
Abstract
We seek an aetiopathogenic model for the spectrum of Parkinson’s disease (PD), functional bowel disease, depression and cognitive impairment. The adopted concept is that systemic immuno-inflammatory processes mediate neuro-inflammation. The model would be based on phenotype, exposome (including gastrointestinal microbiome), milieu (immuno-inflammatory and metabolome), human genetics and their interactions. It would enable a patient’s position, to be understood in terms of drivers, perpetuators and mediators, and a future position, with and without intervention, predicted. Even the cardinal facets of PD may have different drivers: halting one may allow escape down subordinate pathways. Peptic ulceration is prodromal to PD. In our randomised placebo-controlled trial, hypokinesia improved over the year following biopsy-proven Helicobacter pylori eradication and rigidity worsened. This was independent of any (stable, long t½) antiparkinsonian medication. There are pointers to an autoimmune process: for example, surveillance-confirmed hypokinesia effect was indication specific. During surveillance, successive antimicrobial courses, other than for Helicobacter, were associated with cumulative increase in rigidity. Exhibiting laxatives appeared to stem the overall temporal increase, despite antiparkinsonian medication, in rigidity. Thus, intestinal dysbiosis may be a major source of bystander neuronal damage. There are biological gradients of objective measures of PD facets on circulating inflammatory markers and leucocyte subset counts. Moreover, lactulose hydrogen breath test positivity for small-intestinal bacterial overgrowth (present in two thirds of PD patients) is associated with the same subsets: higher natural killer and total CD4+ counts and lower neutrophils. With greater aetiopathogenic understanding, relatively low cost and on-the-shelf medication could have a major impact. A new generation of animal models, based on the gut microbiome, is envisaged.
Similar content being viewed by others
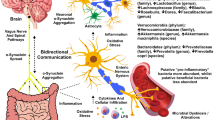
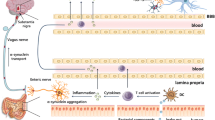

Introduction
Since the shaking palsy, a rigid brady/hypokinetic syndrome with a characteristic tremor and stooped posture was described (Parkinson 1817); there have been few therapeutic milestones. Indeed, the only major advance, dopamine substitution therapy, dates back to description of dopamine deficiency in the basal ganglia (Ehringer and Hornykiewicz 1960).
Simultaneous co-morbidities, of which James Parkinson noted constipation, may have mediators, drivers and perpetuators in common. These include depression and mild cognitive impairment, with or without progression to dementia, as well as functional bowel disease. Unravelling the aetiopathogenesis, in a common disease with a long prodrome, will be jeopardised by not taking into account that individuals can be quantifiably down-the-pathway (Kirollos et al. 1993). A co-morbidity where the core condition is not overt may define an extreme of the spectrum and explain containment. Targeting aetiopathogenesis, rather than just phenotypic descriptors of disease, has the potential to open doors on cost-effective screening, prophylaxis, amelioration of the underlying processes and cure.
An aetiopathogenic model for this disease spectrum and its evolution with time and intervention is, thus, needed. Evolution refers both to change in individual facets of phenotype and shift within the spectrum. The model would enable a patient’s position in the spectrum to be understood in terms of drivers, perpetuators and mediators, and a future position, with and without intervention, predicted. Exposome, milieu (immuno-inflammatory and metabolome), human genetics and their interactions need to be considered as building blocks. Only ‘biomarkers’ which reflect driving or perpetuating forces can be useful in the modelling. Understanding the development of disease-specific pathophysiology requires longitudinal observational study to unmask associations, interventions to home in on cause/effect relationships and a new generation of animal models. A quantifiable aetiopathogenic model can be cross-referenced against quality of life and health economic outcomes.
The challenge requires (i) considering the whole disease spectrum; (ii) including pre-presentation states and attenuated or partial manifestations, not just ‘the tip of the iceberg’; (iii) assembling raw clinical clues without selectivity; (iv) using valid, sensitive, specific and reliable measures (objective where possible) of disease facets, which can track evolution; (v) stratifying paths of evolution into those intrinsic to initiation and the subsidiary; and (vi) animal models of the aetiopathogenesis that is not relying on surgical, chemical or genetic lesioning and so being downstream of, or out-with, environmental driving processes.
Underpinning concepts and main ideas
Like other chronic diseases, Parkinson’s disease (PD) is multi-step and multi-factorial. Even more steps and factors, and biological gradients will be needed to explain co-morbidities and the spectrum of disease. Even the cardinal facets of PD may have different, not necessarily coincident, drivers: halting one may allow escape down subordinate pathways. However, all this does not preclude a systematic explanation.
The core concept is that neuro-inflammation in PD and overlap diseases is mediated by systemic immuno-inflammatory processes (Dobbs et al. 2008, 2012). It is not just reaction to aberrant protein deposition or degenerating neurons. There is indicative evidence that dysbiosis in the alimentary tract is the major driver of these processes (Augustin et al. 2014; Dobbs et al. 2008, 2010, 2012, 2013). Position of a patient within the disease spectrum is further determined by interaction with host genetics (risk and inflammatory), inflammatory and metabolic milieu and the exposome (including environmental factors, such as tobacco smoking). Intervention against drivers, perpetuators or mediators would allow disease modification.
Some indicative models
Influence of microbiome
Germ-free mice move more and take more risks: they have increased striatal synaptogenesis and dopamine/serotonin turnover (Bercik et al. 2011). In specific pathogen-free mice, non-absorbable antimicrobials increase exploratory behaviour and hippocampal brain-derived neurotrophic factor (BDNF). Gavage of caecal contents from a more outgoing mouse strain into a more timid increases both exploratory behaviour and BDNF, and vice versa. Specific probiotics cause behavioural change.
Important associations between stool bacteria microbiota and human health have been identified (Arumugam et al. 2011; Blottiere et al. 2013; Collins et al. 2012; Cotillard et al. 2013; Doré et al. 2013; Le Chatelier et al. 2013; Manichanh et al. 2006; Qin et al. 2014). There is evidence that stool microbial metagenomics can discriminate better for chronic disease than human genomics (Qin et al. 2014; Speliotes et al. 2010): interventions here should clarify cause/effect relationships. Psychiatric illness in irritable bowel syndrome (and inflammatory bowel disease) and autism spectrum disorder have been ascribed to dysbiosis (Doré et al. 2013). Autism recalls the restricted behaviour of PD. An inflammation-associated form of depression is described (Doré et al. 2013). Exhibition of the non-absorbable broad-spectrum antimicrobial rifaximin was accompanied by amelioration of parkinsonism associated with hepatic encephalopathy in three patients with cirrhosis and portosystemic shunting, in whom blood ammonia and electroencephalogram were unchanged (Kok et al. 2013). Imaging features in the globus pallidus, classical of parkinsonism in cirrhosis, were reduced. The only study of microbiota in PD is a cross-sectional comparison of 72 probands (almost all on antiparkinsonian medication) with 72 controls (with markedly increased frequency of cerebro- and cardio-vascular co-morbidities), without reference to dietary differences (Scheperjans et al. 2014).
