
Abstract
On-line coupling of electrochemistry (EC) to mass spectrometry (MS) is a powerful approach for identifying intermediates and products of EC reactions in situ. In addition, EC transformations have been used to increase ionization efficiency and derivatize analytes prior to MS, improving sensitivity and chemical specificity. Recently, there has been significant interest in developing paper-based electroanalytical devices as they offer convenience, low cost, versatility, and simplicity. This report describes the development of tubular and planar paper-based electrochemical cells (P-EC) coupled to sonic spray ionization (SSI) mass spectrometry (P-EC/SSI-MS). The EC cells are composed of paper sandwiched between two mesh stainless steel electrodes. Analytes and reagents can be added directly to the paper substrate along with electrolyte, or delivered via the SSI microdroplet spray. The EC cells are decoupled from the SSI source, allowing independent control of electrical and chemical parameters. We utilized P-EC/SSI-MS to characterize various EC reactions such as oxidations of cysteine, dopamine, polycyclic aromatic hydrocarbons, and diphenyl sulfide. Our results show that P-EC/SSI-MS has the ability to increase ionization efficiency, to perform online EC transformations, and to capture intermediates of EC reactions with a response time on the order of hundreds of milliseconds. The short response time allowed detection of a deprotonated diphenyl sulfide intermediate, which experimentally confirms a previously proposed mechanism for EC oxidation of diphenyl sulfide to pseudodimer sulfonium ion. This report introduces paper-based EC/MS via development of two device configurations (tubular and planar electrodes), as well as discusses the capabilities, performance, and limitations of the technique.

ᅟ
Similar content being viewed by others

Introduction
In recent years, there has been significant interest in developing paper-based devices for analytical and biosensor applications because they are cheap, malleable, disposable, easy-to-use, and simple [1]. Paper-based electroanalytical devices offer the potential for high sensitivity and low limits of detection [1]. The first paper-based electrochemical (EC) device was introduced by Henry and coworkers [2] in 2009 for the detection of glucose, lactate, and uric acid in biological samples. This device utilized a carbon ink/Prussian blue mixture for the working electrode (WE) and counter electrode (CE), and silver/silver chloride ink for the reference electrode (RE). Cui et al. [3] developed a paper EC sensor comprised of solid-contact ion sensing and reference electrodes to determine the concentration of potassium ions in samples absorbed into a paper substrate (one end of the paper substrate was immersed in the sample solution), demonstrating the ability to integrate solid-contact electrodes with the convenience of disposable paper substrates [3]. Online coupling of paper-based EC cells to mass spectrometry (MS) would open for study a new set of analytical techniques and applications. However, to our knowledge, this technological advance has not been demonstrated.
Electrochemistry coupled to MS (EC/MS) was first introduced by Bruckenstein and Gadde in 1971 [4]. Since then, EC/MS has found wide application in many areas of science and industry, such as the characterization of EC processes [5], drug metabolism [6], and biomolecules (e.g., proteins [7–12] and deoxyribonucleic acid [13, 14]), as well as online derivatization of functional groups [15, 16] and chemical imaging [5, 17, 18]. In EC/MS, the mass spectrometer serves as a sensitive detector for identifying products and intermediates generated in EC processes, providing mass-to-charge (m/z) ratios and structural information via tandem MS spectrometry (MS/MS) [5]. Conversely, EC transformations prior to MS can selectively transform analytes to improve ionization efficiency and selectivity [5, 19].
EC is typically coupled to electrospray ionization (ESI) because of its high sensitivity and its ability to transfer intact non-volatile species from solution to the gas phase (viz. ESI is a ‘soft’ ionization method) [20–23]. In ESI, EC processes occur at the solution-capillary interface (‘solution’ in this context refers to the solvent carrying analytes and reactants through the EC cell and ESI emitter) [24–31], which are influenced by the ESI current, capillary material, and composition of the solution. As a result of this inherent complexity in ESI, a distinct EC cell is desired to enable independent physical, electrical, and chemical control [32–35]. Two-electrode [34, 36–41] and three-electrode [33, 35, 42–44] EC/ESI-MS configurations have been developed that achieve electrical decoupling through (a) floating the EC cell on the potential induced by the ESI high voltage or (b) electrically isolating the electronic circuits. An interesting example is an integrated three-electrode EC/ESI-MS device developed by Cole and coworkers [33, 35, 42] in which solution interacts with the EC working electrode a few millimeters before the end of the ESI source, leading to shorter response times (t r < 3 s; time between EC conversion and detection at three times the signal-to-noise (S/N)) compared to typical configurations that have tubing connecting the EC cell and ESI emitter (t r is determined by tubing length, tubing internal diameter, and solution flow rate).
