated (90 °C) for 2 h. HCl was then removed by evaporation in vacuum. The sugar mixtures were diluted with H2O and extracted with EtOAc. The aqueous layer was neutralized with 0.1 M NaOH and eventually dried to yield the monosaccharide mixture which was immediately dissolved in pyridine (2 mL). l-cysteine methyl ester hydrochloride (about 1.5 mg) was added in to the solution of the sugar mixtures in pyridine (2 mL), and the reaction kept at 60 °C for 1 h. Thereafter, trimethylsilylimidazole (2 mL) was added in to the mixtures and reaction kept further at 60 °C for 1 h and finally halted then submitted for GC analysis [23], run on Hewlett Packard (HP) 5890 series II gas chromatography equipped with flame ionization detector (FID) and thermal conductivity detector (TCD). The column used was HP-5: column temperature: 150–280 °C, increasing at the rate of: 3 °C/min; carrier gas: N2 (1.5 mL/min); injector and detector temperature: 250 °C; injection volume: 1.0 μL; and split ratio: 1/50. The retention times of the samples were compared with those of the derivatives of authentic sugars, under the same condition. The sugar moiety of 1 was determined to be d-glucose (tR: 28.537 min), 2 was also resolved to be d-glucose (tR: 28.398 min), by confirming with the standard d-glucose (tR: 28.418 min).
3.3.4 Anti-Inflammatory Activity
The NO production assay with L-NG-monomethyl arginine (L-NMMA) as a positive control was performed as described previously by Chen et al. [24].
References
O.A. Pascal, A.E.V. Bertrand, T. Esaie, H.-A.M. Sylvie, A.Y. Eloi, Pharm. Innov. 6, 34–39 (2017)
M. Ernst, O.M. Grace, S.-L.C. Haris, N. Ronsted, N. Nilsson, H.T. Simonsen, J. Ethnopharm. 176, 90–101 (2015)
S. Ozbilgin, C.G. Saltan, Turkish. J. Pharm. Sci. 9, 241–256 (2012)
Q.W. Shi, X.H. Su, H. Kiyota, Chem. Rev. 108, 4295–4327 (2008)
https://www.rhs.org.uk/Plants/89512/Euphorbia-griffithii-Fireglow/Details. Accessed 15 July 2019
https://www.gardenia.net/plant/euphorbia-griffithii-Fireglow. Accessed 15 July 2019
https://www.finegardening.com/plant/euphorbia-griffithii-fireglow. Accessed 15 July 2019
P. Arapitsas, S. Menichetti, F.F. Vincieri, A. Romani, J. Agri. Food Chem. 55, 48–55 (2007)
G.A.C. Fortes, A.J.R. da Silva, P.H. Ferri, S.C. Santos, Rec. Nat. Prod. 9, 292–296 (2015)
K. Arena, F. Cacciola, D. Mangraviti, M. Zoccali, F. Rigano, N. Marino, P. Dugo, L. Mondello, Anal. Bioanal. Chem. 411, 4819–4829 (2019)
T. Tanaka, H. Fujisaki, G. Nonaka, I. Nishioka, Chem. Pharm. Bull. 40, 2937–2944 (1992)
S. Liu, T. Zhu, Y. Qiu, W. Qi, H. Wu, B. Cai, J. Lin, BioRes. 14, 5355–5364 (2019)
W.Q. Liang, G.J. Xu, D. Weng, B. Gao, X.F. Zheng, Y. Qian, Chem. Nat. Comp. 51, 47–49 (2015)
M. Tsutomu, Y. Takashi, T. Shingu, T. Okuda, Chem. Pharm. Bull. 36, 3849–3856 (1988)
Y.Y. Tang, X.M. He, J. Sun, C.B. Li, L. Li, J.F. Sheng, M. Xin, Z.C. Li, Molecules 24, 1186 (2019)
Z. Li, J. Yang, H. Wang, R. Xu, Z. Rao, C.L. Si, J. Zhang, Chem. Nat. Comp. 55, 144–146 (2019)
T. Okuda, T. Hatano, N. Ogawa, Chem. Pharm. Bull. 30, 4234–4237 (1982)
B.T.D. Trinh, D. Staerk, A.K. Jager, J. Ethnopharm. 186, 189–195 (2016)
M.A.A. Orabi, S. Taniguchi, T. Hatano, Phytochemistry 70, 1286–1293 (2009)
M. Ageta, G. Nonaka, I. Nishioka, Chem. Pharm. Bull. 36, 1646–1663 (1988)
J. Bi, W. Wang, J. Du, K. Chen, K. Cheng, European. J. Med. Chem. 179, 233–245 (2019)
Y.K. Hamad, Y. Abobakr, M.Z.M. Salem, H.M. Ali, BioRes. 14, 4489–4511 (2019)
C.Z. Gu, J.J. Lv, X.X. Zhang, Y.J. Qiao, H. Yan, Y. Li, D. Wang, H.T. Zhu, H.R. Luo, C.R. Yang, J. Nat. Prod. 78, 1829–1840 (2015)
Q.R. Chen, D. Li, X.J. Qin, H. Liu, W. Ni, H. Yan, L. He, Q. Zhao, X. Ma, H.Y. Liu, Phytochem. Lett. 25, 6–11 (2018)
Acknowledgements
The authors appreciate the members of the analytical group at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Science, for measuring the spectroscopic data. This work was partially supported by Yunnan Key Laboratory of Natural Medicinal Chemistry (S2017-ZZ14).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kaunda, J.S., Zhang, YJ. Two New Phenolic Constituents from the Stems of Euphorbia griffithii. Nat. Prod. Bioprospect. 9, 405–410 (2019). https://doi.org/10.1007/s13659-019-00223-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-019-00223-2