Influences on phenotype
Constipation and small-intestinal bacterial overgrowth
In PD, the frequency of defaecation diverges from that of controls three decades before the median age of neurological diagnosis (Charlett et al. 1997). Moreover, infrequent bowel movements are associated with a subsequent diagnosis (Abbott et al. 2001). Morphological and neurochemical changes of PD are found throughout the enteric nervous system and in dorsal vagal nuclei which serve the gastrointestinal tract (for review, see Dobbs et al. 2008).
Sixty-seven per cent of PD probands are lactulose hydrogen breath test (LHBT) positive for small-intestinal bacterial overgrowth (SIBO) on presentation (Dobbs et al. 2012). A likely cause is caeco-ileal bacterial reflux from an overloaded right colon. SIBO influences the immuno-inflammatory milieu, and biological gradients connect milieu to phenotype. Accompanying dysbiosis in the fermentation ‘bioreactor’ of the right colon may reduce production of anti-inflammatory substances such as short-chain fatty acids (Neish 2009).
Clues regarding rigidity
Surveillance of arm rigidity showed a significant temporal increase (7 % per year) in flexor rigidity, with consequent increase in the ratio, flexor to extensor rigidity, denoting simian posture (Augustin et al. 2014). Exhibiting laxatives, in general, was associated with stemming the increase, after adjustment for the various classes of long t½ antiparkinsonian medication (dopaminergic agonists, MAO-B inhibitors, amantadine, anticholinergics) and the limited use of levodopa in low dosage. Exhibiting the guanylate cyclase-C receptor agonist (linaclotide) was associated with reversing the temporal trend.
Successive courses of antimicrobials in PD are associated with cumulative increase in flexor rigidity (Fig. 1), over and above the effect of time and irrespective of indication (Dobbs et al. 2013).

Cumulative worsening of objectively measured rigidity with successive antimicrobial interventions in PD
Clues regarding brady/hypokinesia
That peptic ulceration is prodromal (Strang 1965) paved the way to exploring Helicobacter pylori in PD. In a randomised controlled trial (RCT), biopsy-proven H. pylori eradication reduced hypokinesia of gait in PD (Dobbs et al. 2010). Longitudinal observation showed indication specificity, in that antimicrobials for other indications did not improve hypokinesia (Dobbs et al. 2013). In the trial, whilst hypokinesia improved, rigidity worsened over the year post-eradication, both plateauing over the next (Fig. 2). There was overall clinical benefit. Improved hypokinesia was independent of any (stable, long t½) antiparkinsonian medication. (Receipt of levodopa was an exclusion.) Increased rigidity may flag acquisition of SIBO, since H. pylori and LHBT positivity are inversely related in PD (Dobbs et al. 2012). At present, the level of evidence is 1b since this is an individual RCT (OCEBM 2011).

Schematic representation of the effect of H. pylori eradication on stride length and flexor rigidity in PD. Estimated mean time trends following successful blinded-active (black), open-active (grey) and placebo (dashed). Rejection of null hypothesis was based on double-blind protocol analysis of time trends in the primary outcome, stride length, reinforced by intention-to-treat analysis on its final measurement in blinded phase (p = 0.005), despite inclusion of the two proven eradication failures following blinded-active
In no disease, where H. pylori is causal, is it present in all cases. However, current or past Helicobacter infection may be a necessary though not sufficient player in developing the full syndrome. There is a lack of birth cohort effect for H. pylori in PD, as in gastric cancer and peptic ulcer where causal links with H. pylori are generally accepted (Dobbs et al. 2000). Danish population registers show increased prescription of anti-Helicobacter drugs in the 5 years prior to diagnosis (Nielsen et al. 2012). Dopaminergic agonists can prevent duodenal ulcer relapse in man (Sikiric et al. 1991), but whether by suppressing H. pylori is unknown.
Classical spousal approach to environmental causality
Spouses of PD probands are a short but highly significant ‘distance-down-the-pathway’ with respect to objective measures of PD facets (Kirollos et al. 1993, 1996; O’Neill et al. 1994; Weller et al. 1992). Probands and spouses have relative lymphopenia (with particular effect on B cells) (Charlett et al. 2009). There is a proportional increase in natural killer cell (NK) count in probands, in CD4+ in spouses. Half of the probands and a third of spouses have chronic functional bowel abnormality (Ellis et al. 2007). Like probands, two thirds of spouses are LHBT positive (Dobbs et al. 2012). The whole is difficult to attribute to selective mating or to learned or reactive behaviour. Neither is it explained by H. pylori: spouses had a lower frequency of Helicobacter anti-urease IgG enzyme-linked immunosorbent assay (ELISA) seropositivity than either probands or controls (Charlett et al. 2009), as though an acquired dysbiosis had suppressed it.
Influence of immuno-inflammatory milieu
Biological gradients on circulating leukocyte subtypes
There are gradients of objective measures of facets of PD on blood leucocyte subtype counts (Dobbs et al. 2012). Brady/hypokinesia and flexor rigidity are worse the higher the NK count. Increased brady/hypokinesia was noted with Helicobacter positivity, over and above that explained by NK count and of a magnitude equivalent to that of a levodopa challenge. Association of rigidity with a higher NK count is modulated by the total CD4+ count. The CD4+ subset includes regulatory T cells (T-reg) which inhibit NK effector mechanisms. Tremor is worse with lower neutrophils: this may reflect neutrophil sequestration in the gut.
LHBT positivity is associated with the same blood leucocyte subtypes: (higher) NK and CD4+ counts and (lower) neutrophils. Moreover, clouds of lysosomes seen in duodenal enterocytes in relation to luminal bacteria underline that SIBO is not an innocent bystander in PD (Fig. 3a) (Dobbs et al. 2012). The simplest biologically plausible explanation is that circulating leucocytes represent mediators of neuronal damage, and dysbiosis, flagged here by SIBO, represents a driver.