The field of ambient mass spectrometry (AMS) was started by Cooks and coworkers with the development of desorption electrospray ionization (DESI). AMS provides the ability to perform chemical analyses of systems in their natural state, which has led to impactful discoveries in numerous research areas such as forensics, environmental science, and cancer biochemistry [45–53]. Recently, Chen and coworkers [11, 54–58] demonstrated that DESI can characterize liquid samples (liquid sampling DESI; LS-DESI) such as the effluent from chromatographic columns [59–61] and EC flow-cells [10, 62–64]. When microdroplets from a LS-DESI source impact the outlet of tubular [54] or thin layer [10] EC flow cells, species in the EC effluent are desorbed into secondary microdroplets that travel towards a MS for detection. The electrical, chemical, and physical components of the EC cell and LS-DESI source [54] are separate, which enables independent control of electrical potentials, minimizes deleterious effects of electrolytes on ionization efficiency, prevents unwanted reactions that can potentially occur at ESI electrode surfaces, increases the scope of EC chemical systems available for study by EC/MS, and reduces carry-over effects in the ionization source. Recently, the Zare and Chen laboratories developed an EC/DESI-MS configuration that enables ambient capture of solution-phase intermediates with lifetimes of tens of milliseconds [65], which has significant advantage for elucidating EC mechanisms. In another significant advance, the Cooks and Ouyang laboratories introduced a paper-based spray ionization source (PSI) that provides an elegant, simple, portable, and cheap method for analyzing various samples (e.g. chemicals and cells) and reactions by MS [66–74]. Coupling P-EC to MS would leverage the advantages of paper (e.g. cheap, versatile, and disposable), as well as advance paper electroanalytical technologies through providing mechanistic information about processes occurring in paper.
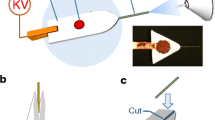
Herein, we harness the benefits of paper for EC/MS applications. We report the development and characterization of a two-electrode paper-based EC cell for coupling to MS (hereafter referred to as P-EC/MS; Figure 1). The P-EC/MS system is composed of (a) a paper substrate sandwiched between two solid-contact stainless steel (SS) mesh electrodes (planar P-EC) and (b) an ambient microdroplet probe directed at P-EC in line with the MS (Figures 1a and 1c). In a typical experiment, primary microdroplets containing reagents/analytes impact the paper substrate saturated with electrolyte after passing through SS mesh electrode E1. It is also possible to use pure solvents in the SSI microdroplet spray and saturate the paper with analytes/reagents and electrolyte. However, unless stated otherwise, the capabilities of P-EC/MS will be described with reagents/analytes added to the microdroplet spray. Application of a potential across the electrodes (ΔV) produces EC intermediates and products, which are extracted into secondary microdroplets that travel through E2 and undergo sonic spray ionization (SSI) [75] to generate gas-phase ions. The flexibility of the SS mesh electrodes and paper substrate make the EC cell malleable, providing multiple configurations for coupling P-EC cells to MS (Figure 1b shows the tubular configuration).

Schematic of a planar (a) and tubular (b) paper-based EC assembly (P-EC) coupled to MS. (c) Photograph showing the actual planar P-EC/MS experimental setup. Figures are not drawn to scale
The capabilities of P-EC/SSI-MS were demonstrated using well-known EC reactions such as oxidation of cysteine (Cys), diphenyl sulfide (PhSPh), and polycyclic aromatic hydrocarbons (PAH). P-EC/SSI-MS produce results similar to previously published EC/MS characterization of these reactions, demonstrating its ability to monitor EC processes, to increase ionization efficiency of nonpolar compounds, to perform online derivatization of specific analytes, and to distinguish isomers based on differing EC properties. One particularly exciting observation is that P-EC/SSI-MS has a short t r , which facilitates detection of transient intermediates generated at the electrode surfaces on the hundreds of milliseconds time scale. The shorter t r of P-EC/SSI-MS allowed detection of a deprotonated diphenyl sulfide species ([PhSC6H4]) during oxidation [42], providing evidence for a mechanism involving a cation or radical [Ph-S-C6H4] species to form pseudodimer sulfonium ion product [Ph2S+C6H4SPh]. In addition, the P-EC cell and SSI source are discrete systems, allowing independent control of electrical and chemical parameters. Finally, using a paper substrate between the electrodes provides a simple, cheap, disposable, and versatile approach for coupling EC to MS.
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. The stainless steel meshes (304 SS woven mesh with an unpolished (mill) finish; wire diameter = 200 μm; 30% open area) were purchased from Amazon.com (Seattle, WA, USA). The paper substrate was made from two sheets of Kimwipe (Kimberly-Clark, Neenah, WI, USA) and purchased from Thermo Fisher Scientific (San Jose, CA, USA). The direct current (DC) power supply (Keysight U8002A; 0–30 V; 0–5 A) was purchased from Testequity, LLC (Moorpark, CA, USA).
Design and Operation of the Paper-Based Electrochemical Cells
In planar P-EC, a paper substrate is placed between and in contact with two SS mesh electrodes (E1 and E2 in Figure 1a; E1: x × y × z (thickness) = 16 mm × 11 mm × 200 μm; E2: x × y × z = 20 mm × 15 mm × 200 μm). The dimensions of E1 < E2 and of the paper substrate > E2 to minimize the possibility of electrical shorting during operation of the EC cell. The x and y dimensions of the paper substrate are relatively similar to the electrode E1 (paper thickness = ~50 μm per sheet) and the E1-paper-E2 components of the P-EC assembly (500 μm total thickness) are held together with two regular office metal binder clips. Plastic insulators (colored green in Figure 1c) were inserted between the binder clips and the electrode surfaces. The P-EC assembly was positioned at 90o relative to the MS inlet and held in place using three-pronged clamps attached to ring stands (Figure 1c). The microdroplet SSI emitter was placed 17 mm from the MS inlet (0o relative) and 2 mm from E1. In the tubular P-EC (Figure 1b) configuration, two sheets of SS mesh (dimensions of E4 are less than E3) separated by a paper substrate of relatively similar dimensions were wrapped around a circular object to produce tubular electrodes (internal diameter (i.d.) × outer diameter (o.d.) × z = 3.2 mm × 3.8 mm × 15 mm; internal surface area (ISA) = [π × (ID/2)2 × z] = 120.6 mm2). The microdroplet emitter is placed 0o relative to the MS inlet, and the emitter-to-P-EC and P-EC-to-inlet distances are 2 mm (Figure 1b). For both the planar and tubular P-EC assemblies, alligator clips were used to connect each electrode to terminals of the DC power supply (Figure 1).
