
Abstract
We present here extensive mass spectrometric studies on the formation of a Tris conjugate with a therapeutic monoclonal antibody. The results not only demonstrate the reactive nature of the Tris molecule but also the sequence and reaction conditions that trigger this reactivity. The results corroborate the fact that proteins are, in general, prone to conjugation and/or adduct formation reactions and any modification due to this essentially leads to formation of impurities in a protein sample. Further, the results demonstrate that the conjugation reaction happens via a succinimide intermediate and has sequence specificity. Additionally, the data presented in this study also shows that the Tris formation is produced in-solution and is not an in-source phenomenon. We believe that the facts given here will open further avenues on exploration of Tris as a conjugating agent as well as ensure that the use of Tris or any ionic buffer in the process of producing a biopharmaceutical drug is monitored closely for the presence of such conjugate formation.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biologics or biopharmaceutical products have seen an exponential growth, in terms of both scientific interest and market value, which is essentially driven by developments in the field of biosimilars and novel biopharmaceuticals [1–6]. Owing to the inherent complexity of biomacromolecules, the physicochemical characterization of biologics poses an analytical challenge. Nonetheless, comprehensive structural and functional characterization is a prerequisite to determine the quality, as it directly affects the product efficacy, safety, and immunogenicity. Amongst the physicochemical attributes, aggregation, charge heterogeneity, sequence variants, post-translational modifications, and process-induced chemical modifications are critical to monitoring, and are done utilizing state-of-art analytical techniques [3, 7–10].
The biopharma industry depends heavily on processes that can be scaled up. This requires extensive research involving various buffers and solvents, both for sample preparation and purification. Peptides and proteins that form core-biologics have groups like secondary amines and hydroxyls that are prone to chemical conjugations and, hence, it becomes imperative to study the interaction of process-related buffering and solvent components with them.
One of the commonly used buffers in the process is Tris buffer, which has the chemical name of 2-amino-2-hydroxymethyl-propane-1,3-diol and is also known as Tris (hydroxy-methyl) amino-methane. Tris has a buffering range of about pH 7 to pH 9. The Tris in aqueous solution can form highly reactive electrophilic aziridinium cation by dissociation. The cation formed can potentially attach to the reactive nucleophilic residues present in the protein sequence via covalent modification. These reactions are related to the classical hard-soft acid-base (HSAB) theory [11]. The nucleophilic residues can be the N-terminus, cysteine’s thiol group, lysine’s ε-amino group, histidine’s secondary nitrogen, tyrosine’s phenolic hydroxyl group, and also the carboxylic acid groups [12, 13] of aspartic and glutamic acid residues. The chemical mediated degradation of the biologicals during the upstream and downstream manufacturing steps as well as storage conditions can lead to several site-specific modifications. These include, but are not limited to, methionine/cysteine/tryptophan oxidation, C-terminal lysine clipping, N-terminal glutamine cyclization (pyro-glutamate formation), asparagine/glutamine deamidation, succinimide formation, and aspartate isomerization. All these modifications can have a substantial effect on their structure and biological activity [3, 9, 14–16].
Immunoglobulin (IgG) antibodies are one of the most pursued classes of biotherapeutics in terms of research and usage. The tetrameric structure of these comprises of more than 1000 amino acids with an average molecular weight ~150 kDa formed by two sets of heavy and light polypeptide chains connected via four inter-chain disulfide bridges and four intra-chain disulfide bridges within each heavy chain and two intra-chain disulfide bridges within each light chain. The surface exposed amino acid residues, which are solvent accessible, are highly prone towards chemical modifications involving electrophilic and nucleophilic reactions. This is commonly observed for amino acids like lysine, arginine, asparagine, glutamine and cysteine with reactive functional nucleophilic group [3, 17]. Furthermore, the asparagine and glutamine residues are highly prone to pH and temperature mediated deamidation [3, 18, 19].
In this background, we present here the investigation results on chemical modification of a tryptic digested IgG peptide that was driven by a widely utilized buffering agent, Tris. It is evident from the studies that the nucleophilic functional group present in Tris reacts with the amino acid residues via a deamidation mediated mechanism that leads to conjugate formation. It was also observed that the reaction exhibits pH dependency. This Tris-conjugated IgG peptide modification led to an extra peak in the total ion chromatogram (TIC) profile of the tryptic IgG peptide, which exhibited an additional mass of 104 Da compared with the native tryptic IgG peptide. The data presented in this study provide meaningful insight towards the possible modification in the sequence due to sample preparation that are sometime misinterpreted as translational errors. Furthermore, it differentiates the in-solution/in-process induced stabilized intermediates/species from the tangible impurities produced in the manufacturing process.