a Electron micrographs illustrating a cloud of irregular lysosomes in a duodenal enterocyte in a PD patient with SIBO, at low magnification (right) and higher (left). b Electron micrographs illustrating protein arrays encapsulated in a double membrane, at low magnification (left: multiple bodies) and higher (right: body amongst (normal) mitochondria), where arrays are seen longitudinally and in transverse section
Biological gradients on circulating immune-inflammatory markers
There are gradients of objective measures of PD facets on serum cortisol and tumour necrosis factor alpha (TNF-α), and of global motor scores on peripheral blood mononuclear cell production of cytokines and nuclear factor-kappa B (NFκB) expression (Charlett et al. 1998; Dobbs et al. 1999; Reale et al. 2009). Cortisol is elevated by, on average, 17 % in PD. Serum interleukin-6 (IL-6) increases with age: it is elevated in PD by an amount equivalent to 10 years of ageing. Moreover, a higher concentration of IL-6, in blood collected 4 years previously, is predictive of incident PD (Chen et al. 2008).
Immuno-inflammatory activation can increase homocysteine production (Lazzerini et al. 2007). Hyperhomocysteinemia in PD (43 %) is explained, in small part, by the serum concentration of vitamin B12 (cobalamin), with no complementary effect of folate (Charlett et al. 2009). (Methyltetrahydrofolate acts as a methyl donor, and cobalamin is a co-factor, in remethylation of homocysteine to methionine by methionine synthetase.) Hyperhomocysteinemia is not explained by Helicobacter status or gastric atrophy. Impaired terminal ileal B12 absorption, associated with dysbiosis, might contribute. Although there was no evidence of frank B12 deficiency in PD, 16 % of probands had concentrations within the ‘equivocal range’ (Charlett et al. 2009). (Serum folate distribution was platykurtic.) Immuno-inflammatory activation may increase demand for B12 to such an extent that a concentration in the ‘equivocal’ range is pathological. Since SIBO both provokes an inflammatory response and increases bacterial utilisation of B12, it is likely to contribute to hyperhomocysteinemia in PD.
Hyperhomocysteinemia is associated with an increased risk of development of dementia and Alzheimer’s disease (AD) (Seshadri et al. 2002). Low and equivocal serum B12 concentrations, and the metabolically active fraction of serum cobalamin, have been implicated (Clarke et al. 1998; Refsum and Smith 2003; Seshadri et al. 2002), but the contribution of gastric atrophy and impaired ileal absorption is unknown. The low serum folate of AD (Clarke et al. 1998) appears to be an argument against SIBO (associated with increased synthesis of folate) being the main player, but demand for folate to detoxify homocysteine may be increased.
Using the western blot profile of IgG antibodies against electrophoretically separated H. pylori antigens, the predicted probability of being labelled as having PD was greatest with cytotoxin-associated gene product (CagA) positivity and vacuolating toxin negativity, and urease B negativity (Weller et al. 2005). With this pattern, the odds for having PD were increased fivefold at age 80 years. The predictive ability was not confined to those with current infection.
Metabolome
From an aetiopathogenic standpoint, a key question, then, is whether the metabolome regulates systemic inflammation. In mice, short-chain fatty acids (SCFA), such as butyrate, produced by colonic bacterial fermentation, promote colonic T-reg cells and thereby suppress pro-inflammatory T cells (Smith et al. 2013). This suggests a mechanism for CD4+ modulation (T-reg component) of rigidity and fits with PD probands’ spouses having a proportional (protective) increase in total CD4+ count. However, whilst oral administration of SCFA to germ-free mice to address their deficit increased colonic T-regs, it had no effect on mesenteric lymph node, splenic or thymic T-regs. Human studies have linked fermentation products to behaviour (Collins et al. 2012). For example, high faecal propionic acid concentrations correlate with anxiety in irritable bowel syndrome.
Metabolomic interest in PD and AD has concentrated on markers of damage (e.g. hypoxia, oxidative stress and membrane lipid remodelling) in blood (Bogdanov et al. 2008; Orešič et al. 2011) and breath (Nakhleh et al. 2015).
Bystander damage and cross-reactivity
The biological gradients described suggest bystander damage to the central nervous system in PD, driven by dysbiosis. Dysbiosis could account for the continuing substantia nigra microglial activation of PD (Pfeiffer 2009). This does not exclude added insults by intercurrent infection/its treatment.
In PD, nigral microglia express major histocompatibility antigens, including HLA-DR (McGeer et al. 1998). They secrete TNF-α, whilst dopaminergic neurons express its receptors and upregulate NFκB (Boka et al. 1994; Hunot et al. 1997). Nigral and cerebrospinal fluid (CSF) concentrations of other cytokines associated with innate immune response, IL-1β and IL-6, are elevated (Mogi et al. 1994, 1996). Although pro-inflammatory polymorphisms have not been identified as risks for PD in genome-wide studies, they might act as a conditional dependency of an effect modifier.
Adaptive immunity and autoimmunity may also have a role, separated in time from bystander damage or concurrent (Dobbs et al. 2008). Nigral dopaminergic neurons bind IgG, adjacent microglia express HLA-DR and high-affinity Fcγ antibody receptors, whilst peripheral lymphocytes are seen in relation to degenerating neurons (Orr et al. 2005). Serum, CSF and purified IgG from PD probands selectively inhibit dopamine uptake of rodent nigral dopaminergic neurons and selectively destroy neurons in vivo and in vitro (Chen et al. 1998; Dahlström et al. 1990; Defazio et al. 1994; McRae et al. 1986). Toxicity is dependent on complement and microglial Fcγ receptors (Defazio et al. 1994; He et al. 2002). Associations of PD with HLA-DR loci suggest classical autoimmunity (Ahmed et al. 2012; Hamza et al. 2010; International Parkinson Disease Genetics Consortium et al. 2011). Indeed, antinuclear antibody seropositivity flags poor response of hypokinesia to H. pylori eradication therapy (Dobbs et al. 2010). That H. pylori has been associated with hypokinesia when the infection load is low (detected by PCR only, not culture) is compatible with autoimmunity (Dobbs et al. 2008, 2010). There could also be cross-reactivity through innate pattern recognition of Helicobacter at the genus level (Dobbs et al. 2005) or of a broader microbial community.
The adaptive immune response in the substantia nigra in PD (Orr et al. 2005) and the presence of peripheral immune cells (as well as Lewy bodies) in therapeutically useful dopamine cell brain implants (Kordower et al. 2008) fit with a peripheral immune process driving neuronal damage (Lewy bodies are intracytoplasmic neuronal inclusions of misfolded α-synuclein, considered the gold standard for designation of PD). Indeed, in more advanced PD, the proportion of IgG-labelled nigral neurons decreases, but the activated microglia persist (Orr et al. 2005), suggesting that the immune-inflammatory process is driving neuronal damage, not vice versa. Elevated cortisol and cytokines in PD (Charlett et al. 1998; Dobbs et al. 1999) would increase permeability of the blood–brain barrier to peripheral immune cells, antigen, antibodies or products. There might also be vagal afferent signalling to microglia (Watkins 1995).