Construction of the microdroplet sprayer has been previously described by Cooks and coworkers [53]. Briefly, the sprayer consisted of a Swagelok (Swagelok, Fremont, CA, USA) 1/16′′ SS tee through which a FS capillary (250 μm i.d.; 360 μm o.d.); Polymicro Technologies, Lisle, IL, USA) delivered liquid from a Harvard Apparatus Standard Pump 22 (Holliston, MA, USA) syringe pump. FS passed through both 180o openings, and was held in place with a SS ferrule and SS nut at the end of the tee. The other end of FS passed through a SS capillary (length = 5 cm; i.d. = 0.020′′), which was held in place with a second SS ferrule and SS nut. FS capillary protruded from the end of the SS capillary by ~1 mm. Different solution compositions were sprayed with flow rate at 20 μL/min. Sheath gas (nitrogen; N2) entered the sprayer at the 90o opening of the tee (line pressure = ~200 PSI) and exited SS capillary, generating the microdroplet spray.
In a typical P-EC/SSI-MS experiment, the paper substrate is saturated with electrolyte using a Pasteur pipet and kept saturated throughout the analysis. Then, 4 V is supplied to the electrodes using a DC power supply [for the planar configuration, the positive electrode (E2) was closest to the MS; for tubular P-EC, the positive electrode (E3) forms the internal surface of the assembly]. Analyte is then delivered to the P-EC assembly via the microdroplet spray emitter. In the planar configuration, microdroplets impact the paper substrate after passing through E1. Electrochemical species generated in the paper substrate are extracted by subsequent impacting microdroplets, which carry the analytes to the mass spectrometer. In tubular P-EC/MS, the primary microdroplets deliver the analyte to the paper substrate after passing through E3 (Figure 1b). Generated electrochemical species are then desorbed and extracted into secondary microdroplets. An important difference between the planar and tubular designs is that the secondary microdroplets can either travel to the mass spectrometer or undergo subsequent collisions with the paper substrate leading to additional reactions. As a result, the length and ISA of the tubular electrodes influence parameters such as the reaction time, ionization efficiency, and sensitivity.
Mass Spectrometry
The microdroplets entering the mass spectrometer undergo desolvation using a capillary temperature of 275oC on an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) [76–81]. Unless specified otherwise, the Orbitrap MS was typically operated using the following parameters: single-stage m/z range = m/z 60–500, m/z resolution setting = 100,000 at m/z 400, mass accuracy = 2–5 ppm, microscans = 1, ion injection time = 500 ms, tube lens voltage = 110 V, spray voltage = 0 kV (SSI). For experiments that measure t r , ion detection was performed using the linear ion trap of the hybrid mass spectrometer (ITMS) with similar settings as described above, except that ion injection time = 100 ms.
Results and Discussion
Characterization of Planar P-EC/SSI-MS
The well-known EC formation of disulfides from thiols (e.g., Cys in Figure 2a) [10] was used to characterize features of planar P-EC/SSI-MS (Figure 1a). SSI was used to avoid voltage-induced reaction pathways such as the formation of reactive oxygen species, which can occur with water (H2O) or acetonitrile (CH3CN) as ESI solvents [82]. When a solution of Cys (MW = 121 gmol–1; 10 mM in 1:1 water:methanol (H2O:CH3OH)) was used as the microdroplet spray and KCl (10 mM) as the electrolyte added to the paper substrate, application of a potential difference (ΔV) = 4 V across electrodes E1 and E2 yielded mass spectra (Figure 2b and c) showing ion signals corresponding to [Cys + H]+ (m/z 122.026), [Cys + Na]+ (m/z 144.008), [Cys + 2Na – H]+ (m/z 165.990), [Cys-S-S-Cys + H]+ (m/z 241.030), [Cys-S-S-Cys + Na]+ (m/z 263.012), and [Cys-S-S-Cys + 2Na – H]+ (m/z 284.994). The ion signals corresponding to the disulfide product were not observed when ΔV = 0 V (Figure 2c), successfully demonstrating P-EC oxidation followed by online SSI-MS.