Experimental
Materials
The recombinant DNA-derived humanized monoclonal IgG1 kappa type antibody analyzed in this manuscript was produced at Biocon Ltd., Bangalore, India. This IgG was expressed using Chinese hamster ovary (CHO) cell line followed by standard manufacturing steps that included utilization of protein-A and ion-exchange (IEX)-based chromatography to obtain the highly purified IgG protein. The proteolytic enzymes utilized included sequencing grade modified trypsin (part no. V511A; Promega Corporation, Madison, WI, USA) and PNGase F (Cat.No.11365193001; Roche Diagnostics GmbH, Mannheim, Germany). All the chemicals used were reagent grade or above. The chemicals, including Tris (hydroxymethyl) aminomethane, ammonium bicarbonate, guanidine hydrochloride, trifluroacetic acid (TFA), hydrochloric acid (HCl), formic acid (FA), dithiothreitol (DTT), and iodoacetamide (IAA) were procured from Sigma-Aldrich (St. Louis, MO, USA). Acetonitrile - HPLC grade (ACN) was purchased from J.T. Baker (Phillipsburg, NJ, USA).
Endoprotease Tryptic Digestion
The IgG samples were first denatured by treatment with 6 M guanidine hydrochloride (1:1 v/v) at 37 °C for 30 min followed by reduction and alkylation with 1 M DTT (protein:DTT::100:1 v/v) and IAA (protein:IAA::50:1 v/v) at 37 °C for 60 min each. The reduced and alkylated samples were desalted and concentrated using Amicon centrifuge filters (Amicon Ultra centrifugal filters; Millipore Corporation, Bedford, MA, USA). The pH of the concentrated samples was adjusted to 8.0 ± 0.2 using Tris-HCl buffer, and trypsin digestion was performed by addition of trypsin at a ratio of 1:25 (protein to trypsin w/w) at 37 °C for 16 h. The digestion was arrested using 1% FA.
Deglycosylation of IgG Samples
The deglycosylation was performed on all the IgG samples using peptide-N-glycosidase F (PNGase F) in the enzyme to protein ratio of 4:100 w/w at 37 °C for 5 h in Tris-HCl buffer of pH 8.0 ± 0.2.
Incubation Experiments with Tris buffer
The deglycosylated IgG samples were individually buffer exchanged and concentrated with 1 M ammonium bicarbonate and 0.1 M to 1 M Tris-HCl buffers using Amicon centrifuge filters. Both intact mass and PMF experiments were performed on the IgG samples thus generated.
Mass Spectrometry Data Acquisition and Analysis
Intact mass analysis was done by analyzing the deglycosylated IgG using ACE C4 reverse phase chromatography column (RP) (Advanced Chromatography Technologies Ltd., Scotland, UK) on an Acquity UHPLC liquid chromatography system coupled to a Synapt G2-HDMS electrospray-time-of-flight (ESI-TOF) mass spectrometer (Waters MS-Technologies, Manchester, UK). The RP-HPLC method used a gradient program involving two mobile phases, (1) solvent A: acidified MilliQ water (Cat.No. ZMQS5V001; Miili-Q, Millipore, Molsheim, France) with 0.1% FA and (2) solvent B: 100% ACN with 0.1% FA. The gradient started from 10% B and moved towards 90% B, over a run time of 10 min, with a flow rate of 0.3 mL/min at column temperature of 55 °C. The ESI-TOF was set to run in sensitive positive ion mode with capillary voltage of 3800 V, sample cone voltage of 80 V, mass to charge (m/z) range of 700–4000 at 1 s per scan, and with mass resolution of 15,000. Sodium iodide solution in propanol water mixture, from the Waters Q-TOF Product Sample Kit (part no. 700003276-2; Waters MS-Technologies, Manchester, UK) was used for m/z calibration for mass range 500–7000 m/z in ES+ mode and the same was applied for all the experiments. For the deconvolution, four charge states were considered from the charge states envelope distribution, with consideration of an error window of ±0.5 Da. The deconvoluted molecular mass was obtained using MaxEnt 1 (Maximum Entropy) algorithm from the software Waters MassLynx 4.1 (Waters MS-Technologies).
For the protein mass fingerprints, the IgG fragments obtained after tryptic digestion were separated on RP ACE 5 C18-300 chromatography column (Advanced Chromatography Technologies Ltd., Scotland, UK) on a Shimadzu UFLC system (Shimadzu Corporation, Kyoto, Japan), using MilliQ water with 0.1% TFA (solvent A) and 90% ACN with 0.09%TFA (solvent B) as mobile phases. A linear gradient set from 2% to 98% of solvent B over a runtime of 160 min with a flow rate of 0.8 mL/min and column temperature of 40 °C. The separated IgG fragments were analyzed for MS and MS/MS using a tandem ESI LTQ-Orbitrap MS Platform (Thermo Fisher Scientific, Bremen, Germany). The data was acquired in positive ionization mode with a resolution of 15,000. CID MSn data was acquired with normalized collision energy of 35. The standard calibration mix containing n-butylamine, caffeine, MRFA, and Ultramark 1621 (Pierce LTQ Velos ESI Positive Ion Calibration Solution, Cat.No. #88323; ThermoFisher Scientific, Rockford, USA) was used to calibrate m/z range 50–2000 in ES+ mode, and the same was applied for all the experiments. The data analysis was done using the Thermo Xcalibur software ver. 3.0 from Thermo Fisher Scientific Inc. (Thermo Fisher Scientific, Bremen, Germany).
