
Abstract
Cancer cachexia is defined as a multifactorial syndrome of involuntary weight loss characterized by an ongoing loss of skeletal muscle mass and progressive functional impairment. It is postulated that cardiac dysfunction/atrophy parallels skeletal muscle atrophy in cancer cachexia. Cardiotoxic chemotherapy may additionally result in cardiac dysfunction and heart failure in some cancer patients. Heart failure thus may be a consequence of either ongoing cachexia or chemotherapy-induced cardiotoxicity; at the same time, heart failure can result in cachexia, especially muscle wasting. Therefore, the subsequent heart failure and cardiac cachexia can exacerbate the existing cancer-induced cachexia. We discuss these bilateral effects between cancer cachexia and heart failure in cancer patients. Since cachectic patients are more susceptible to chemotherapy-induced toxicity overall, this may also include increased cardiotoxicity of antineoplastic agents. Patients with cachexia could thus be doubly unfortunate, with cachexia-related cardiac dysfunction/heart failure and increased susceptibility to cardiotoxicity during treatment. Cardiovascular risk factors as well as pre-existing heart failure seem to exacerbate cardiac susceptibility against cachexia and increase the rate of cardiac cachexia. Hence, chemotherapy-induced cardiotoxicity, cardiovascular risk factors, and pre-existing heart failure may accelerate the vicious cycle of cachexia-heart failure. The impact of cancer cachexia on cardiac dysfunction/heart failure in cancer patients has not been thoroughly studied. A combination of serial echocardiography for detection of cachexia-induced cardiac remodeling and computed tomography image analysis for detection of skeletal muscle wasting would appear a practical and non-invasive approach to develop an understanding of cardiac structural/functional alterations that are directly related to cachexia.
Similar content being viewed by others
1 Introduction
Cancer cachexia is a multifactorial syndrome of involuntary weight loss defined by an ongoing loss of skeletal muscle, fat mass, and progressive functional impairment [1-3]. Cachexia is a major cause of morbidity and mortality, occurring in up to 80 % of patients with progressive cancer, and suggested to be responsible for death in up to 20 % of the patients [4]. Cachexia-associated clinical manifestations include skeletal muscle wasting, anemia, anorexia, and altered immune function which contribute to fatigue, impaired quality of life, and reduced survival [5]. Patients with severe features of cachexia/skeletal muscle wasting are generally unable to react appropriately to stress, and have increased susceptibility to infections, complications during hospitalization, and chemotherapy toxicity [6, 7].
Cachexia can be found in several pathological conditions in humans such as heart failure (HF), chronic obstructive pulmonary disease, acquired immunodeficiency syndrome, cancer, and renal failure, and the presence of cachexia is associated with poor prognosis [8].
Weight loss in cachexia involves muscle and fat mass as well as multiple organs including liver, kidney, spleen, and lung [9]. A new finding in animal studies is that cardiac dysfunction and atrophy parallels skeletal muscle atrophy in cancer cachexia [10, 11]. Effects of cancer-induced cachexia on cardiac function and structure have not been widely studied in human. Wilens et al. [12] performed necropsies on unselected men (n = 1,375) and suggested that weight loss due to disseminated cancer was the most common cause of cardiac atrophy. Wilens appears to have first used the term cardiac atrophy in cancer patients. Burch et al. [13] reported that cancer patients have smaller hearts and cardiac dysfunction based on electrocardiogram and X-ray imaging.
Heart failure is by itself and in the absence of any other disease associated with cardiac cachexia. Cardiac cachexia is characterized by involuntary weight loss, reduced anthropometric indices of muscle mass, and disturbed homeostasis of several body systems [14]. Since HF is an independent cause of cachexia, cancer cachexia-induced cardiac atrophy and HF may appear as an additional contributing factor to cachexia that consequently exacerbates wasting in the cancer patient.
The purpose of this paper is to review findings which suggest that patients with cancer cachexia may develop a vicious cycle of progressive HF and cachexia (Fig. 1).

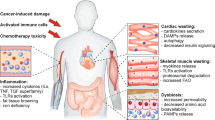
Bilateral effects of cachexia and heart failure in the cancer context. I cancer cachexia is postulated to result in cardiac atrophy/heart failure leading to loss of cardiac function. II, III pre-existing cardiovascular risk/morbidity as well as cardiotoxic chemotherapy are additional factors that contribute to heart failure in some cancer patients. IV heart failure can be initialized/exacerbated by both of cancer cachexia and cardiotoxic chemotherapy. V developed heart failure by itself is demonstrated to result in cachexia (cardiac cachexia), augments the severity of the existing cancer cachexia, and potentially increases the susceptibility to chemotherapy-induced cardiotoxicity. These effects could sequentially worsen with cachexia driving heart failure and heart failure contributing to augmented cachexia. CV cardiovascular
2 Underlying mechanism of cancer muscle wasting/cachexia
Cachexia is caused by complex interactions between pro-inflammatory cytokines, hypermetabolism, catabolism of muscle protein, neurohormonal changes, and proteolytic and lipolytic factors produced by the host and tumor [1-3]. Cancer cachexia is also associated with a decrease in protein synthesis that might be a consequence of, at least in part, alteration in the activation of the 5′ AMP-activated protein kinase, protein kinase B (Akt), and mammalian target of rapamycin (mTOR) signaling pathways [15, 16].