(a) EC oxidation of cysteine (Cys). (b) Mass spectrum showing P-EC/SSI-MS of Cys using ΔV = 4 V. (c) Mass spectrum showing P-EC/SSI-MS of Cys when no voltage is applied to the EC cell
The paper substrate is sufficiently porous such that ion signal intensities are not significantly affected when the EC cell is placed between the SSI and the MS inlet. When various solutions of Cys (10–4 M to 10–2 M in increments of 5 × 10–4 M) were directed at the mass spectrometer inlet capillary, the sum of the absolute intensities of Cys ionic species ([Cys + H]+ (m/z 122.026), [Cys + Na]+ (m/z 144.008), and [Cys + 2Na – H]+ (m/z 165.990) represented as Σ[M + n]+, where M is the neutral molecule and n represents H, Na, or (2Na – H)) at each concentration was relatively similar with and without the EC cell (Figure 3a; [KCl] = 0 M; ΔV = 0 V). In addition, application of 4 V to the EC cell did not significantly change Σ[Cys + n]+ (Figure S1a). Unless stated otherwise, error bars represent the standard deviation of three values and a fresh paper substrate was used for each measurement. In addition, it is important to note that the position of P-EC (angles and distances) has a significant effect on ion signal intensity, so the same parameters were used for all measurements. The ratio (Σ[Cys + n]+ with P-EC)/(Σ[Cys + n]+ without P-EC) across all Cys concentrations = 1.1 ± 0.3, which indicates that the paper EC cell does not significantly affect sampling efficiency (S E ) = ion transmission efficiency (T E ) + ionization efficiency (I E ) when placed between the emitter and the MS inlet.

Characterization of planar P-EC/MS. (a) Sum of the ion signal intensities corresponding to Cys (Σ[M + n]+, where n = H, Na, and (2Na – H); M = Cys) as a function of [Cys] in the SSI microdroplet spray. (b) Plot showing the influence of [KCl] on ion signals corresponding to Cys and Cys-S-S-Cys. (c) Plot showing the effect of EC potential on the signal intensities of the protonated, sodiated, and disodiated species of Cys (left axis). The plot also shows ΔV effect on the sum of all ion signals corresponding to Cys and the disulfide product (right axis)
The concentration of KCl affects ionization efficiency (I E ) from the paper substrate, whereas ΔV has no effect on I E . When the paper substrate was saturated with various concentrations of KCl (1, 10, and 100 mM) at ΔV = 0 V, the ratio (Σ[Cys + n]+ for [KCl] = 100 mM)/(Σ[Cys + n]+ for [KCl] = 1 mM) = 3.5 ± 0.5 for [Cys] = 10 mM (Figure 3b), indicating a slight increase in ion signal intensities. When the KCl(aq) concentration is greater than 10 mM, KCl(s) gradually accumulates on the MS inlet, eventually obstructing ion transfer into the mass spectrometer. For this reason, the remaining P-EC/MS experiments were performed at [KCl] = 10 mM. When ΔV was increased from 0 to 5 V in 1 V increments while holding the KCl and Cys concentrations constant, Σ[Cys + n]+ = (7.0 ± 0.6) × 104 with relative standard deviation (RSD) of only 9% (Figure S1b). All these results show that (1) potentials applied to the EC cell (0–7 V) have no observable effect on the average charge of the secondary microdroplets, and (2) higher electrolyte concentrations increase I E , which is in agreement with the proposed statistical charge distribution mechanism for SSI (average charge of the microdroplets is proportional to the square root of ion concentration) [83].
The optimum voltage for EC conversion of Cys was determined by varying ΔV and monitoring product/reactant intensity ratios, which increase from 2 to 4 V and then plateaus at 4 V (Figure 3c). These observations indicate that 2 V is the threshold for appearance of Cys-S-S-Cys ion signals in acquired mass spectra ([Cys] = [KCl] = 10 mM) and that 4 V produces maximum conversion under P-EC/SSI-MS conditions. Incidentally, [Cys] = 1 mM is the threshold concentration for observing oxidation using P-EC/SSI-MS when ΔV = 4 V and [KCl] = 10 mM (Figure S1a). This voltage range (2 - 4 V) agrees with previous flow-EC/LS-DESI-MS oxidation of analogous compound glutathione [10]. From the equation of the reaction (Figure 2a), 2 mol of Cys are converted for every mol of disulfide product, which agrees with [Cys-S-S-Cys + H]+/ [Cys + H]+ = 0.7 ± 0.1 for ΔV > 4 V ( plot in Figure 3c). Interestingly, Σ[Cys-S-S-Cys + n]+/Σ[Cys + n]+ = 1.2 ± 0.2 (
plot in Figure 3c). Interestingly, Σ[Cys-S-S-Cys + n]+/Σ[Cys + n]+ = 1.2 ± 0.2 ( plot in Figure 3c; the values for Σ[Cys + n]+ and Σ[Cys-S-S-Cys + n]+ can be obtained from the
plot in Figure 3c; the values for Σ[Cys + n]+ and Σ[Cys-S-S-Cys + n]+ can be obtained from the  and
and  plots in Figure 3b, respectively), suggesting that the disulfide product has higher sodiation efficiency compared with Cys under these conditions. In addition, at ΔV = 4 V, the measured values for Σ[Cys + n]+, Σ[Cys-S-S-Cys + n]+, and Σ[Cys-S-S-Cys + n]+/Σ[Cys + n]+ increase from [KCl] = 1 to 10 mM and then plateaus from 10 to 100 mM, indicating that [KCl] = 10 mM is the optimum concentration for Cys oxidation without causing significant salt deposition on the inlet capillary (see
plots in Figure 3b, respectively), suggesting that the disulfide product has higher sodiation efficiency compared with Cys under these conditions. In addition, at ΔV = 4 V, the measured values for Σ[Cys + n]+, Σ[Cys-S-S-Cys + n]+, and Σ[Cys-S-S-Cys + n]+/Σ[Cys + n]+ increase from [KCl] = 1 to 10 mM and then plateaus from 10 to 100 mM, indicating that [KCl] = 10 mM is the optimum concentration for Cys oxidation without causing significant salt deposition on the inlet capillary (see  and
and  plots in Figure 3b and Figure S1c).
plots in Figure 3b and Figure S1c).