Results and Discussion
Over the last two decades, mass spectrometry has emerged as the preferred tool for deeper insights into peptides and proteins at a molecular level, essentially due to lower sample requirement and ease of data analysis and inference. In this regard, the top-down and bottom-up approaches are the vital tools in deciphering the molecular profile of hitherto unknown proteins and peptides. However, for molecules that are used as therapeutic biopharmaceuticals, since the protein or peptide of interest is known, the peptide mass fingerprinting (PMF) methodology becomes a powerful tool because of the confidence in the generation of qualitatively holistic and quantitatively interrogative data for the overall structural characterization.
PMFs are thus extensively used, not only for the characterization of biopharma products and their related substances but also for the assessment of quality of such products from a primary structure perspective. Further, it helps to identify sequence-related variants, decode post-translation modifications, and acts as an evaluating tool for in-process testing.
Monoclonal Antibodies; IgG, Form the Core of Anti-Cancer Treatments Amongst the Biopharmaceutics
Since these are complex entities, thorough investigation of the PMF dataset becomes all the more imperative. For an IgG, a tryptic protein digest is the most commonly used sample for generating and analyzing a PMF, especially to establish the primary sequence integrity across multiple batches produced at industrial scale. During such an analysis for one of the IgGs, the observation of an unknown peptide in comparison with the theoretical peptides triggered this investigation. This unknown peptide peak observed in the tryptic PMF is marked by an asterisk (*) in Figure 1a, which shows the zoomed TIC profile. The complete TIC profile is shown in the Supplementary Figure S1. The said peptide eluted at a retention time (RT) of 122.9 min and corresponded to a mass of 1912.08 [M + H]1+ Da (z = 2; 956.54[M + 2H]2+).

Mass spectra of the tryptic digest of the IgG sample: (a) zoomed TIC profile from RT 120 to 128 min. The TIC peak corresponding to the peptide, PFc, is marked by a hash (#) whereas the peak for PFc+104 is marked by an asterisk (*). The isotope distributions for PFc and PFc+104, respectively, at 904.51 Da [M + 2H]2+ and 956.54 Da [M + 2H]2+, are shown as insets and are marked by arrows from the corresponding peptide peaks. (b) CID MS2 peptide fragmentation spectra of the unknown peptide corresponding to the mass of 956.54 Da (doubly charged, [M + 2H] 2+ ion). The high intensity daughter ions are annotated in the profile showing the peptide sequence of DWL
De-Novo Sequencing Confirms 104 Da Mass Addition to a Tryptic Digested IgG Peptide that Appertains to the Fc Domain
The IgG samples are analyzed by tryptic PMF followed by MS/MS based sequencing analysis. The tryptic PMF profile followed by MS/MS based sequencing analysis of the IgG sample gave the complete coverage with respect to the primary sequence of the IgG as shown by the mapping of the CID-MS2-based sequences with the theoretical tryptic IgG peptide sequences. The unknown peptide eluting at 122.9 min RT, however, did not match with any tryptic digested IgG peptide sequence.
The strategy of de-novo sequencing methodology [20, 21] of this unknown peptide using the CID-MS2 peptide fragmentation spectra provided the partial sequence of DWL as part of the peptide under investigation. The said sequencing was done manually starting with 1490.68 m/z, which was the most abundant ion in the higher m/z regime and building up the sequence towards the low m/z like 1377.76 (L), 1191.64 (W), and 1076.60 (D). The same is depicted in Figure 1b.
The partial sequence DWL was used to perform sequence match by alignment with the tryptic peptides of the IgG sequence and was found to match a single Fc peptide VVSVLTVLHQDWLNGK, named PFc+104 henceforward. Further, the RT of the native Fc peptide, PFc, with the mass of 904.51 Da [M + 2H]2+ was found to be eluting at RT 123.6 min, which is close to the elution RT (122.9 min) of PFc+104 peptide. The peptides are, respectively, marked by a hash (#) and an asterisk (*) in Figure 1a. This observation not only corroborates that the PFc+104 and PFc are related but also shows that the PFc+104 formation is not formed in-source as in that case we should have observed PFc+104 at the same RT as that of PFc.
The PFc+104 peptide sequence was confirmed by b and y ions based sequencing of this peptide’s CID-MS2 fragmentation spectra. The analysis also revealed that the site-specific modification occurs at the asparagine residue in this peptide. This is evident from the fact that the b4 to b13 ions from all the MS2 mass spectra were identical to the unmodified peptide, PFc, whereas the y3* to y15* and b14* showed an addition of 104 Da. The comparative b and y ion annotations are shown in Figure 2a and b that are the MS2 spectral expansions for PFc and PFc+104, respectively, where the relevant b and y ions are marked.