Activation of the ubiquitin–proteasome system seems to be crucially important in cachexia-induced muscle wasting, resulting in degradation of intracellular proteins including myofibrillar proteins [17]. Several studies showed the importance of pro-inflammatory cytokines (interleukin [IL]-1β, IL-6, and tumor necrosis factor-α [TNF-α]), which activate their receptors on muscle and subsequently activate the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). NF-κB activation up-regulates the ubiquitin-dependent degradation of the myofibrillar proteins [18-20]. Furthermore, increased oxidative stress and reduced activity of antioxidant enzyme contribute to anorexia and cachexia [21, 22].
It is believed that insulin resistance may play a potential role in pathogenesis of cancer cachexia through multiple mechanisms [23, 24]. Overlap exists between insulin signaling and ubiquitin–proteasome pathways in both insulin sensitive and insulin resistant states. Due to the resistance against binding of insulin to its receptor, phosphoinositide 3-kinase activity is decreased, leading to decreased phosphorylation of Akt. Lower levels of pAkt release the inhibition of forkhead box transcription factors O (FoxO) and caspase-3, resulting in increased proteolytic activity [24]. Cancer cachexia substantially impacts on fast twitch skeletal fibers. FoxO and NF-κB affect fast, glycolytic fibers more than slow, oxidative fibers [25].
3 Cancer cachexia and cardiac alterations: animal models
Mechanisms by which cancer cachexia causes cardiac dysfunction or HF are becoming clearer (Fig. 2). Sjöström et al. [26] investigated a sarcoma model of cachexia in mice and showed significant cardiac atrophy [almost 9 % reduction in heart dry weight (p < 0.01)] and a reduced amount of myofibrillar, collagen, and soluble proteins 11 days after tumor implantation, compared to control animals. Tian et al. [27, 28] investigated the effects of colon-26 (C26) tumor-induced cachexia on cardiac function and structure. They showed echocardiography-defined evidence of functional impairment [(decreased heartbeat per minute, 528 ± 8 in control mice vs 418 ± 13 in tumor-bearing mice; p < 0.05) and (decreased fractional shortening [FS], 33 % difference; p < 0.05)] and decreased posterior wall thickness (PWT) (30 % difference at systole) which is a feature of cardiac atrophy. Gene expression analysis also indicated increased brain natriuretic peptide and c-fos, reduced peroxisome proliferator-activated receptor α, and its responsive gene muscle-type carnitine palmitoyltransferase 1 β. A decreased amount of cardiac myofibrillar proteins and troponin I and increased protein ubiquitination were also consistent with cardiac atrophy and impaired cardiac contractility in cachectic mice. Tian et al. suggested that disturbance in p44/42 mitogen-activated protein kinase plays an important role in initiation and progression of cancer-associated cardiac atrophy [27, 28]. Xu et al. [10] in a similar study reported the significant adverse effect of C26 tumor on systolic function/contractility (decreased %FS, 28.4 ± 4.18 vs 41.2 ± 5.01 in controls, p < 0.01). They also showed significant decrease in diastolic PWT in tumor-bearing mice (0.5997 ± 0.090 vs 0.7575 ± 0.1147 mm in controls, p < 0.05) as evidence of atrophy.

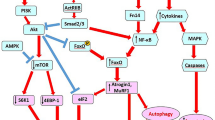
Cardiac atrophy parallels skeletal muscle wasting occurring in cancer cachexia. Gray arrow shows the effects of tumor on peripheral muscle and myocardium which results in peripheral muscle wasting as well as myocardial atrophy, White arrow biochemical pathways, Black arrows up-regulation and down-regulation. FOXO forkhead box O3, IL interleukin, mTOR mammalian target of rapamycin, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, PI3K phosphoinositide 3 kinase, TNFα tumor necrosis factor α
Cosper et al. [11] claimed that cardiac atrophy caused by C26 adenocarcinoma in mice is more prominent in males due to lack of the protective effects of estrogen. Unlike Xu et al. [10], Cosper et al. [11] did not find any significant change in ejection fraction (EF) or %FS. Preserved EF along with increased rate of cardiac fibrosis as reported by Cosper et al. [11] perhaps suggests an association between cancer cachexia and diastolic HF with preserved EF. There is no evidence regarding diastolic cardiac function in Cosper et al.’s study. Cosper et al. [11] also indicated that cardiac atrophy is due to a decrease in myocyte size and not an increase in cell death which was again more prominent in male mice. Based on Cosper et al.’s [11] findings, autophagy especially after a long period of cachexia is the main underlying mechanism of cardiac atrophy in tumor-bearing mice [11]. Manne et al. [29] also confirmed increased autophagy, not protein ubiquitination or cardiomyocyte apoptosis, in cachectic ApcMin/+ mice atrophic hearts.
Muhlfeld et al. [30] studied Lewis lung carcinoma in mice and did not find any significant functional and structural changes in echocardiographic parameters. This inconsistency with other results [27] may be due to different types of tumor (i.e., Lewis lung carcinoma vs C26). However, in this study, only a few parameters of systolic function were reported and diastolic function was not reported. They showed robust metabolic changes of cardiomyocytes in tumor-bearing animals: decreased myofibrillar volume (p = 0.06), increased sarcoplasmic volume (p < 0.01), and increased volume of lipid droplets (p < 0.01). Muhlfeld et al. [30] showed increased lipid content of cardiomyocytes in tumor-bearing mice (triglycerides per unit myocardium (mg/mg), 12.12 ± 3.75 vs 19.5 ± 7.91; p < 0.05), but markers of lipid peroxidation and apoptosis were not different in tumor-bearing vs control mice. Interestingly, they found a reduction in expression of various innervation-related targets such as neuropeptide Y and nerve growth factor as well as reduced length of axons, in tumor-bearing mice. This hypo-innervation is suggested to contribute to cardiac atrophy in tumor-bearing mice [30].