Outside the context of inflammation, effects of some probiotic bacteria on behaviour of experimental animals appear vagal dependent (Collins et al. 2012). Other workers (Bercik et al. 2011) have produced behavioural and neurochemical changes, apparently independent of the autonomic system and circulating cytokines, by manipulating the microbiota. They propose the pathway may involve neurally active substances (e.g. an antidepressant effect of butyrate).
Abnormal mitochondria
Nigral and platelet mitochondrial dysfunction is described in PD (Dobbs et al. 2008). There is dysmorphology in duodenal enterocytes. Long, thin mitochondria, associated with the rough endoplasmic reticulum, are commonly seen, in the presence of SIBO but absence of recent H. pylori infection (Charlett et al. 2009). The complex branching also seen may result from failure to divide (Dobbs et al. 2008).
With current or recent H. pylori infection, arrays encapsulated in a double membrane are found in half of cases (Fig. 3b), lying among normal mitochondria (Dobbs et al. 2008; Ellis et al. 2007). There is a report of similar mitochondrial inclusions in cerebral neurons in Creutzfeldt–Jakob-like disease (Lewin and Edwards 1991). Alternatively, these bodies might be viroplasm, not mitochondria. Our electron microscopists (personal communication: Ellis D and Curry A) had not previously observed similar bodies but subsequently found examples in archived duodenal biopsies from two patients with human immunodeficiency virus (HIV) infection.
If these findings are replicated in enteric neurons and/or myocytes and are associated with mitochondrial dysfunction, they may provide a mechanism behind slow gastrointestinal transit. Indeed, cardiac and skeletal muscle mitochondrial hypofunction may contribute to hypotension and brady/hypokinesia.
A viral primer as a supplementary explanation?
Faecal overload and SIBO are predisposed to by the slow GI transit of PD and may have detrimental feedback on it, but what initiates that slow transit? Could there be a viral primer? Enteroviruses infect via the gastrointestinal tract and are associated with neurological syndromes. Indeed, there is recent evidence of an enterovirus as a cause of encephalitis lethargica and post-encephalitic parkinsonism (Dourmashkin et al. 2012), and in our pilot study, using faecal samples taken at the start of a diarrhoeal episode, the frequency of enterovirus genogroup B RNA appeared high in PD patients and their spouses (personal communication: Appleton H).
A viral primer could also be involved in the relative lymphopenia seen in both PD patients and their spouses, compared with controls. This robust finding was not explained by antiparkinsonian medication, Helicobacter status or breath hydrogen. A relatively benign retrovirus might explain this and the slow transit. The comparatively high frequency of Dientamoeba fragilis (18 % of PD probands and their spouses cf. 2.6 % of routine parasitology requests) (Ellis et al. 2007) could flag mild acquired immunodeficiency. The epidemiology of IP and HIV is distinct, but parkinsonism is seen in uncomplicated HIV infection (as well as with opportunistic infections in acquired immunodeficiency syndrome) (Koutsilieri et al. 2002; Karlsen et al. 1992; Berger and Arendt 2000). Moreover, jejunal autonomic denervation is described with HIV infection (Dourmashkin et al. 2012). Although Lewy bodies are not reported in HIV (Koutsilieri et al. 2002), motor dysfunction compatible with basal ganglia damage is found in early and basal ganglia dopaminergic cell loss is seen without clinical parkinsonism. In simian immunodeficiency virus-infected monkeys, nigrostriatal dopamine is halved within 2 months (Koutsilieri et al. 2002).
Time sequence: a process initiated by, driven from, the gastrointestinal tract?
The time sequence of constipation and peptic ulceration fits with the misfolded protein theory of PD pathogenesis. Aggregates of misfolded α-synuclein are found throughout the enteric nervous system and in dorsal vagal nuclei (Braak et al. 2006). A subpopulation of α-synuclein-expressing myenteric neurons, synaptically connected to vagal efferents, has been described in rodents (Phillips et al. 2008). As PD evolves, aggregation spreads from the brainstem to the substantia nigra, areas of the midbrain and basal forebrain, eventually reaching the neocortex (Braak et al. 2003).
Although misfolded proteins can aggregate by template replication in a prion-like manner (Luk et al. 2009), it is not known what initiates or ‘seeds’ misfolding, converts containment to progression or drives progression. Enteric α-synuclein aggregates are described with local inflammation in the gut, both clinical and experimental colitis (Grathwohl et al. 2013). In early PD, inflammation, measured by imaging microglial activation, in the affected nigrostriatal pathway accompanies loss of presynaptic dopamine transporter (Ouchi et al. 2005) and becomes more widespread on follow-up (Ouchi et al. 2009). Central neuro-inflammation may have peripheral mediators (see ‘Influence of immuno-inflammatory milieu’).
Effective therapeutic strategies to prevent, contain and clear α-synuclein deposition may be jeopardised by a persistent driver presented by gut microbiota or by its immuno-inflammatory or metabolomic mediator(s).
Effecting a paradigm shift
‘The best science often emerges from situations where results carefully obtained do not fit within accepted paradigms’ (Prusiner 1982). Whereas an array of stances is legitimate in the humanities, in science, a paradigm shift (Kuhn 1970) is all-or-nothing. It may rest on a single discovery, or, as here, unify a network of novel findings. Practicality can be as important as explanation in effecting a shift. The hypothesis that H. pylori is the usual cause of peptic ulcer (Marshall and Warren 1984) was dismissed until it became evident that eliminating Helicobacter effected cure. Until then, the success of H2 receptor antagonists in healing an ulcer had made the new paradigm unnecessary. Although requiring no change in disciplinary ownership, just in focus, from modifying gastric physiology to antimicrobial therapy, shifting that paradigm took more than a decade. In our current example, there is a need for new disciplinary interfaces in order to yield disease-modifying therapies. Considering PD only in terms of progressive, self-perpetuating, degeneration relegates any environmental influence to being, at best, remote hit-and-run. Nosological classification as ‘non-communicable’ (WHO) reinforces this. Regarding any systemic illness in PD as an ‘intercurrent event’, as opposed to co-morbidity, compounds the problem.