MS2 mass spectra for the peptide (a) PFc and (b) PFc+104. The b ion series coverage of b7 to b15 and y ion series coverage of y3 to y12 are annotated on the peaks in the individual spectra. The modified b ion (b14*) and y ions (y3* to y15*) are marked by an asterisk (*) in the MS2 spectra of PFc+104 peptide in (b). The representative theoretical b and y ions corresponding to the MS2 spectrum of the peptides PFc and PFc+104 are given on top of each panel
Tris Conjugate Formation Leads to the 104 Da Mass Addition
To decode the identity of the 104 Da mass addition in the PFc+104 peptide, the masses of all the solvent components, such as guanidine hydrochloride, dithiothreitol, iodoacetamide, and Tris, that are used to generate the tryptic digest were studied. This revealed that Tris, which has a mass of 121 Da, with a loss of 17 Da, came closest to the addition of 104 Da. The high resolution mass spectrum of the PFc+104 peptide revealed that the mass error between the theoretical and observed mass of the said peptide, if considered as a Tris conjugate, was less than 15 ppm, thus indicating that the PFc+104 peptide may indeed be formed because of the interaction with the Tris molecule. To investigate this observation further, the IgG sample was incubated with various concentrations of Tris under the tryptic digestion conditions (see Method section). Analysis of these samples, wherein the area of the extracted ion chromatogram (EIC) of the PFc+104 peptide was monitored, showed that the EIC area increased with increasing concentration of the Tris buffer as well as relative to the EIC percent area of the PFc peptide (Supplementary Table S1). The same is depicted in Figure 3a, which shows the EIC for the peptide PFc+104 in samples that were subjected to incubation with increasing concentration of Tris. A plot of the same shown as empty circles in Figure 3b shows a linear correlation, with R2 = 0.99, between the generation of the PFc+104 peptide and the increasing concentration of Tris. The trend for the PFc peptide was opposite as expected (Supplementary Table S1; Figure 3b).

EIC profile, linearity plots and PMF TIC spectra: (a) EIC profiles for peptide PFc+104 from the PMF spectra generated using Tris buffer (pH 8.0) at strengths of 0 mM (i; negative control; ammonium bicarbonate buffer was used for adjusting the pH to 8.0), 100 mM (ii), 250 mM (iii), 500 mM (iv), 750 mM (v), and 1000 mM (vi). (b) Plots of the normalized EIC area of peptides PFc (filled triangle), PFc-D (filled circle), PFc+104 (empty circle), and PFc-succ (empty square) against different strengths of Tris. (c) TIC mirror profile overlay of the tryptic PMF generated using ammonium bicarbonate and Tris-HCl buffer as the digestion buffers annotated as I and M, respectively. The peptide PFc+104 is marked by an asterisk (*) in the profile M. The peptides PFc, PFc-D, and PFc-succ are marked in the profile I to indicate their respective positions in the PMF
Formation of the Tris conjugate was further confirmed by replacing Tris with ammonium bicarbonate buffer as a negative control, where it was observed that the peak that corresponded to the PFc+104 peptide was not present (Figure 3c). Figure 3c, which shows a mirror overlay of the variants of PFc peptide from the tryptic PMF of the IgG using Tris-HCl (panel marked M in Figure 3c) with image from ammonium bicarbonate buffer (panel marked I in Figure 3c), clearly demonstrates that Tris leads to the 104 Da mass increase as the PFc+104 [marked by an asterisk (*)] is observed only in the tryptic PMF data generated in Tris buffer. Thus, the 104 Da mass addition was attributed to Tris conjugate formation with the native PFc peptide.
To further strengthen our interpretation, experimental conditions were designed at the intact mass level to investigate the effect of Tris on the IgG sample. This also helped to demonstrate the conjugation if Tris is used in the downstream purification steps of the IgG. The experimental design for the intact mass analysis essentially included deglycosylating the IgG followed by mass data acquisition. The deglycosylation was done to ensure that the presence of the glycan moiety does not lead to interference and, hence, artefactual results with respect to concluding the presence or absence of the 104 Da mass addition to the intact IgG. Thus, the intact mass analysis by LC-ESI-TOF MS (Supplementary Figure S2), showed that the average molecular mass of a deglycosylated IgG is 145164.00 Da. We also observed a mass of 145,326.00 Da that corresponds to IgG with 162 Da additional mass, which is due to the nonenzymatic glycation wherein a hexose sugar can attach with a basic amino acid [22]. We, however, did not observe any mass corresponding to IgG with 104 Da addition. The charge state distribution along with the deconvoluted mass using MaxEnt 1 algorithm is depicted in Supplementary Figure S2. This confirmed that IgG with 104 Da mass addition is absent in intact IgG samples. Furthermore, to determine the effect of Tris on intact IgG, enrichment of Tris conjugate was performed by incubating the deglycosylated IgG sample with various concentrations of Tris (see Method section). This resulted in 104 Da mass addition to the IgG and is depicted in the Supplementary Figure S3, demonstrating the Tris reactivity to the IgG molecule. This experimental result in conjunction with the ammonium bicarbonate buffer experiment, which essentially served as a negative control (Figure 3c), reveals that whilst the IgG sample inherently did not show the presence of the Tris conjugate, such samples are, nonetheless, prone to conjugating with the Tris molecule. Thus it becomes all the more imperative that any addition of Tris during sample purification or preparation should be monitored stringently, especially in the context of biotherapeutics.