There are a variety of potential sources of variation which could contribute to differences in the magnitude of heart structure and functional changes. Skeletal muscle atrophy in rodent cancer models is affected by tumor primary type, degree of tumor burden, tumor-associated metabolic changes, and host animal type and sex, and it seems likewise plausible that these factors influence the heart as well. The specific measures which were made on the hearts in rodent models of cancer have also been somewhat heterogeneous. Finally, it is also noteworthy that the animal models lack the distinctive profiles of comorbidity, including cardiac comorbidity of human cancer patients (Table 1).
3.1 Modulation of cancer-induced cardiac alterations
Wysong et al. [31] confirmed the cardiac atrophy in C26 adenocarcinoma model of cachexia in mice and were able to block it using systemic administration of compounds that can specifically inhibit NF-κB (compound A and NEMO-binding domain (NBD) peptide). Furthermore, Shadfar et al. [32] proved protective effects of resveratrol against C26-induced cardiac atrophy in mice through NF-κB inhibition.
Palus et al. [33] reported overall cardiac atrophy in rats with cancer cachexia, induced by Yoshida AH-130 hepatoma cells, which was seen in the heart weight (752 ± 9 versus 496 ± 15 mg) as well as a reduction of the end-diastolic diameter compared to sham. They showed that treatment with simvastatin somewhat can improve the cardiac function in cancer rats (cardiac output in untreated sham, 78.9 mL/min vs tumor-bearing rats, 42.4 mL/min and improved by 1, 10, or 20 mg/kg/day simvastatin 62.2, 59.0, and 57.0 mL/min, respectively, all p < 0.05 vs placebo). Partial normalization of cardiac atrophy due to simvastatin treatment is another interesting finding of Palus et al. [33].
Zhou et al. [34] showed that in both the cachectic C26 tumor-bearing mice and cachectic inhibin-deficient mice, heart weights were decreased by 20–29 % compared to the normal controls (i.e., cardiac atrophy) and a considerable reduction in ventricular wall thickness. They found that treating the mice with ActRIIB antagonist can completely block the cardiac atrophy in both C26 mice and inhibin-deficient mice.
Springer et al. [35] showed that the xanthine oxidase inhibitor, oxypurinol, partially recovered left ventricular (LV) mass (p < 0.05) and LVEF (p < 0.05) in Yoshida AH-130 hepatoma cachexia rat model.
These findings in rodents further support the idea that cancer cachexia results in atrophy of the myocardium by mechanisms similar to those described for skeletal muscle wasting. However, whether cardiac atrophy occurs in humans with cancer cachexia is still a subject of debate. We cannot conclude any relation between the rate of cachexia and severity of cardiac remodeling in rodent studies. It is postulated that based on the rate of cachexia, a range of HF severity can be resulted from diastolic HF with preserved EF to pure systolic HF.
Further research should be performed to investigate the effects of cachexia on the ability of the heart to respond appropriately to physiologic and pathologic stressors. For instance, cachexia effects on a rodent model of pressure overload (transverse aortic constriction [TAC]) may uncover the interaction between TAC model, which results in cardiac hypertrophy, and atrophy which might be the consequence of cachexia.
4 Heart failure and cachexia development: cardiac cachexia
Cardiac cachexia is a frequent finding in classical HF patients with impaired systolic function [14]. Piepoli et al. [36] found cachexia features/marked muscle mass wasting in HF patients compared with matched healthy controls using dual energy X-ray absorptiometry. Significant computed tomography (CT)-defined reduction of muscle cross-sectional area of the thigh as well as impaired maximal quadriceps muscle strength were noticeable signs of cachexia in HF patients compared with age-matched healthy controls [37].
The possible mechanism of cachexia development in HF includes increased energy requirements, decreased nutrient absorption, decreased energy intake, increased inflammatory cytokines, neurohormonal activation, and impairment of skeletal muscle growth hormones [38, 39], similar to mechanisms proposed for cancer cachexia.
Although baseline echocardiographic and cardiac magnetic resonance imaging (MRI) measurements did not show any difference in LV mass between the patients with and without cardiac cachexia, overtime assessments after 6 months (echocardiography) and mean of 15 months (MRI) showed a significant reduction [40, 41]. Both of these studies showed that cardiac atrophy developed as cachexia progressed [40, 41].
5 Bilateral effects of cachexia and heart failure
Heart failure clearly results in cachexia in humans and if, as suggested by animal studies, cancer cachexia leads to HF, then it is possible to hypothesize that there may exist bilateral effects of the two conditions (Fig. 1). The suggestion that cancer cachexia may lead to the development of HF requires new investigations. Some individuals with cancer lose skeletal muscle very intensely (i.e., >5 kg of muscle mass in 90 days) [42], and these would be obvious candidates for developing concurrent cardiac atrophy with development of cardiac dysfunction.
A simple model (Fig. 1) would have a primary interaction between the development of cachexia and HF in cancer patients. There are two additional factors which would serve to exacerbate the primary interaction, the use of cardiotoxic chemotherapy and cardiovascular morbidity that pre-existed the development of the malignancy.
6 Chemotherapy-induced cardiotoxicity: postulated association with cachexia
Different classes of chemotherapy, targeted therapy drugs, and chemoprevention regimens showed cardiotoxic side-effects in a subgroup of patients [43]. Cardiac toxicities are thought to be under-reported [44]. Since cachectic patients are more susceptible to anticancer agent-induced toxicity [6], this may also include increased cardiotoxicity of antineoplastic agents. A wide range of cardiac disorders such as acute coronary syndrome and dysrhythmia have been associated with chemotherapy-induced cardiotoxicity [45]. Anthracyclines and tyrosine kinase inhibitors (TKIs) are two major examples.