What is sure, in a disease peculiar to man, is that re-evaluating the patient is a good starting point. Detective work is needed, where subtle clues are uncovered and statistical analysis builds on meticulous clinical observation. In such exploratory studies, it is necessary to understand what is measured and what influences it, explore effect modification, examine biological plausibility and seek corroborative evidence. Not until a large number of clues have been assimilated will their position within a causal scheme become more certain. Pragmatic studies can then be conducted for the testable cause/effect hypotheses generated. This is the antithesis of one-step pragmatism. A scientifically challenging causal pathway does not preclude a clinical solution sufficiently simple to be assimilated into practice.
Opposition in principle is the great delayer. ‘Student’ of the t test wrote of RA Fisher’s concise statistical/mathematical text ‘When I came to “evidently” I know that it means two hours hard work at least before I can see why’ (Bodmer 2003). Getting over ‘why not’ in the face of inertia of consensus opinion is far more time consuming. ‘Consensus is always conditioned by the antecedent knowledge and its interpretation, and hence is time dependent….it should be associated with permanent criticism, which hopefully will induce corrective changes’ (Vonka 2000). Grassroot opinion, from people with the chronic disease who want a cure, is important in influencing professional consensus.
Conclusion
Biological plausibility of the underpinning concept and main ideas lies in the fit of a constellation of observational and intervention studies. In particular, biological gradients (Hill 1965) of repeated (antimicrobial) interventions and circulating inflammatory markers on measures of facets of PD add to the RCT evidence for the gastrointestinal microbiome being involved in its causality. Metagenomics can be used to address the Bradford Hill predicament of although ‘The clear dose–response curve admits of a simple explanation’ (causality), ‘Often the difficulty is to secure some satisfactory quantitative measure of the environment which will permit us explore this dose–response.’
It is envisaged that PD will be reclassified as a systemic condition in response to immuno-inflammatory activation, influenced by microbiota, tempered by human genetics, within a spectrum of neuropsychiatric and gastroenterological conditions. Dichotomous classification of PD is misconceived. Since facets may have different, non-coincident, driving forces, defining phenotype by their objective quantification is a sine qua non for progress. Lumping facets together (global clinical scores) presumes they progress in parallel within-subject, in set proportion between, and share driving forces.
A ‘depth-in-breadth’ approach maximises the potential of yielding new targets, such as optimising microbiota, eradicating pathogens, immune modulation (including cross-talk at the mucosal level and signalling to/activity of local, systemic and brain immune system) and optimising the metabolome. Relevant animal models, driven by gastrointestinal microbiome and autoimmunity, would consolidate this approach. Elucidating the cause of slow transit and relative lymphopenia may reveal a trigger event. The potential is as a catalyst for change in approach to chronic disease in general, with major social, health and financial implications.
Where drivers, perpetuators and mediators remain active, attempts at neuronal replacement, repair and regeneration and symptomatic treatment may underperform. A complex causal pathway does not preclude interim clinical solutions.
References
Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS, Ross GW (2001) Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 57:456–462. doi:10.1212/WNL.57.3.456
Ahmed I, Tamouza R, Delord M, Krishnamoorthy R, Tzourio C, Mulot C, Nacfer M, Lambert JC, Beaune P, Laurent-Puig P, Loriot MA, Charron D, Elbaz A (2012) Association between Parkinson’s disease and the HLA-DRB1 locus. Mov Disord 27:1104–1110. doi:10.1002/mds.25035
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T (2011) Enterotypes of the human gut microbiome. Nature 473:174–178. doi:10.1038/nature09944
Augustin A, Charlett A, Weller C, Dobbs SM, Taylor D, Ibrahim MAA, Bjarnason I, Dobbs RJ (2014) Osmotic laxatives are associated with lower rigidity in idiopathic parkinsonism. United Eur Gastroenterol J 2(suppl 1):A248
Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J, Deng Y, Blennerhassett P, Macri J, McCoy KD, Verdu EF, Collins SM (2011) The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 141:599–609. doi:10.1053/j.gastro.2011.04.052
Berger JR, Arendt G (2000) HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol 14:214–221. doi:10.1080/13550280290049831
Blottiere H, de Vos WM, Ehrlich SD, Doré J (2013) Human intestinal metagenomics: state of the art and future. Curr Opin Microbiol 16:232. doi:10.1016/j.mib.2013.06.006
Bodmer W (2003) RA Fisher, statistician and geneticist extraordinary: a personal view. Int J Epidemiol 32:938–942
Bogdanov M, Matson WR, Wang L, Matson T, Saunders-Pullman R, Bressman SS, Beal MF (2008) Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain 131:389–396. doi:10.1093/brain/awm304
Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC (1994) Immunocytochemical analysis of tumour necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett 172:151–154. doi:10.1016/0304-3940(94)90684-X
Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. doi:10.1016/S0197-4580(02)00065-9
Braak H, de Vos RA, Bohl J, Del Tredici K (2006) Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396:67–72. doi:10.1016/j.neulet.2005.11.012
Charlett A, Dobbs RJ, Weller C, Dobbs SM (1997) Stasis in the gut: the source of xenobiotic in idiopathic parkinsonism. Eur J Clin Pharmacol 52(suppl):168
Charlett A, Dobbs RJ, Purkiss AG, Wright DJ, Peterson DW, Weller C, Dobbs SM (1998) Cortisol is higher in parkinsonism and associated with gait deficit. Acta Neurol Scand 97:77–85. doi:10.1111/j.1600-0404.1998.tb00614.x