Formation of the Tris Conjugate Occurs via Asparagine-Succinimide Intermediate
In order to decipher the mechanistic details of the Tris conjugate formation, the masses around the PFc+104 peptide were further scrutinized. It was found that the increase in the PFc+104 peptide peak’s EIC area correlated well with the decrease in the EIC area of the peak corresponding to the asparagine-succinimide intermediate (PFc-succ) peptide (Supplementary Table S1). This correlation, shown by empty circles and squares in Figure 3b, indicates that the chemical reaction of the Tris molecule with the asparagine-succinimide intermediate leads to the formation of the Tris conjugate.
Further, considering the fact that asparagine-succinimide is an intermediate for the deamidated species, we also evaluated the formation of the same with respect to the Tris conjugate. As seen in Figure 3b, the rate of formation for both the deamidation (filled circles) PFc-D and the Tris conjugate (empty circles) PFc+104 are similar, considering that the slopes are in the same order of magnitude. However, the deamidated PFc-D ensemble is present at about two log order higher magnitude compared with the Tris conjugate (PFc+104), as seen by the EIC area values shown in Supplementary Table S1.
Comparative data analysis and cross interpretation across all the deamidated variants of the native PFc and modified PFc+104 peptide was performed, and the y ions from y3* to y15* and also the b14* ions confirmed the modified PFc+104 peptide to have the 104 Da mass addition to the asparagine residue in the native PFc peptide (Figure 2a, b). Clearly, deamidation event resulting in modification of the asparagine to asparagine-succinimide intermediate with a loss of 17 Da supports the 104 Da addition mass, which relates to the Tris conjugate. The EIC profile with the MS spectra for variants of the PFc peptides are shown in Figure 4. The asparagine deamidation-derived aspartic acid and iso-aspartic acid, which show same mass characteristics but elute with slightly different retention time, have been marked in Figure 4b assuming that the iso-aspartic specie is present at a lower abundance than the aspartic acid specie. This correlates well with the previously published literature [23–26]. The corresponding MS2 mass spectra for the native PFc and modified PFc+104, PFc-D, PFc-succ peptides along with their b and y ions annotated for the confirmation are depicted, respectively, in the Figure 2a, b; Supplementary Figure S4a, b.

EIC profile with MS spectra for variants of the PFc peptide (a) PFc - with unmodified asparagine (Asn) eluting at RT 123.6 min, (b) PFc-D - with aspartic acid (Asp) eluting at RT 123.3 min and isoaspartic acid (isoAsp) eluting at RT 124.4 min, (c) PFc-succ - with asparagine-succinimide intermediate (Asu) eluting at RT 126.2 min, (d) PFc+104 - with 104 Da mass addition at asparagine residue eluting at RT 122.9 min. The mass spectra of the doubly charged ion for each peptide showing its individual isotopic distribution are shown as an insert on the right hand side corner in each panel. The arrow marks indicate the presence of each PFc variant
Specific Polypeptide Sequence Motif governs Tris Conjugate Formation
It is well known in literature that an asparagine residue that is succeeded by a glycine residue, forming the motif NG, is more prone to deamidation than NX, where X is any other amino acid than glycine [27, 28]. This happens because of the less sterically hindered local environment that the glycine molecule gives to its neighboring residues compared with other amino acids.
Considering the observation that the Tris conjugate formation is mediated by asparagine-succinimide formation, which essentially is the intermediate step for asparagine deamidation, and the fact described in the previous paragraph, one may hypothesize that the Tris conjugate formation will require the presence of sequence specific NG motif. In order to prove this hypothesis, all the asparagine-containing peptides in the Tryptic digest of the IgG sample were analyzed, and it was observed that indeed it is correct to state that the Tris conjugate formation requires the NG motif in the polypeptide sequence.
Figure 5a and b show, respectively, the EIC profiles and corresponding mass spectral expansions of two such peptides, LSCAASGFNIK (PNX) and GFYPSDIAVEWESNGQPENNYK (PNG). The expansions mark the presence of PNG+104, whereas no such peptide was seen with an addition of 104 Da to the mass of PNX (shown by the absence). Further, Figure 5c shows EIC and the mass spectral expansion for FNWYVDGVEVHNAK (PXG) peptide as a negative control. Absence of an EIC peak with 104 Da mass addition to PXG (Figure 5c) peptide not only corroborates the prerequisite of NG motif for Tris conjugate formation but also adds to the proof for asparagine-succinimide mediated conjugation. Further, peptides carrying the motif “NG” in the IgG sequence and showing the said modification have been captured in Supplementary Table S2 and Supplementary Figure S5, thus adding strength to the hypothesis that this modification must be governed by sequence specificity. Furthermore, we have shown in Supplementary Figure S6 that the MS2 spectra for the IgG peptide P3 (Supplementary Table S2) carrying the “NG” motif confirms the 104 Da addition or Tris conjugation at the asparagine residue. In addition to this, the PENNY peptide (PNG) exhibited deamidation but the Tris conjugation at NN motif was found to be absent.