New concerns arise regarding the unexpected cardiac events following TKIs, in particular sunitinib therapy. Di Lorenzo et al. [46] conducted a multicenter study and showed a 6.9 % incidence of HF following sunitinib therapy. Apart from LVEF reduction and HF, other cardiac abnormalities are also observed subsequent to sunitinib therapy. Acute coronary syndrome, atrial fibrillation [47], decreased heart rate, and dose-dependent QT interval changes [48] have also been associated with sunitinib therapy. Cho et al. [49] evaluated the cardiac events of 23 patients with renal cell carcinoma (RCC) who received salvage IL-2 therapy and reported severe cardiac events in 6 patients who all had the prior use of TKIs (sorafenib or sunitinib).
Anthracyclines such as doxorubicin can also lead to cardiomyocyte injury. Roughly 10 % of patients treated with doxorubicin or its derivatives will present with cardiac side-effects up to 10 years after the cessation of chemotherapy [23]. Several underlying mechanisms have been proposed for doxorubicin cardiotoxicity; however, no clinically proven treatment has been found for doxorubicin cardiomyopathy [50].
Generally, cardiotoxicity of any kind and its severity due to anticancer therapy is multifactorial in nature, determined by the interaction between genetic and environmental factors [43]. Individual genetic background is known to be important in anthracycline cardiotoxicity [51]. Several predisposing factors have been mentioned to be related to chemotherapy-induced cardiotoxicity. For instance, a history of hypertension [46], coronary artery disease [46, 52], and HF [52] seem to be associated with sunitinib-induced cardiotoxicity. Cochet et al. [53] reported that impaired LV diastolic function before treatment is an independent predictor of trastuzumab-induced cardiotoxicity after adjuvant anthracycline therapy in the patients with breast cancer, while Serrano et al. [54] confirmed that age, history of cardiac disease, and/or diabetes are risk factors for trastuzumab-related cardiotoxicity in breast cancer patients.
Severe muscle wasting is suggested to predispose patients to dose-limiting toxicity (DLT) characteristic of different chemotherapies and regimens. Antoun et al. [6] reported a significant association between low body mass index and skeletal muscle wasting and sorafenib DLT in patients with RCC. Similar associations were found for fluoropyrimidines in metastatic breast and colorectal cancer, adjuvant multidrug regimens in breast cancer, and sorafenib in hepatocellular carcinoma settings [55]. One question that needs to be asked, however, is whether any relation exists between cancer cachexia and cardiotoxicity specifically. No study so far specifically investigated the impact of cachexia on the degree and progression of cardiac dysfunction/cardiotoxicity following potentially cardiotoxic chemotherapy agents. More extensive research in regard to chemotherapy-induced cardiotoxicity is required, including its potential interaction with cachexia.
Generally, a wide range of chemotherapy-induced HF has been reported: acute HF, chronic HF with impaired systolic function, and diastolic HF with preserved EF.
Currently, there is no robust evidence of any association between diastolic HF with preserved EF and cancer cachexia. Cardiac follow-up for the cancer patients undergoing chemotherapy should definitely include the techniques which can elucidate diastolic function (e.g., tissue Doppler imaging [TDI]). TDI to complement conventional echocardiography has been shown to be beneficial in recent studies regarding chemotherapy-induced cardiotoxicity follow-up [56, 57]. Furthermore, adding strain and strain-rate measurements are highly sensitive in early precise detection of diastolic HF with preserved EF [58]. Strain imaging is highly sensitive for early detection of chemotherapy-induced cardiotoxicity [59, 60].
In conclusion, patients with cachexia could thus be doubly unfortunate, with both cachexia-related HF and increased susceptibility to cardiotoxicity during treatment.
7 Cachexia and pre-existing cardiovascular risk
A preliminary report suggests that cancer and HF patients both have clinical manifestation of tachycardia and reduced LVEF, dyspnea, fatigue, and reduced exercise capacity [61]. Indeed, it has been suggested that cancer fatigue syndrome may reflect a presentation of non-overt HF [62]. However, beyond studies looking specifically at cardiotoxic chemotherapy, there is a lack of detailed assessment of cardiac function in cancer patients. Von Haehling et al. [63] reported that patients with cancer tend to have higher values for blood pressure, stroke volume, cardiac output, and dP/dtmax at rest which may represent a higher cardiovascular risk in cancer patients compared to control subjects. A recent cross-sectional analysis of data of 93,663 patients (Gourin et al.) who had head and neck cancer surgery showed that after controlling for all other variables, patients with weight loss (i.e., evidence of cachexia) had an increased risk of acute cardiac events compared with patients without weight loss (relative risk ratio, 1.32; p = 0.016) [64].
Heart disease is one of many categories of comorbidity that affect cancer patients. Table 1 shows the prevalence of cardiac disorders in a population of 16,500 patients who died of cancer in Alberta, Canada, 1993–2000. These disorders were noted in administrative health data (hospital discharge abstracts) encompassing all hospitalizations occurring in the 365 days preceding the death of each patient. This time encompasses the part of the disease trajectory when cachexia is the most prominent [42]. Overall, a diagnosis of HF was noted in 7.5 % of patients; however, this was especially prevalent in certain subsets (14.4 % in multiple myeloma, 10.4 % in leukemia, 9.7 % in lymphoma, 8.9 % in male genital in urinary system cancers, 8.5 % in lung cancer, and 7.6 % in female breast cancer). By contrast, relatively few (1.5–3 %) patients with cancers of the brain, endocrine system, oral cavity, pharynx, and skin had a diagnosis of HF. Smoking and cardiovascular atherosclerotic diseases in some cancer types may exacerbate consequent cardiac complications. This variation in cardiac comorbidity as well as the inherent individual and tumor-specific variation in the evolution of cachexia will contribute to variation in the cachexia–cardiac interactions.