An important parameter in P-EC/SSI-MS is the polarity of ΔV with respect to direction of the microdroplet spray and MS inlet. All experiments described above were performed with E2 connected to the positive terminal of the DC power supply (Figure 1a). When the polarity is switched so that E2 is the cathode, [Cys-S-S-Cys + n]+ ion signals were not observed (Figure S2). In addition, [Cys + n]+ signal intensities remain relatively unchanged when E2 is the cathode. These results suggest that disulfide species generated at the anode (E1) are reduced at the cathode (E2) prior to exiting the EC assembly, producing mass spectra containing primarily [Cys + n]+ species.
P-EC/SSI-MS characterization of the oxidation of dopamine (DOP) to dopamine o-quinone (DOPQ) was used to estimate t r (Figure 4). Application of 4 V to the EC cell yielded mass spectra containing ion signals at m/z 137.060 [DOP – NH3 + H]+, m/z 154.086 [DOP + H]+, m/z 176.068 [DOP + Na]+, m/z 307.166 [2DOP + H]+, m/z 329.148 [2DOP + Na]+, and oxidation product at m/z 152.071 [DOPQ + H]+ (Figure 4b). The extracted ion chromatogram (XIC) for [DOPQ + H]+ shows that oxidation only occurs when a potential is applied (ON state) to the EC cell. Now, t r = delay from voltage onset (delay time) + time for signal intensity to increase to S/N = 3 (rise time). Signal intensity at the base (t base ) of the first peak is 295 and at the point where the slope of the peak decreases is 3638 (these points are connected by a red line, Figure 4c). Thus, rise time = 500(2 × 295)/(3638 – 295) = 90 ms (IT = 500 ms; total scan time = 3 s). Similar calculations for the first three peaks in Figure 4c gives an average rise time of 120 ± 40 ms (for peak 3, t base is equal to the average noise intensity of ~30; note that the slope of peaks 3 and 4 decrease after two MS scans relative to t base ). For these experiments, the temporal resolution using IT = 500 ms is not sufficient to measure the delay time. ITMS XIC (IT = 100 ms for [PhSPh]•+ (m/z 186; reaction mechanism discussed later in the article) generated from EC oxidation of PhSPh, yielded a rise time of 100 ± 100 ms and delay time of 330 ± 10, which gives t r = 500 ± 100 ms (Figure 4d).

Response time of planar P-EC/SSI-MS. (a) EC oxidation of dopamine (DOP) to form dopamine o-quinone (DOPQ). (b) Average Orbitrap mass spectrum across peak 1 in the extracted ion chromatogram (XIC) shown in (c). (c) XIC for [DOPQ + H]+ with (ON) and without voltage applied to the EC cell. Red lines indicate the rising edge of each peak. (d) ITMS XIC for [PhSPh]•+ (m/z 186; see Figure 6 for reaction mechanism and Orbitrap mass spectra; arrows indicate the time when the DC power supply is switched ON for each trial, which have distinct colors)
In summary, experiments characterizing planar P-EC/SSI-MS show that (1) the paper substrate in the EC cell does not affect ion signal intensity (viz. T E ), (2) [electrolyte] influences I E owing to the square root dependence of average microdroplet charge on ion concentration, (3) the magnitude of ΔV applied to the EC cell has no effect on I E or T E , (4) this P-EC cell configuration has threshold [KCl] = 10 mM and [Cys] = 1 mM at ΔV = 4 V (threshold potential = 2 V) for observing disulfide product, (5) P-EC allows the use of relatively high [electrolyte] ≤ 10 mM without fouling of the MS inlet (deposition of KCl was observed at 100 mM), (6) the polarity of ΔV relative to the SSI microdroplet spray direction and MS inlet determine the species observed in acquired mass spectra (viz. the mass spectrometer primarily samples species generated at E2 in the P-EC cell), and (g) the response time is on the order of hundreds of milliseconds.
Ionization and Differentiation of Polycyclic Aromatic Hydrocarbons (Planar P-EC/SSI-MS)
It is often necessary to characterize samples containing nonpolar molecules such as polycyclic aromatic hydrocarbons (PAHs), which are components of crude oil [84]. Many PAHs such as the isomers of benzo[α]pyrene (BAP) are potent carcinogens [33] that require environmental monitoring. Under certain conditions, ESI can extract an electron from PAHs when the number of aromatic rings in the molecule is greater than two [84, 85]. Direct detection of PAHs by SSI-MS is more challenging because a high voltage is not applied to the microdroplet spray solution. In addition, MS is unable to resolve isomers, thus requiring additional derivatization steps for characterization of PAH.
We utilized P-EC/SSI-MS to ionize and differentiate BAP and perylene (PER) isomers (C20H12; 252 Da) based on the intensities of species generated in EC reactions with H2O. When mixtures of BAP:PER (9 mM:1 mM and 1 mM:9 mM in CH2Cl2/ACN) were placed in the SSI microdroplet spray, [C20H12]•+ (m/z 252.091) was not observed (Figure 5a and b), indicating that the SSI mechanism is not sufficient to remove an electron from this aromatic ring system. This observation agrees with previous studies showing that the ESI potential ionizes PER with very low efficiency and does not yield any detectable signal for BAP [33]. Application of ΔV = 4 V to the EC cell yielded ion signals at m/z 252.091 ([BAP]•+ and [PER]•+) for both the 9:1 and 1:9 solutions (Figure 5c and d).