EIC profile of peptide, (a) PNX, (b) PNG, and (c) PXG. In each figure, the bottom panel shows the EIC for the same peptide with an addition of 104 Da at the asparagine residue. The sequence information, charge state, and retention times for each peptide have been marked in individual panels. The mass spectra of the doubly charged ion for each peptide showing its individual isotopic distribution are shown as an insert on the right hand side corner in each panel. Addition of 104 Da observed for the peptide shown in the bottom panel of (b) was confirmed from MS2 data analysis. For peptides that did not show the addition of 104 Da modification, the panel is annotated by the word “Absent”
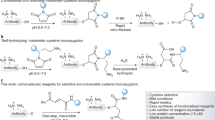
The NG motif sequence is prone to deamidation, especially in the solvent-exposed regions of a protein molecule. Presence of a glycine residue at the C-terminal part of the asparaginyl residue further aids the deamidation since absence of a side chain in glycine creates less steric hindrance, thus favoring higher flexibility to undergo intramolecular cyclization followed by formation of isoaspartyl residue [18, 29–33]. Moreover, in acidic conditions, the relative abundance of asparagine-succinimide intermediate is found to be higher as the acidic condition stabilizes the intermediate. In comparison, in alkaline conditions, they are highly reactive and are also prone towards nucleophilic attack due to which they are not stable beyond 48 h [34–37].
The nucleophilic functional group with free amine in the chemical molecule like hydrazines and hydroxylamines can potentially react with the cyclic succinimide intermediate formed as part of degradation pathway in the asparagine residue with all the possible factors leading to a covalent adduct [35, 38–46]. Tris with its free amine, a potential nucleophilic functional group, reacts with the cyclic asparagine-succinimide intermediate leading to the formation of in-solution covalent adduct that gives the mass of 104 Da addition at the asparagine site adjacent to the glycine residue (Scheme 1). The mechanism is essentially governed by SN2 nucleophilic substitution reaction that leads to formation of the Tris conjugate.

Mechanism showing the process of asparagine deamidation with loss of 17 Da via hydrolysis under mild acidic conditions to asparagine-succinimide intermediate and later the formation of aspartic acid and isoaspartic acid by nucleophilic water attack. In the presence of Tris in the solution, the amine nucleophilic group present in Tris molecule reacts with the asparagine-succinimide intermediate leading to the formation of the Tris conjugate
Conclusion
Proteins and peptides that form the core of biologics or biopharmaceutics have many reactive moieties within the molecule because of the presence of the lone pairs on the nitrogen in the amide backbone bond as well as in the amino groups present in the side chains. This in particular is enhanced in the asparagine residues having glycine as neighboring residues that makes the said residue vulnerable towards deamidation pathway via the asparagine-succinimide intermediate formation. This intermediate is highly unstable and can undergo chemical modification in the presence of a reactive substrate. In this background, this article, to our knowledge, is the first to extensively document the effect of Tris as a conjugating agent to a biologic and its effect on the degradation profile of the biologic. Further, from the perspective of sequence specificity and mechanism of the Tris conjugate formation via the asparagine-succinimide intermediate as well, to our knowledge, this is the first experimental evidence of the formation of the said conjugate. Tris being a common reagent used as buffer in process, this study will have enormous implications on studies pertaining to formation of potentially immunogenic degradation products in a biologic. It will thus be important that usage of Tris is monitored in terms of potential conjugate formation.
We envisage that the results discussed in this manuscript will open up avenues on studies to obtain deeper insights into the conjugate formations by ionic buffers and their roles in the potency and efficacy of a biologic drug.