Although the association between HF and cancer has been established, there has been little discussion about the effects of cachexia on cardiac alterations in the presence of cardiovascular risk factors and morbidity. In other words, the effects of cachexia on the heart of the patients with either pre-existing risk factors or HF need to be studied in the near future. Likewise, in non-malignant disease, researchers are beginning to probe the complex interactions among HF, cachexia, and comorbid conditions [65].
8 An argument for more detailed assessments in cardio-oncology research and practice
A global guideline of assessments (imaging or biomarkers) for the early detection, management, and prevention of cancer-induced cardiac disorders does not currently exist. Cardiac management may have been ignored in part owing to the poor prognosis of some patients. However, with improvement in the management of cardiac comorbidity and tolerance of cancer therapy, research in cardio-oncology is needed.
In animal studies, development of cardiac atrophy subsequent to cancer cachexia is clearly proven [10, 11, 26-34]. Prospective studies are needed to uncover the possibility of association between cachexia and HF in human patients. It may be somewhat complicated to separately evaluate the effects of cardiotoxicity and the effects of cancer cachexia on cardiac function and structure. Also, coexistence of cardiovascular morbidities (e.g., hypertension) makes the interpretation problematic.
Further investigations should be undertaken to clarify the association between cancer cachexia and cardiac structural and functional alterations in human patients. Application of cardiac imaging techniques combined with skeletal CT scan in longitudinal studies may elucidate the parallel wasting of skeletal and cardiac muscle. CT is extensively used for routine oncology-related clinical assessments, and these images can be efficiently used to detect skeletal muscle wasting/cachexia as well as other features of clinical importance (accumulation of visceral adipose tissue, pathological accumulation of lipids in tissues) [66-68]. A detailed treatment of methods can be found elsewhere [67-73]. For assessment of cardiac functional and structural alterations, advanced echocardiographic methods appear to be suitable. Magnetic resonance imaging offers better image quality in some patients and would also provide additional structural information of the myocardium. But the limited availability and the relatively high costs would not allow serial measurements in larger cohorts of patients.
It will be of interest to evaluate plasma biomarkers in detection of cardiac alterations in cancer patients. The utility of brain natriuretic peptide (BNP) and pro-BNP in detection of HF patients has been proven [74]. Promising evidence exists in regard to high sensitivity troponin I (hs-TnI) and BNP in detection of chemotherapy-induced cardiotoxicity [75, 76]. Some inflammatory biomarkers including C-reactive protein, TNF-α, and IL-6 seem to be acceptable predictors of cancer cachexia/muscle wasting [77, 78] as well as HF progression in cardiac (i.e., non-cancer) patients [79, 80]. Hs-TNI and BNP suggested to be tested in further longitudinal cancer studies with both cardiotoxic and non-cardiotoxic agents.
We are proposing that there is a group of cancer patients who have elevated risk of cardiac impairments that reduce their fitness to tolerate treatment, reduce their quality of life, and potentially limit their survival. This group of patients is not currently receiving cardiac investigations as part of standard care, and thus, their cardiac problems could be underestimated. Oncologists have existing indications for cardiac investigation and follow up; however, these are restricted to investigational new drugs and drugs in current use that are cardiotoxic (Fig. 3). Regulatory agencies require electrocardiogram in all cancer patients in all clinical trials, and multi gated acquisition (MUGA) scan and echocardiography are used in trials of new drugs with potential cardiotoxicity. For doxorubicin and epirubicin, which have established cardiotoxicity, cardiac evaluation is part of clinical practice guidelines [81]. MUGA scan is a standard of care for patients receiving these agents at specified doses. Aside from these specific instances, there is no mandated cardiac investigation in cancer patients and no basis to make recommendations without new evidence. Collaboration between medical oncologists and cardiologists is essential to develop this area [43]. We must develop a clearer idea of which cancer patients could benefit from cardiac therapies. The new clinical investigations should be focused in patients with multiple risk factors as discussed here (comorbidity, sarcopenia, cachexia risk factors, cardiovascular risk factors, presenting with severe fatigue/exercise intolerance) but whose quality and quantity of life over the disease trajectory is likely to be significantly compromised if their heart condition remained untreated. The deployment of interventions is at this time entirely speculative, but it is of interest in rodent studies that HF medications such as statins, beta blockers, and aldosterone antagonists could attenuate both skeletal and heart muscle wasting in cancer cachexia models [33, 82].

Suggested cardio-oncology evaluations for cancer patients undergoing cardiotoxic treatment or are at high risk of cardiac disorder development. ECG electrocardiogram, MUGA multi gated acquisition scan
9 Conclusion
It is postulated that over time, during development of cancer cachexia, significant cardiac dysfunction and progressive cardiac muscle wasting may occur. Also, developed HF as a consequence of cachexia itself or pre-existing cardiovascular disease and/or anticancer drug cardiotoxicity may play a role as a further source of cachexia. Possible bilateral effects between cancer-induced cachexia and subsequent HF require investigation in human studies. Although a large and growing body of literature has investigated the cardiotoxic effects of several types of chemotherapy agents, whether cachexia aggravates chemotherapy-induced cardiotoxicity requires investigation. Moving forward, identification of skeletal muscle loss in cancer patients with regular CT scan as well as parallel cardiac assessments with feasible tools (i.e., echocardiography) will contribute to development of novel knowledge in human patients.