Planar P-EC/SSI-MS (electrolyte [KNO3] = 10 mM) spectra of solutions containing benzo[α]pyrene (BAP) and perylene (PER) in CH2Cl2/ACN. Mass spectra for (a) BAP:PER (1 mM:9 mM) and (b) BAP:PER (9 mM:1 mM) solutions using ΔV = 0 V (background signals shown with red labels). Mass spectra for (c) BAP:PER (1:9) and (d) BAP:PER (9:1) solutions using ΔV = 4 V. Chemical structures for BAP and PER are shown in panel (c). (e) EC oxidation of BAP in the presence of water
Interestingly, a peak at m/z 267.078 is also observed, which corresponds to the formation of BAP and PER oxidation products ([BAP – H + O]+ and [PER – H + O]+) from reaction with H2O (Figures 5c, d and e shows the proposed mechanism for oxidation of BAP). The intensity of m/z 267.078 relative to m/z 252.078 is as follows: 1 mM PER = ~5% (data not shown); 9 mM BAP:1 mM PER > 100%; (Figure 5d); and, 1 mM BAP:9 mM PER = ~10% (Figure 5c). These results suggest that EC oxidation of BAP has faster kinetics and/or [BAP – H + O] has higher ionization efficiency compared to [PER – H + O], which agrees with previous EC/ESI-MS analyses showing no [PER – H + O]+ in acquired mass spectra [33, 34, 54]. The significant difference in the signal intensities of the oxidation products provides a means to distinguish a mixture of the two isomers using EC/MS. Previous EC/ESI-MS spectra of H2O-mediated oxidation of BAP also show a peak at nominal m/z 283 corresponding to [BAP – H + 2O]+ with higher relative intensity compared with nominal m/z 267 and nominal m/z 252 [33]. The ion signal at m/z 283.073 is observed at very low relative intensities of ~0.9%, ~0.3%, and ~0.4% (compared with m/z 252.091) for 1 mM PER, 1:9 BAP:PER, and 9:1 BAP:PER solutions, respectively, when 4 V was applied to the EC cell (Figure 5c and d). The lower relative intensity (<1%) of the ion signal at nominal m/z 283 suggests that planar P-EC/SSI-MS (t r = 500 ± 100 ms) is accessing an earlier reaction time point compared with the EC/ESI-MS (t r < 3 s [33]). These results highlight the ability of planar P-EC/SSI-MS to increase I E , and sensitivity of nonpolar analytes, differentiate isomers based on distinct EC properties and reactions, and intercept EC intermediates and products on short time scales.
Elucidation of Electrochemical Reaction Mechanisms (Planar P-EC/SSI-MS)
The characterization of EC reactions is important for understanding many important chemical and biological processes [5]. One well-known reaction is the anodic oxidation of diphenyl sulfide (PhSPh) to produce species containing sulfoxide and sulfone functional groups (Figure 6a) [42]. The proposed EC reaction mechanism [42] involves oxidation of neutral [PhSPh] to yield radical cation [PhSPh]•+ (m/z 186.050), which is unstable and may undergo further oxidation to diphenyl sulfone ([PhS(O)2Ph + H]+ at m/z 219.048 with ~0.1% relative intensity compared with m/z 203.035) via diphenyl sulfoxide (observed as [PhS(O)Ph + H]+ at m/z 203.053, [PhS(O)Ph + Na]+ at m/z 225.035, and [2PhS(O)Ph + Na]+ at m/z 427.080) in the presence of H2O (Figure 6). Alternatively, [PhSPh]•+ may react to yield pseudodimer sulfonium ion [Ph2S+C6H4SPh] at m/z 371.093 and other oxidation products ([Ph2S+C6H4S(O)Ph] at m/z 387.087 with ~0.3% relative intensity compared with m/z 203.035 and [Ph2S+C6H4S(O)2Ph], which was not observed). All of these species, except [Ph2S+C6H4S(O)2Ph], were observed via P-EC/SSI-MS (Figure 6b) including predominant background ions at nominal m/z 289, 301, and 317, which were also observed at ΔV = 0 V (Figure 6c).