Charlett A, Dobbs RJ, Dobbs SM Weller C, Weller C, Ibrahim MA, Dew T, Sherwood R, Oxlade NL, Plant JM, Bowthorpe J, Lawson AJ, Curry A, Peterson DW, Bjarnason IT (2009) Blood profile holds clues to role of infection in a premonitory state for idiopathic parkinsonism and of gastrointestinal infection in established disease. Gut Pathogens 1:20. doi:11186/1757-4749-1-20
Chen S, Le WD, Xie WJ et al (1998) Experimental destruction of substantia nigra initiated by Parkinson’s disease immunoglobulins. Arch Neurol 55:1075–1080. doi:10.1001/archneur.55.8.1075
Chen H, O’Reilly EJ, Schwarzschild MA, Ascherio A (2008) Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am J Epidemiol 167:90–95
Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM (1998) Folate, vitamin B12 and serum total homocysteine levels in confirmed Alzheimer’s disease. Arch Neurol 55:1449–1455. doi:10.1001/archneur.55.11.1449
Collins SM, Surette M, Bercik P (2012) The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 10:735–742. doi:10.1038/nrmicro2876
Cotillard A, Kennedy SP, Ling CK, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, Gougis S, Rizkalla S, Batto J-M, Renault P, ANR MicroObes consortium, Doré J, Zucker J-D, Clement K, Ehrlich SD (2013) Dietary intervention impact on gut microbial gene richness. Nature 500:585–590. doi:10.1038/nature12480
Dahlström A, Wigander A, Lundmark K, Gottfries C-G, Carvey PM, McRae A (1990) Investigations on auto-antibodies in Alzheimer’s and Parkinson’s diseases using defined neuronal cultures. J Neural Transm 29(suppl):195–206
Defazio G, Dal Toso R, Benvegnù D, Minozzi MC, Cananzi AR, Leon A (1994) Parkinson serum carries complement-dependent toxicity for rat mesencephalic dopaminergic neurons in culture. Brain Res 633:206–212. doi:10.1016/0006-8993(94)91541-5
Dobbs RJ, Charlett A, Purkiss AG, Dobbs SM, Weller C, Peterson DW (1999) Association of circulating TNF-α and IL-6 with ageing and parkinsonism. Acta Neurol Scand 100:34–41. doi:10.1111/j.1600-0404.1999.tb00721.x
Dobbs RJ, Charlett A, Dobbs SM, Weller C, Peterson DW (2000) Parkinsonism: differential age-trend in Helicobacter pylori antibody. Aliment Pharmacol Ther 14:1199–1205. doi:10.1046/j.1365-2036.2000.00815.x
Dobbs RJ, Dobbs SM, Weller C, Bjarnason IT, Oxlade NL, Charlett A, Al-Janabi MA, Kerwin RW, Mahler RF, Price AB (2005) Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism part 1: eradication of Helicobacter in the cachexia of idiopathic parkinsonism. Helicobacter 10:267–275. doi:10.1111/j.1523-5378.2005.00331.x
Dobbs RJ, Dobbs SM, Weller C, Charlett A, Bjarnason IT, Curry A, Ellis DS, Ibrahim MA, McCrossan MV, O’Donohue J, Owen RJ, Oxlade NL, Price AB, Sanderson JD, Sudhanva M, Williams J (2008) Helicobacter hypothesis for idiopathic parkinsonism: before and beyond. Helicobacter 13:309–322. doi:10.1111/j.1523-5378.2008.00622.x
Dobbs SM, Dobbs RJ, Weller C, Charlett A, Bjarnason IT, Lawson AJ, Letley D, Harbin L, Price AB, Ibrahim MAA, Oxlade NL, Bowthorpe J, Leckstroem D, Smee C, Plant JM, Peterson DW (2010) Differential effect of Helicobacter pylori eradication on time trends in brady/hypokinesia and rigidity in idiopathic parkinsonism. Report on completion of a randomised, double-blind, placebo-controlled efficacy study. Helicobacter 15:279–294. doi:10.1111/j.1523-5378.2010.00768.x
Dobbs RJ, Charlett A, Dobbs SM, Weller C, Ibrahim MAA, Iguodala O, Smee C, Plant JM, Lawson AJ, Taylor D, Bjarnason I (2012) Leukocyte-subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth. A surveillance study. Gut Pathogens 4:12. doi:10.1186/1757-4749-4-12
Dobbs SM, Charlett A, Dobbs RJ, Weller C, Iguodala O, Smee C, Lawson AJ, Taylor D, Bjarnason I (2013) Antimicrobial surveillance in idiopathic parkinsonism: indication-specific improvement in hypokinesia following Helicobacter pylori eradication and non-specific effect of antimicrobials for other indications in worsening rigidity. Helicobacter 18:187–196. doi:10.1111/hel.12035
Doré J, Simrén M, Buttle L, Guarner F (2013) Hot topics in gut microbiota. United Eur Gastroenterol J 1:311–318. doi:10.1177/2050640613502477
Dourmashkin RR, Dunn G, Castano V, McCall SA (2012) Evidence for an enterovirus as the cause of encephalitis lethargica. BMC Infect Dis 12:136. doi:10.1186/1471-2334-12-136
Ehringer H, Hornykiewicz O (1960) Distribution of noradrenaline and dopamine (3-hydroxytyramine) in human brain and its relation to diseases of the extrapyramidal system. Wien Klin Wschr 38:1236–1239. doi:10.1016/S1353-8020(98)00012-1
Ellis D, Dobbs RJ, Dobbs S, Curry A, Bjarnason I, Williams J, McCrossan MV, Weller C, Charlett A (2007) Duodenal enterocyte mitochondrial involvement and abnormal bowel function in idiopathic parkinsonism. In: Hanin I, Windisch M, Poewe W, Fisher A (eds) ADPD 2007 new trends in Alzheimer and Parkinson related disorders. Medimond S.r.l, Bologna, pp 269–272
Grathwohl SA, Steiner JA, Britschgi M, Brundin P (2013) Mind the gut: secretion of α-synuclein by enteric neurons. J Neurochem 125:487–490. doi:10.1111/jnc.12191
Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott W, Nutt J, Factor SA, Payami H (2010) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42:781–785. doi:10.1038/ng.642
He Y, Le W-D, Appel SH (2002) Role of Fcγ receptors in nigral cell injury induced by Parkinson disease immunoglobulin injection into mouse substantia nigra. Exp Neurol 176:322–327. doi:10.1006/exnr.2002.7946
Hill AB (1965) The environment and disease: association or causation? Proc R Soc Med 58:295–330
Hunot S, Brugg B, Ricard D, Michel PP, Muriel M-P, Ruberg M, Faucheux BA, Agid Y, Hirsch EC (1997) Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson’s disease. Proc Natl Acad Sci U S A 94:7531–7536. doi:10.1073/pnas.94.14.7531
International Parkinson Disease Genetics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simón-Sánchez J, Schulte C, Lesage S, Sveinbjörnsdóttir S, Stefánsson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW (2011) Imputation of sequence variants for identification of genetic risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377:641–649. doi:10.1016/S0140-6736(10)62345-8