References
Lucia S, Esposito M, Rossi Fanelli F, Muscaritoli M. Cancer cachexia: from molecular mechanisms to patient’s care. Crit Rev Oncog. 2012;17:315–21.
Baracos VE. Pitfalls in defining and quantifying cachexia. J Cachexia Sarcopenia Muscle. 2011;2:71–3.
Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–95.
Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862–71.
Dodson S, Baracos VE, Jatoi A, Evans WJ, Cella D, Dalton JT, et al. Muscle wasting in cancer cachexia: clinical implications, diagnosis, and emerging treatment strategies. Annu Rev Med. 2011;62:265–79.
Antoun S, Baracos VE, Birdsell L, Escudier B, Sawyer MB. Low body mass index and sarcopenia associated with dose-limiting toxicity of sorafenib in patients with renal cell carcinoma. Ann Oncol. 2010;21:1594–8.
Lieffers JR, Bathe OF, Fassbender K, Winget M, Baracos VE. Sarcopenia is associated with postoperative infection and delayed recovery from colorectal cancer resection surgery. Br J Cancer. 2012;107:931–6.
Sukhanov S, Semprun-Prieto L, Yoshida T, Michael Tabony A, Higashi Y, Galvez S, et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci. 2011;342:143–7.
Fukuda T, Sumi T, Nobeyama H, Yoshida H, Matsumoto Y, Yasui T, et al. Multiple organ failure of tumor-bearing rabbits in cancer cachexia is caused by apoptosis of normal organ cells. Int J Oncol. 2009;34:61–7.
Xu H, Crawford D, Hutchinson KR, Youtz DJ, Lucchesi PA, Velten M, et al. Myocardial dysfunction in an animal model of cancer cachexia. Life Sci. 2011;88:406–10.
Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011;71:1710–20.
Wilens SL, Dische MR, Henderson D. The low incidence of terminal myocardial infarction and the reversibility of cardiac hypertrophy in cachexia. Am J Med Sci. 1967;253:651–60.
Burch GE, Phillips JH, Ansari A. The cachectic heart. A clinico-pathologic, electrocardiographic and roentgenographic entity. Dis Chest. 1968;54:403–9.
von Haehling S, Lainscak M, Springer J, Anker SD. Cardiac cachexia: a systematic overview. Pharmacol Ther. 2009;121:227–52.
White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One. 2011;6:e24650.
Schmitt TL, Martignoni ME, Bachmann J, Fechtner K, Friess H, Kinscherf R, et al. Activity of the Akt-dependent anabolic and catabolic pathways in muscle and liver samples in cancer-related cachexia. J Mol Med (Berl). 2007;85:647–54.
Baracos VE. Hypercatabolism and hypermetabolism in wasting states. Curr Opin Clin Nutr Metab Care. 2002;5:237–9.
Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–84.
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
Zhou W, Jiang ZW, Tian J, Jiang J, Li N, Li JS. Role of NF-kappaB and cytokine in experimental cancer cachexia. World J Gastroenterol. 2003;9:1567–70.
Mantovani G, Madeddu C, Maccio A. Cachexia and oxidative stress in cancer: an innovative therapeutic management. Curr Pharm Des. 2012;18:4813–8.
Laviano A, Meguid MM, Preziosa I, Rossi Fanelli F. Oxidative stress and wasting in cancer. Curr Opin Clin Nutr Metab Care. 2007;10:449–56.
Asp ML, Tian M, Wendel AA, Belury MA. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int J Cancer. 2010;126:756–63.
Honors MA, Kinzig KP. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J Cachexia Sarcopenia Muscle. 2012;3:5–11.
Wang Y, Pessin JE. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care. 2013;16:243–50.
Sjostrom M, Wretling ML, Karlberg I, Eden E, Lundholm K. Ultrastructural changes and enzyme activities for energy production in hearts concomitant with tumor-associated malnutrition. J Surg Res. 1987;42:304–13.
Tian M, Asp ML, Nishijima Y, Belury MA. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int J Oncol. 2011;39:1321–6.
Tian M, Nishijima Y, Asp ML, Stout MB, Reiser PJ, Belury MA. Cardiac alterations in cancer-induced cachexia in mice. Int J Oncol. 2010;37:347–53.
Manne ND, Lima M, Enos RT, Wehner P, Carson JA, Blough E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int J Oncol. 2013;42:2134–40.
Muhlfeld C, Das SK, Heinzel FR, Schmidt A, Post H, Schauer S, et al. Cancer induces cardiomyocyte remodeling and hypoinnervation in the left ventricle of the mouse heart. PLoS One. 2011;6:e20424.
Wysong A, Couch M, Shadfar S, Li L, Rodriguez JE, Asher S, et al. NF-kappaB inhibition protects against tumor-induced cardiac atrophy in vivo. Am J Pathol. 2011;178:1059–68.
Shadfar S, Couch ME, McKinney KA, Weinstein LJ, Yin X, Rodriguez JE, et al. Oral resveratrol therapy inhibits cancer-induced skeletal muscle and cardiac atrophy in vivo. Nutr Cancer. 2011;63:749–62.
Palus S, von Haehling S, Flach VC, Tschirner A, Doehner W, Anker SD, et al. Simvastatin reduces wasting and improves cardiac function as well as outcome in experimental cancer cachexia. Int J Cardiol. 2013;168:3412–8.
Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–43.
Springer J, Tschirner A, Hartman K, von Haehling S, Anker SD, Doehner W. The xanthine oxidase inhibitor oxypurinol reduces cancer cachexia-induced cardiomyopathy. Int J Cardiol. 2013.