Three mechanisms have been proposed for the formation of the pseudodimer sulfonium ion involving three distinct intermediates (bolded in the scheme below) [42, 86]:
-
Mechanism I
$$ \begin{array}{l}\begin{array}{l}2\left[\mathrm{PhSPh}\right]\to 2{{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}+2{\mathrm{e}}^{-}\\ {}\kern5em \left(m/z186.050\right)\end{array}\hfill \\ {}\begin{array}{l}{{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}\rightleftarrows\ {\left[\mathbf{P}\mathbf{h}\mathbf{S}{\mathbf{C}}_{\mathbf{6}}{\mathbf{H}}_{\mathbf{4}}\right]}^{\bullet }+{\mathrm{H}}^{+}\\ {}\kern5em \left(185.042\ \mathrm{Da}\right)\end{array}\hfill \\ {}\begin{array}{l}{{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}+{\left[{\mathrm{Ph}\mathrm{SC}}_6{\mathrm{H}}_4\right]}^{\bullet}\to \left[{\mathrm{Ph}}_2{\mathrm{S}}^{+}{\mathrm{C}}_6{\mathrm{H}}_4\mathrm{S}\mathrm{P}\mathrm{h}\right]\\ {}\kern1em \left(m/z371.093\right)\end{array}\hfill \end{array} $$ -
Mechanism II
$$ \begin{array}{l}\left[\mathrm{PhSPh}\right]\to {{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}+{\mathrm{e}}^{-}\hfill \\ {}\begin{array}{l}{{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}\to {\left[\mathbf{P}\mathbf{h}\mathbf{S}{\mathbf{C}}_{\mathbf{6}}{\mathbf{H}}_{\mathbf{4}}\right]}^{+}+{\mathrm{e}}^{-}+{\mathrm{H}}^{+}\\ {}\kern5em \left(m/z185.042\right)\end{array}\hfill \\ {}\begin{array}{l}{\left[{\mathrm{Ph}\mathrm{SC}}_6{\mathrm{H}}_4\right]}^{+}+\left[\mathrm{PhSPh}\right]\to \left[{\mathrm{Ph}}_2{\mathrm{S}}^{+}{\mathrm{C}}_6{\mathrm{H}}_4\mathrm{S}\mathrm{P}\mathrm{h}\right]\\ {}\kern12em \left(m/z371.093\right)\end{array}\hfill \end{array} $$ -
Mechanism III
$$ \begin{array}{l}\left[\mathrm{PhSPh}\right]\to {{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}+{\mathrm{e}}^{-}\hfill \\ {}\begin{array}{l}{{\left[\mathrm{PhSPh}\right]}^{\bullet}}^{+}+\left[\mathrm{PhSPh}\right]\to {{\left[\mathbf{P}{\mathbf{h}}_{\mathbf{2}}\mathbf{S}{\mathbf{C}}_{\mathbf{6}}{\mathbf{H}}_{\mathbf{5}}\mathbf{S}\mathbf{P}\mathbf{h}\right]}^{\bullet}}^{+}\\ {}\kern10.5em \left(m/z372.101\right)\ \end{array}\hfill \\ {}\begin{array}{l}{{\left[{\mathrm{Ph}}_2{\mathrm{SC}}_6{\mathrm{H}}_5\mathrm{S}\mathrm{P}\mathrm{h}\right]}^{\bullet}}^{+}\to {\left[{\mathrm{Ph}}_2{\mathrm{SC}}_6{\mathrm{H}}_4\mathrm{S}\mathrm{P}\mathrm{h}\right]}^{+}+{\mathrm{e}}^{-}+{\mathrm{H}}^{+}\\ {}\kern10.5em \left(m/z371.093\right)\ \end{array}\hfill \end{array} $$
Previous EC/ESI-MS studies (ΔV = 1.5 – 1.75 V) using t r = 3 s, identified [PhSPh]•+ at nominal m/z 186 and the pseudodimer sulfonium ion at nominal m/z 371, but ion signals for the intermediates at nominal m/z 372 and 185 were not observed, which led to the conclusion that mechanism I is predominant [42]. Using P-EC/SSI-MS (t r = 500 ± 100 ms), an ion signal was observed at m/z 185.042 for intermediate [PhSC6H4]+ indicating that mechanism II is also involved in the formation of the pseudodimer sulfonium ion. The Orbitrap mass spectrometer used in this study is not capable of resolving the 13C isotope of pseudodimer sulfonium ions (m/z 372.096) and [Ph2SC6H5SPh]•+ (m/z 372.101) (Figure 6b). However, the relative intensity of nominal m/z 372 (~26%) agrees with the expected value for the 13C pseudodimer sulfonium isotope, suggesting that mechanism III is not predominant. These results demonstrate that P-EC/SSI-MS can intercept intermediates generated at electrode surfaces with temporal resolution of hundreds of milliseconds, making it a powerful technique for elucidating fast EC processes.
Adding Reagents to the Paper Substrate in Planar and Tubular P-EC/SSI-MS
Addition of [PhSPh] and electrolyte to the paper substrate of planar P-EC/SSI-MS yielded more complex mass spectra having the same base peak ([PhS(O)Ph + H]+ at m/z 203.053) and with significantly lower relative intensities of relevant EC species (Figure 6a and mechanism II) compared with placing reagent in the microdroplet spray (compare Figure 6b and Figure S3a). The relative intensities of species relevant to anodic oxidation of [PhSPh] are 5% [PhSPh]•+ (m/z 186.050), 0.05% [PhSC6H4]+ (m/z 185.042), 25% [PhS(O)Ph + Na]+ (m/z 225.035), 0.4% [Ph2S+C6H4SPh] (m/z 371.093), 3% [2PhS(O)Ph + Na]+ (m/z 427.080), and 1% [Ph2S+C6H4S(O)Ph] at m/z 387.087 (viz. all species in Figure 6 and mechanism II observed except [PhS(O)2Ph + H]+ at m/z 219.048). These results suggest that placing reagent in the paper causes unwanted competitive reaction pathways leading to numerous unidentified ion signals in acquired mass spectra. These ion signals may originate from additional [PhSPh]-related redox processes and/or from reaction with components of the paper matrix.