References
Aggarwal, S.R.: What's fueling the biotech engine [mdash] 2012 to 2013. Nat. Biotechnol. 32, 32–39 (2014)
Ecker, D.M., Jones, S.D., Levine, H.L.: The therapeutic monoclonal antibody market. MAbs 7, 9–14 (2015)
Sandra, K., Vandenheede, I., Sandra, P.: Modern chromatographic and mass spectrometric techniques for protein biopharmaceutical characterization. J. Chromatogr. A 1335, 81–103 (2014)
Udpa, N., Million, R.P.: Monoclonal antibody biosimilars. Nat. Rev. Drug Discov. 15, 13–14 (2016)
Walsh, G.: Biopharmaceutical benchmarks 2010. Nat. Biotechnol. 28, 917–924 (2010)
Walsh, G.: Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 32, 992–1000 (2014)
Beck, A., Debaene, F., Diemer, H., Wagner‐Rousset, E., Colas, O., Dorsselaer, A.V., Cianférani, S.: Cutting‐edge mass spectrometry characterization of originator, biosimilar, and biobetter antibodies. J. Mass Spectrom. 50, 285–297 (2015)
Beck, A., Sanglier-Cianférani, S., Van Dorsselaer, A.: Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal. Chem. 84, 4637–4646 (2012)
Beck, A., Wagner-Rousset, E., Ayoub, D., Van Dorsselaer, A., Sanglier-Cianférani, S.: Characterization of therapeutic antibodies and related products. Anal. Chem. 85, 715–736 (2012)
Fekete, S., Guillarme, D., Sandra, P., Sandra, K.: Chromatographic, electrophoretic, and mass spectrometric methods for the analytical characterization of protein biopharmaceuticals. Anal. Chem. 88, 480–507 (2015)
Swain, C.G., Scott, C.B.: Quantitative correlation of relative rates. Comparison of hydroxide ion with other nucleophilic reagents toward alkyl halides, esters, epoxides, and acyl halides. 1. J. Am. Chem. Soc. 75, 141–147 (1953)
Noort, D., Hulst, A.G., Jansen, R.: Covalent binding of nitrogen mustards to the cysteine-34 residue in human serum albumin. Arch. Toxicol. 76, 83–88 (2002)
Rubino, F.M., Pitton, M., Di Fabio, D., Colombi, A.: Toward an “omic” physiopathology of reactive chemicals: thirty years of mass spectrometric study of the protein adducts with endogenous and xenobiotic compounds. Mass Spectrom. Rev. 28, 725–784 (2009)
Yan, Y., Wei, H., Fu, Y., Jusuf, S., Zeng, M., Ludwig, R., Krystek, S.R., Chen, G., Tao, L., Das, T.K.: Isomerization and oxidation in the complementarity-determining regions of a monoclonal antibody: a study of the modification–structure–function correlations by hydrogen–deuterium exchange mass spectrometry. Anal. Chem. 88, 2041–2050 (2016)
Perdivara, I., Deterding, L.J., Przybylski, M., Tomer, K.B.: Mass spectrometric identification of oxidative modifications of tryptophan residues in proteins: chemical artifact or post-translational modification? J. Am. Soc. Mass Spectrom. 21, 1114–1117 (2010)
Liu, H., Ponniah, G., Zhang, H.-M., Nowak, C., Neill, A., Gonzalez-Lopez, N., Patel, R., Cheng, G., Kita, A.Z., Andrien, B.: In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs 6, 1145–1154 (2014)
Hao, P., Adav, S.S., Gallart‐Palau, X., Sze, S.K.: Recent advances in mass spectrometric analysis of protein deamidation. Mass Spectrom. Rev. 9999, 1–16 (2016)
Catak, S., Monard, G., Aviyente, V., Ruiz-López, M.F.: Deamidation of asparagine residues: direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 113, 1111–1120 (2009)
Liu, S., Moulton, K.R., Auclair, J.R., Zhou, Z.S.: Mildly acidic conditions eliminate deamidation artifact during proteolysis: digestion with endoprotease Glu-C at pH 4.5. Amino Acids. 1–9 (2016)
Liu, M., Zhang, Z., Zang, T., Spahr, C., Cheetham, J., Ren, D., Zhou, Z.S.: Discovery of undefined protein cross-linking chemistry: a comprehensive methodology utilizing 18O-labeling and mass spectrometry. Anal. Chem. 85, 5900–5908 (2013)
Liu, M., Zhang, Z., Cheetham, J., Ren, D., Zhou, Z.S.: Discovery and characterization of a photo-oxidative histidine-histidine cross-link in IgG1 antibody utilizing 18O-labeling and mass spectrometry. Anal. Chem. 86, 4940–4948 (2014)
Quan, C., Alcala, E., Petkovska, I., Matthews, D., Canova-Davis, E., Taticek, R., Ma, S.: A study in glycation of a therapeutic recombinant humanized monoclonal antibody: where it is, how it got there, and how it affects charge-based behavior. Anal. Biochem. 373, 179–191 (2008)
Dai, S., Ni, W., Patananan, A.N., Clarke, S.G., Karger, B.L., Zhou, Z.S.: Integrated proteomic analysis of major isoaspartyl-containing proteins in the urine of wild type and protein L-isoaspartate O-methyltransferase-deficient mice. Anal. Chem. 85, 2423–2430 (2013)
Ni, W., Dai, S., Karger, B.L., Zhou, Z.S.: Analysis of isoaspartic acid by selective proteolysis with Asp-N and electron transfer dissociation mass spectrometry. Anal. Chem. 82, 7485–7491 (2010)
Winter, D., Pipkorn, R., Lehmann, W.D.: Separation of peptide isomers and conformers by ultra performance liquid chromatography. J. Sep. Sci. 32, 1111–1119 (2009)
Krokhin, O.V., Antonovici, M., Ens, W., Wilkins, J.A., Standing, K.G.: Deamidation of-Asn-Gly-sequences during sample preparation for proteomics: consequences for MALDI and HPLC-MALDI analysis. Anal. Chem. 78, 6645–6650 (2006)