Piepoli MF, Kaczmarek A, Francis DP, Davies LC, Rauchhaus M, Jankowska EA, et al. Reduced peripheral skeletal muscle mass and abnormal reflex physiology in chronic heart failure. Circulation. 2006;114:126–34.
Schulze PC, Linke A, Schoene N, Winkler SM, Adams V, Conradi S, et al. Functional and morphological skeletal muscle abnormalities correlate with reduced electromyographic activity in chronic heart failure. Eur J Cardiovasc Prev Rehabil. 2004;11:155–61.
Freeman LM. The pathophysiology of cardiac cachexia. Curr Opin Support Palliat Care. 2009;3:276–81.
Mijan-de-la-Torre A. Recent insights on chronic heart failure, cachexia and nutrition. Curr Opin Clin Nutr Metab Care. 2009;12:251–7.
Florea VG, Henein MY, Rauchhaus M, Koloczek V, Sharma R, Doehner W, et al. The cardiac component of cardiac cachexia. Am Heart J. 2002;144:45–50.
Florea VG, Moon J, Pennell DJ, Doehner W, Coats AJ, Anker SD. Wasting of the left ventricle in patients with cardiac cachexia: a cardiovascular magnetic resonance study. Int J Cardiol. 2004;97:15–20.
Prado CM, Sawyer MB, Ghosh S, Lieffers JR, Esfandiari N, Antoun S, et al. Central tenet of cancer cachexia therapy: do patients with advanced cancer have exploitable anabolic potential? Am J Clin Nutr. 2013;98:1012–9.
Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardio-oncological prevention. J Natl Cancer Inst. 2010;102:14–25.
Schieszer J. The underreported cardiac toxicity of anticancer drugs. Solid Tumors. 2012;07:06.
Gillespie HS, McGann CJ, Wilson BD. Noninvasive diagnosis of chemotherapy related cardiotoxicity. Curr Cardiol Rev. 2011;7:234–44.
Di Lorenzo G, Autorino R, Bruni G, Carteni G, Ricevuto E, Tudini M, et al. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann Oncol. 2009;20:1535–42.
Mego M, Reckova M, Obertova J, Sycova-Mila Z, Brozmanova K, Mardiak J. Increased cardiotoxicity of sorafenib in sunitinib-pretreated patients with metastatic renal cell carcinoma. Ann Oncol. 2007;18:1906–7.
Bello CL, Mulay M, Huang X, Patyna S, Dinolfo M, Levine S, et al. Electrocardiographic characterization of the QTc interval in patients with advanced solid tumors: pharmacokinetic- pharmacodynamic evaluation of sunitinib. Clin Cancer Res. 2009;15:7045–52.
Cho DC, Puzanov I, Regan MM, Schwarzberg T, Seery V, Lee MY, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother. 2009;32:181–5.
Shi Y, Moon M, Dawood S, McManus B, Liu PP. Mechanisms and management of doxorubicin cardiotoxicity. Herz. 2011;36:296–305.
Menna P, Paz OG, Chello M, Covino E, Salvatorelli E, Minotti G. Anthracycline cardiotoxicity. Expert Opin Drug Saf. 2012;11 Suppl 1:S21–36.
Telli ML, Witteles RM, Fisher GA, Srinivas S. Cardiotoxicity associated with the cancer therapeutic agent sunitinib malate. Ann Oncol. 2008;19:1613–8.
Cochet A, Quilichini G, Dygai-Cochet I, Touzery C, Toubeau M, Berriolo-Riedinger A, et al. Baseline diastolic dysfunction as a predictive factor of trastuzumab-mediated cardiotoxicity after adjuvant anthracycline therapy in breast cancer. Breast Cancer Res Treat. 2011;130:845–54.
Serrano C, Cortes J, De Mattos-Arruda L, Bellet M, Gomez P, Saura C, et al. Trastuzumab-related cardiotoxicity in the elderly: a role for cardiovascular risk factors. Ann Oncol. 2012;23:897–902.
Prado CM, Antoun S, Sawyer MB, Baracos VE. Two faces of drug therapy in cancer: drug-related lean tissue loss and its adverse consequences to survival and toxicity. Curr Opin Clin Nutr Metab Care. 2011;14:250–4.
Di Lisi D, Bonura F, Macaione F, Cuttitta F, Peritore A, Meschisi M, et al. Chemotherapy-induced cardiotoxicity: role of the conventional echocardiography and the tissue Doppler. Minerva Cardioangiol. 2011;59:301–8.
Di Lisi D, Bonura F, Macaione F, Peritore A, Meschisi M, Cuttitta F, et al. Chemotherapy-induced cardiotoxicity: role of the tissue Doppler in the early diagnosis of left ventricular dysfunction. Anticancer Drugs. 2011;22:468–72.
Wang J, Khoury DS, Thohan V, Torre-Amione G, Nagueh SF. Global diastolic strain rate for the assessment of left ventricular relaxation and filling pressures. Circulation. 2007;115:1376–83.
Stoodley PW, Richards DA, Boyd A, Hui R, Harnett PR, Meikle SR, et al. Altered left ventricular longitudinal diastolic function correlates with reduced systolic function immediately after anthracycline chemotherapy. Eur Heart J Cardiovasc Imaging. 2013;14:228–34.
Stoodley PW, Richards DA, Meikle SR, Clarke J, Hui R, Thomas L. The potential role of echocardiographic strain imaging for evaluating cardiotoxicity due to cancer therapy. Heart Lung Circ. 2011;20:3–9.
Cramer L, Kung T, Hildebrandt B, Nicolaou A, Doehner W, Anker SD, et al. Common cardiac symptoms in chronic diseases: a comparison between patients with heart failure and colorectal cancer concerning heart rate variability, cardiac function and exercise capacity. Abstracts of the 6th Cachexia Conference, Milan, Italy, December 8–10, 2011 (Part 2). J Cachexia Sarcopenia Muscle. 2012;3:57–8.