Karlsen NR, Reinvang I, Frøland SS (1992) Slowed reaction time in asymptomatic HIV-positive patients. Acta Neurol Scand 86:242–246. doi:10.1111/j.1600-0404.1992.tb05078.x
Kirollos C, O.’Neill CJA, Dobbs RJ, Charlett A, Bowes SG, Purkiss AG, Hunt WB, Dobbs SM (1993) Quantification of the cardinal signs of parkinsonism and of associated disability in spouses of sufferers. Age Ageing 22:20–26. doi:10.1093/ageing/22.1.20
Kirollos C, Charlett A, O’Neill CJA, Kosik R, Mozol K, Purkiss AG, Bowes SG, Nicholson PW, Hunt WB, Weller C, Dobbs SM, Dobbs RJ (1996) Objective measurement of activation of rigidity: diagnostic, pathogenetic and therapeutic implications in parkinsonism. Br J Clin Pharmacol 41:557–564. doi:10.1046/j.1365-2125.1996.38313.x
Kok B, Foxton MR, Clough C, Shawcross DL (2013) Rifaximin is an efficacious treatment for the parkinsonian phenotype of hepatic encephalopathy. Hepatology 58:1516–1517. doi:10.1002/hep.26364
Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB (2008) Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord 23:2303–2306. doi:10.1002/mds.22369
Koutsilieri E, Sopper S, Scheller C, ter Meulen V, Reiderer P (2002) Parkinsonism in HIV dementia. J Neural Transm 109:767–775. doi:10.1007/s007020200063
Kuhn TS (1970) The structure of scientific revolutions, 2nd edn. The University of Chicago Press, Chicago, Enlarged
Lazzerini PE, Capecchi PL, Selvi E, Lorenzini S, Bisogno S, Galeazzi M, Pasini FL (2007) Hyperhomocysteinemia, inflammation and autoimmunity. Autoimmun Rev 6:503–509. doi:10.1016/j.autrev.2007.03.008
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto J-M, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546. doi:10.1038/nature12506
Lewin PK, Edwards V (1991) Mitochondrial inclusions in neurons of Creutzfeldt-Jakob-like disease. Lancet 337:236–237. doi:10.1016/0140-6736(91)92199-C
Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106:20051–20056. doi:10.1073%2Fpnas.0908005106
Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J (2006) Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55:205–211. doi:10.1136/gut.2005.073817
Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315. doi:10.1016/SO140-6736(84)91816-6
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1998) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1289. doi:10.1212/WNL.38.8.1285
McRae A, Degueurce A, Gottfries C-G, Karlsson I, Svennerholm L, Dahlström A (1986) Antibodies in the CSF of a Parkinson patient recognizes neurons in rat mesencephalic regions. Acta Physiol Scand 126:313–315. doi:10.1111/j.1748-1716.1986.tb07821
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin-1-beta, interleukin-6, epidermal growth-factor-alpha and transforming growth-factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180:147–150. doi:10.1016/0304-3940(94)90508-8
Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T (1996) Interleukin (IL)-1-beta, IL-2, IL-4, Il-6 and transforming growth-factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett 211:13–16. doi:10.1016/0304-3940(96)12706-3
Nakhleh MK, Badarny S, Winer R, Jeries R, Finberg J, Haick H (2015) Distinguishing idiopathic Parkinson’s disease from other parkinsonian syndromes by breath test. Parkinsonism Relat Disord 21:150–153. doi:10.1016/j.parkreldis.2014.11.023
Neish AS (2009) Microbes in gastrointestinal health and disease. Gastroenterology 136:65–80. doi:10.1053/j.gastro.2008.10.080
Nielsen HH, Qiu J, Friis S, Wermuth L, Ritz B (2012) Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol 19:864–869. doi:10.1111/j.1468-1331.2011.03643.x
OCEBM Levels of Evidence Working Group. The Oxford 2011 Levels of Evidence. Oxford Centre for Evidence-Based Medicine. http://www.cebm.net/index.aspx/o=5653
O’Neill CJA, Richardson MD, Charlett A, McHugh L, Bowes SG, Purkiss AG, Weller C, Dobbs SM, Dobbs RJ (1994) Could seborrhoeic dermatitis be implicated in the pathogenesis of parkinsonism? Acta Neurol Scand 89:252–257. doi:10.1111/j.1600-0404.1994.tb01675.x
Orešič M, Hyötyläinen T, Herukka S-K, Sysi-Aho M, Mattila I, Seppänan-Laakso T, Julkunen V, Gopalacharyulu PV, Hallikainen M, Koikkalainen J, Kivipelto M, Helisalmi S, Lötjönen J, Soininen H (2011) Metabolome in progression to Alzheimer’s disease. Transl Psychiatry 1, e57. doi:10.1038/tp.2011.55
Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM (2005) A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 28:2665–2674. doi:10.1093/brain/awh625
Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T (2005) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 2005(57):168–175. doi:10.1002/ana.20338
Ouchi Y, Yagi S, Yokokura M, Sakamoto M (2009) Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat Disord 15(S3):S200–S204. doi:10.1016/S1353-8020(09)70814-4
Parkinson J (1817) An essay on the shaking palsy. Sherwood, Neely and Jones, London
Pfeiffer RF (2009) Neuroinflammation and Parkinson disease: the silent battleground. Neurology 73:1434–1435. doi:10.1212/WNL.0b013e3181c2f07d
Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL (2008) Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson’s disease? Neuroscience 153:733–750. doi:10.1016/j.neuroscience.2008.02.074
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. doi:10.1126/science.6801762
Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, Zhou J, Ni S, Liu L, Pons N, Batto JM, Kennedy SP, Leonard P, Yuan C, Ding W, Chen Y, Hu X, Zheng B, Qian G, Xu W, Ehrlich SD, Zheng S, Li L (2014) Alterations of the human gut microbiome in liver cirrhosis. Nature 513:59–64. doi:10.1038/nature13568
Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Nicola MD, Onofrj M (2009) Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun 23:55–63. doi:10.1016/j.bbi.2008.07.003
Refsum H, Smith AD (2003) Low vitamin B-12 status in confirmed Alzheimer’s disease as revealed by serum holotranscobalamin. J Neurol Neurosurg Psychiatry 74:959–961. doi:10.1136/jnnp.74.7.959