Wright, H.T.: Sequence and structure determinants of the nonenzymatic deamidation of asparagine and glutamine residues in proteins. Prot. Eng. 4, 283–294 (1991)
Chelius, D., Rehder, D.S., Bondarenko, P.V.: Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal. Chem. 77, 6004–6011 (2005)
Bischoff, R., Lepage, P., Jaquinod, M., Cauet, G., Acker-Klein, M., Clesse, D., Laporte, M., Bayol, A., Van Dorsselaer, A., Roitsch, C.: Sequence-specific deamidation: isolation and biochemical characterization of succinimide intermediates of recombinant hirudin. Biochemistry 32, 725–734 (1993)
Daugherty, A.L., Mrsny, R.J.: Formulation and delivery issues for monoclonal antibody therapeutics. Adv. Drug Deliv. Rev. 58, 686–706 (2006)
Oliyai, C., Patel, J.P., Carr, L., Borchardt, R.T.: Chemical pathways of peptide degradation. VII. Solid state chemical instability of an aspartyl residue in a model hexapeptide. Pharm. Res. 11, 901–908 (1994)
Radkiewicz, J.L., Zipse, H., Clarke, S., Houk, K.: Neighboring side chain effects on asparaginyl and aspartyl degradation: an ab initio study of the relationship between peptide conformation and backbone NH acidity. J. Am. Chem. Soc. 123, 3499–3506 (2001)
Sinha, S., Zhang, L., Duan, S., Williams, T.D., Vlasak, J., Ionescu, R., Topp, E.M.: Effect of protein structure on deamidation rate in the Fc fragment of an IgG1 monoclonal antibody. Prot. Sci. 18, 1573–1584 (2009)
Chu, G.C., Chelius, D., Xiao, G., Khor, H.K., Coulibaly, S., Bondarenko, P.V.: Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm. Res. 24, 1145–1156 (2007)
Kumar, M., Chatterjee, A., Khedkar, A.P., Kusumanchi, M., Adhikary, L.: Mass spectrometric distinction of in-source and in-solution pyroglutamate and succinimide in proteins: a case study on rhG-CSF. J. Am. Soc. Mass Spectrom. 24, 202–212 (2013)
Violand, B., Schlittler, M., Kolodziej, E., Toren, P., Cabonce, M., Siegel, N., Duffin, K., Zobel, J., Smith, C., Tou, J.: Isolation and characterization of porcine somatotropin containing a succinimide residue in place of aspartate129. Prot. Sci. 1, 1634 (1992)
Yu, X.C., Joe, K., Zhang, Y., Adriano, A., Wang, Y., Gazzano-Santoro, H., Keck, R.G., Deperalta, G., Ling, V.: Accurate determination of succinimide degradation products using high fidelity trypsin digestion peptide map analysis. Anal. Chem. 83, 5912–5919 (2011)
Athmer, L., Kindrachuk, J., Georges, F., Napper, S.: The influence of protein structure on the products emerging from succinimide hydrolysis. J. Biol. Chem. 277, 30502–30507 (2002)
Darrington, R.T., Anderson, B.D.: Effects of insulin concentration and self-association on the partitioning of its A-21 cyclic anhydride intermediate to desamido insulin and covalent dimer. Pharm. Res. 12, 1077–1084 (1995)
DeHart, M.P., Anderson, B.D.: The role of the cyclic imide in alternate degradation pathways for asparagine‐containing peptides and proteins. J. Pharm. Sci. 96, 2667–2685 (2007)
DeHart, M.P., Anderson, B.D.: Kinetics and mechanisms of deamidation and covalent amide-linked adduct formation in amorphous lyophiles of a model asparagine-containing peptide. Pharm. Res. 29, 2722–2737 (2012)
Desfougères, Y., Jardin, J., Lechevalier, V., Pezennec, S., Nau, F.: Succinimidyl residue formation in hen egg-white lysozyme favors the formation of intermolecular covalent bonds without affecting its tertiary structure. Biomacromolecules 12, 156–166 (2010)
Geiger, T., Clarke, S.: Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 262, 785–794 (1987)
Stephenson, R.C., Clarke, S.: Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 264, 6164–6170 (1989)
Alfaro, J.F., Gillies, L.A., Sun, H.G., Dai, S., Zang, T., Klaene, J.J., Kim, B.J., Lowenson, J.D., Clarke, S.G., Karger, B.L.: Chemo-enzymatic detection of protein isoaspartate using protein isoaspartate methyltransferase and hydrazine trapping. Anal. Chem. 80, 3882–3889 (2008)
Klaene, J.J., Ni, W., Alfaro, J.F., Zhou, Z.S.: Detection and quantitation of succinimide in intact protein via hydrazine trapping and chemical derivatization. J. Pharm. Sci. 103, 3033–3042 (2014)
Acknowledgment
The authors thank Biocon Research Limited, Bangalore, Karnataka, India for providing the infrastructure support required to perform the research work described in this manuscript. The authors declare no competing financial interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Pradeep G. Kabadi and Praveen Kallamvalliillam Sankaran contributed equally to this work. All authors have given approval to the final version of the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1.31 mb)
Rights and permissions
About this article

Cite this article
Kabadi, P.G., Sankaran, P.K., Palanivelu, D.V. et al. Mass Spectrometry Based Mechanistic Insights into Formation of Tris Conjugates: Implications on Protein Biopharmaceutics. J. Am. Soc. Mass Spectrom. 27, 1677–1685 (2016). https://doi.org/10.1007/s13361-016-1447-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-016-1447-4