Schunemann M, Anker SD, Rauchhaus M. Cancer fatigue syndrome reflects clinically non-overt heart failure: an approach towards onco-cardiology. Nat Clin Pract Oncol. 2008;5:632–3.
von Haehling S, Lainscak M, Kung T, Cramer L, Fulster S, Pelzer U, et al. Non-invasive assessment of cardiac hemodynamics in patients with advanced cancer and with chronic heart failure: a pilot feasibility study. Arch Med Sci. 2013;9:261–7.
Der-Torossian H, Gourin CG, Couch ME. Translational implications of novel findings in cancer cachexia: the use of metabolomics and the potential of cardiac malfunction. Curr Opin Support Palliat Care. 2012;6:446–50.
von Haehling S, Lainscak M, Doehner W, Ponikowski P, Rosano G, Jordan J, et al. Diabetes mellitus, cachexia and obesity in heart failure: rationale and design of the Studies Investigating Co-morbidities Aggravating Heart Failure (SICA-HF). J Cachexia Sarcopenia Muscle. 2010;1:187–94.
Baracos V, Caserotti P, Earthman CP, Fields D, Gallagher D, Hall KD, et al. Advances in the science and application of body composition measurement. JPEN. 2012;36:96–107.
Prado CM, Birdsell LA, Baracos VE. The emerging role of computerized tomography in assessing cancer cachexia. Curr Opin Support Palliat Care. 2009;3:269–75.
Mourtzakis M, Prado CM, Lieffers JR, Reiman T, McCargar LJ, Baracos VE. A practical and precise approach to quantification of body composition in cancer patients using computed tomography images acquired during routine care. Appl Physiol Nutr Metab. 2008;33:997–1006.
Lieffers JR, Mourtzakis M, Hall KD, McCargar LJ, Prado CM, Baracos VE. A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: contributions of organ and tumor mass to whole-body energy demands. Am J Clin Nutr. 2009;89:1173–9.
Prado CM, Lieffers JR, McCargar LJ, Reiman T, Sawyer MB, Martin L, et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population-based study. Lancet Oncol. 2008;9:629–35.
Parsons HA, Baracos VE, Dhillon N, Hong DS, Kurzrock R. Body composition, symptoms, and survival in advanced cancer patients referred to a phase I service. PLoS One. 2012;7:e29330.
Parsons HA, Tsimberidou AM, Pontikos M, Fu S, Hong D, Wen S, et al. Evaluation of the clinical relevance of body composition parameters in patients with cancer metastatic to the liver treated with hepatic arterial infusion chemotherapy. Nutr Cancer. 2012;64:206–17.
Tan BH, Birdsell LA, Martin L, Baracos VE, Fearon KC. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin Cancer Res. 2009;15:6973–9.
Maisel A, Mueller C, Adams Jr K, Anker SD, Aspromonte N, Cleland JG, et al. State of the art: using natriuretic peptide levels in clinical practice. Eur J Heart Fail. 2008;10:824–39.
Feola M, Garrone O, Occelli M, Francini A, Biggi A, Visconti G, et al. Cardiotoxicity after anthracycline chemotherapy in breast carcinoma: effects on left ventricular ejection fraction, troponin I and brain natriuretic peptide. Int J Cardiol. 2011;148:194–8.
Sawaya H, Sebag IA, Plana JC, Januzzi JL, Ky B, Tan TC, et al. Assessment of echocardiography and biomarkers for the extended prediction of cardiotoxicity in patients treated with anthracyclines, taxanes, and trastuzumab. Circ Cardiovasc Imaging. 2012;5:596–603.
DeJong CH, Busquets S, Moses AG, Schrauwen P, Ross JA, Argiles JM, et al. Systemic inflammation correlates with increased expression of skeletal muscle ubiquitin but not uncoupling proteins in cancer cachexia. Oncol Rep. 2005;14:257–63.
Op den Kamp CM, Langen RC, Minnaard R, Kelders MC, Snepvangers FJ, Hesselink MK, et al. Pre-cachexia in patients with stages I-III non-small cell lung cancer: systemic inflammation and functional impairment without activation of skeletal muscle ubiquitin proteasome system. Lung Cancer. 2012;76:112–7.
Fulster S, Tacke M, Sandek A, Ebner N, Tschope C, Doehner W, et al. Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur Heart J. 2013;34:512–9.
Lubrano V, Pingitore A, Carpi A, Iervasi G. Relationship between triiodothyronine and proinflammatory cytokines in chronic heart failure. Biomed Pharmacother. 2010;64:165–9.
Jannazzo A, Hoffman J, Lutz M. Monitoring of anthracycline-induced cardiotoxicity. Ann Pharmacother. 2008;42:99–104.
Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J. 2013. doi:10.1093/eurheartj/eht302.
Acknowledgments
SMR Kazemi-Bajestani, Harald Becher, Konrad Fassbender, Qunicy Chu, and Vickie Baracos declare that they have no conflict of interest. SMRKB is supported by the Alberta Innovates Health Solutions Graduate Studentship award. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle 2010; 1:7–8 (von Haehling S, Morley JE, Coats AJ, and Anker SD).
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Kazemi-Bajestani, S.M.R., Becher, H., Fassbender, K. et al. Concurrent evolution of cancer cachexia and heart failure: bilateral effects exist. J Cachexia Sarcopenia Muscle 5, 95–104 (2014). https://doi.org/10.1007/s13539-014-0137-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-014-0137-y