Scheperjans F, Aho V, Pereira PAB, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola-Rautio J, Pohja M, Kinnunen E, Murros K, Auvinen P (2014) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord. doi:10.1002/mds.26069
Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PWF, Wolf PA (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346:476–483. doi:10.1056/NEJMoa011613
Sikiric P, Rotkvic I, Mise S, Petek M, Rucman R, Seiwerth S, Zjacic-Rotkvic V, Duvnjak M, Jagic V, Suchanek E, Grabarevic Z, Tomislav A, Brkic T, Djermanovic Z, Dodig M, Marovic A, Hernandez DE (1991) Dopamine agonists prevent duodenal ulcer relapse. Dig Dis Sci 36:905–910
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573. doi:10.1126/science.1241165
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Mägi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segrè AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpeläinen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, Campbell H, Cappuccio FP, Cavalcanti-Proença C, Chen YD, Chen CM, Chines PS, Clarke R, Coin L, Connell J, Day IN, den Heijer M, Duan J, Ebrahim S, Elliott P, Elosua R, Eiriksdottir G, Erdos MR, Eriksson JG, Facheris MF, Felix SB, Fischer-Posovszky P, Folsom AR, Friedrich N, Freimer NB, Fu M, Gaget S, Gejman PV, Geus EJ, Gieger C, Gjesing AP, Goel A, Goyette P, Grallert H, Grässler J, Greenawalt DM, Groves CJ, Gudnason V, Guiducci C, Hartikainen AL, Hassanali N, Hall AS, Havulinna AS, Hayward C, Heath AC, Hengstenberg C, Hicks AA, Hinney A, Hofman A, Homuth G, Hui J, Igl W, Iribarren C, Isomaa B, Jacobs KB, Jarick I, Jewell E, John U, Jørgensen T, Jousilahti P, Jula A, Kaakinen M, Kajantie E, Kaplan LM, Kathiresan S, Kettunen J, Kinnunen L, Knowles JW, Kolcic I, König IR, Koskinen S, Kovacs P, Kuusisto J, Kraft P, Kvaløy K, Laitinen J, Lantieri O, Lanzani C, Launer LJ, Lecoeur C, Lehtimäki T, Lettre G, Liu J, Lokki ML, Lorentzon M, Luben RN, Ludwig B, MAGIC, Manunta P, Marek D, Marre M, Martin NG, McArdle WL, McCarthy A, McKnight B, Meitinger T, Melander O, Meyre D, Midthjell K, Montgomery GW, Morken MA, Morris AP, Mulic R, Ngwa JS, Nelis M, Neville MJ, Nyholt DR, O’Donnell CJ, O’Rahilly S, Ong KK, Oostra B, Paré G, Parker AN, Perola M, Pichler I, Pietiläinen KH, Platou CG, Polasek O, Pouta A, Rafelt S, Raitakari O, Rayner NW, Ridderstråle M, Rief W, Ruokonen A, Robertson NR, Rzehak P, Salomaa V, Sanders AR, Sandhu MS, Sanna S, Saramies J, Savolainen MJ, Scherag S, Schipf S, Schreiber S, Schunkert H, Silander K, Sinisalo J, Siscovick DS, Smit JH, Soranzo N, Sovio U, Stephens J, Surakka I, Swift AJ, Tammesoo ML, Tardif JC, Teder-Laving M, Teslovich TM, Thompson JR, Thomson B, Tönjes A, Tuomi T, van Meurs JB, van Ommen GJ, Vatin V, Viikari J, Visvikis-Siest S, Vitart V, Vogel CI, Voight BF, Waite LL, Wallaschofski H, Walters GB, Widen E, Wiegand S, Wild SH, Willemsen G, Witte DR, Witteman JC, Xu J, Zhang Q, Zgaga L, Ziegler A, Zitting P, Beilby JP, Farooqi IS, Hebebrand J, Huikuri HV, James AL, Kähönen M, Levinson DF, Macciardi F, Nieminen MS, Ohlsson C, Palmer LJ, Ridker PM, Stumvoll M, Beckmann JS, Boeing H, Boerwinkle E, Boomsma DI, Caulfield MJ, Chanock SJ, Collins FS, Cupples LA, Smith GD, Erdmann J, Froguel P, Grönberg H, Gyllensten U, Hall P, Hansen T, Harris TB, Hattersley AT, Hayes RB, Heinrich J, Hu FB, Hveem K, Illig T, Jarvelin MR, Kaprio J, Karpe F, Khaw KT, Kiemeney LA, Krude H, Laakso M, Lawlor DA, Metspalu A, Munroe PB, Ouwehand WH, Pedersen O, Penninx BW, Peters A, Pramstaller PP, Quertermous T, Reinehr T, Rissanen A, Rudan I, Samani NJ, Schwarz PE, Shuldiner AR, Spector TD, Tuomilehto J, Uda M, Uitterlinden A, Valle TT, Wabitsch M, Waeber G, Wareham NJ, Watkins H, Procardis C, Wilson JF, Wright AF, Zillikens MC, Chatterjee N, McCarroll SA, Purcell S, Schadt EE, Visscher PM, Assimes TL, Borecki IB, Deloukas P, Fox CS, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, Mohlke KL, O’Connell JR, Peltonen L, Schlessinger D, Strachan DP, van Duijn CM, Wichmann HE, Frayling TM, Thorsteinsdottir U, Abecasis GR, Barroso I, Boehnke M, Stefansson K, North KE, McCarthy MI, Hirschhorn JN, Ingelsson E, Loos RJ (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42:937–948. doi:10.1038/ng.686
Strang RR (1965) The association of gastro-duodenal ulceration with Parkinson’s disease. Med J Aust 52:842–843
Vonka V (2000) Causality in medicine: the case of tumours and viruses. Philos Trans R Soc Lond B 335:1831–1841. doi:10.1098/rstb.2000.0738
Watkins AD (1995) Perceptions, emotions and immunity: an integrated homeostatic network. Q J Med 88:283–294. doi:10.1016/S1443-8461(02)80004-1
Weller C, Nicholson PW, Dobbs SM, Bowes SG, Purkiss A, Dobbs RJ (1992) Reduced axial rotation in the spouses of sufferers from idiopathic Parkinsonism. Age Ageing 21:189–194. doi:10.1093/ageing/21.3.189
Weller C, Oxlade NL, Dobbs SM, Dobbs RJ, Peterson DW, Bjarnason IT (2005) Role of chronic infection and inflammation in the gastrointestinal tract in the aetiology and pathogenesis of idiopathic parkinsonism. Part 3: predicted probability and gradients of severity of idiopathic parkinsonism based on H. pylori antibody profile. Helicobacter 10:288–297. doi:10.1111/j.1523-5378.2005.00329
Acknowledgments
Our grateful thanks go to the Psychiatry Research Trust, London; the Cecil Pilkington Charitable Trust; and the Cyril Corden Trust. Thanks are also go to Brian Newman and Louise Barton, and Richard and Diana Gloyn for their generous donations. Barclays Corporate Social Responsibility Ambassador Nicholas Smith co-ordinated a fundraising programme with the help of patients and carers.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dobbs, S.M., Dobbs, R.J., Weller, C. et al. Peripheral aetiopathogenic drivers and mediators of Parkinson’s disease and co-morbidities: role of gastrointestinal microbiota. J. Neurovirol. 22, 22–32 (2016). https://doi.org/10.1007/s13365-015-0357-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-015-0357-8