When tubular P-EC/SSI-MS (Figure 1b) was used to characterize oxidation of [PhSPh] in the microdroplet spray, acquired mass spectra (Figure S3b) contained many of the same unidentified ion signals observed in planar P-EC/SSI-MS with [PhSPh] added to the paper electrolyte layer (Figure S3a). In addition, only the [PhS(O)Ph + H]+ (m/z 203.053), [PhS(O)Ph + Na]+ (m/z 225.035), and [PhSPh]•+ (m/z 186.050) species relevant to anodic [PhSPh] oxidation were observed. Importantly, pseudodimer sulfonium ion was not observed and [PhS(O)Ph + H]+ was no longer the base peak (Figure S3b). When [PhSPh] was added to the paper in tubular P-EC/SSI-MS, ion signals for [Ph2S+C6H4SPh] (m/z 371.093) and oxidation product [Ph2S+C6H4S(O)Ph] (m/z 387.087) were also observed, indicating EC conversion (Figure S3c). Thus, the four configurations can be placed in order of ([PhSPh] oxidation):(unwanted competitive pathways) ratio as follows: planar-spray > planar-paper > tubular-paper > tubular-spray (EC cell shape-reagent location). This order suggests that competitive pathways are more pronounced at longer reaction times, which is controlled by the tube dimensions and reagent residence time in the paper substrate.
Current results demonstrate that (1) reagents added to the paper substrate undergo EC conversion with potential reaction with components of the paper matrix and longer residence times, (2) EC transformation can be achieved using a tubular P-EC/SSI-MS design, and (3) tube dimensions significantly impact reactivity and performance characteristics such as ionization efficiency.
Proposed Mechanism for P-EC/SSI-MS
The results outlined above have provided some preliminary insights into the mechanism of P-EC/SSI-MS. When a voltage is applied to the EC cell, oxidation and reduction regions extend a short distance from the electrode surfaces into the paper matrix. Microdroplets from the SSI emitter impact the paper substrate, delivering reagents to the EC cell. These reagents may undergo reduction at E1 as they diffuse through the paper towards the anode E2, where they are oxidized. Owing to the thinness of the electrodes (200 μm) and paper (~100 μm), the speed of impacting microdroplets (~150 m/s [87]) rapidly transfers species generated at the E2-paper interface into the gas-phase, leading to a response time on the hundreds of milliseconds times scale (t r = 500 ± 100; Figure 4). The short t r allows detection of transient EC species such as [PhSC6H4]+ formed in the oxidation of [PhSPh] (Figure 6). When reagents are added to the paper, there is a longer time between application of the voltage and sampling by the microdroplet spray (viz. t r is longer), which may cause unwanted processes such as reaction with chemical components of the paper. When the polarity is switched, reagents in the microdroplet spray may be oxidized at E1 and then reduced at E2 (cathode) prior to entering the gas phase. As a result, ion signals corresponding to oxidized intermediates and products were not observed in positive mode mass spectra (Figure S2). For tubular P-EC/SSI-MS, t r should be longer because of multiple collisions with the paper as microdroplets travel through the tube. In addition, extraction efficiency from the inner surfaces of the tube will be lower compared with the direct 90o impact of the microdroplet spray in the planar configuration. Similar to the planar configuration, the anode should be the inner electrode for characterization of oxidation reactions.
An important point to note is that relatively high concentrations of analyte (mM) were required to observe EC conversion. This concentration regime agrees with previous observations by Cui et al. [3] that solid-contact K+-selective electrodes had detection limits three orders of magnitude higher in a nitrocellulose paper matrix compared to bulk solution. A possible reason for the lower sensitivity is that ion transport through the paper matrix and at the paper–electrode interface is slower compared with solution phase. Further characterization experiments are required to identify the processes occurring in the paper matrix and inside the tube during EC cell operation, as well as elucidate their effect on EC reactivity. In addition, the impact of parameters such as tube dimensions (e.g., length and diameter), composition of the paper matrix, electrode material, and paper porosity on P-EC/MS performance needs to be evaluated.
Conclusions
This report describes coupling of paper-based electrochemical cells (P-EC) to sonic spray ionization mass spectrometry (P-EC/SSI-MS). The EC cells consist of a paper substrate sandwiched between two mesh stainless steel electrodes. In P-EC/SSI-MS, the EC cell is physically decoupled from the ionization source, allowing independent control of electrical and chemical parameters. The flexibility of paper and mesh electrodes allowed development of tubular and planar P-EC/SSI-MS configurations, demonstrating high versatility and modularity. P-EC/SSI-MS was used to successfully characterize EC oxidation of cysteine, dopamine, polycyclic aromatic hydrocarbons, and diphenyl sulfide. Perhaps one of the most exciting features of P-EC/SSI-MS is a demonstrated response time on the order of hundreds of milliseconds, which allowed online detection of transient EC species generated at the electrode surfaces in contact with the paper substrate. The high temporal resolution makes P-EC/SSI-MS a powerful tool for elucidating the mechanisms of EC processes in situ. In addition, the benefits and convenience of paper provide a low cost, disposable, and simple approach for coupling EC processes to MS. P-EC/MS has the potential to open new areas of research and to facilitate the development of P-EC devices via advancing current understanding of molecular EC mechanisms operating in the paper substrate.
References
Cate, D.M., Adkins, J.A., Mettakoonpitak, J., Henry, C.S.: Recent developments in paper-based microfluidic devices. Anal. Chem. 87, 19–41 (2015)
Dungchai, W., Chailapakul, O., Henry, C.S.: Electrochemical detection for paper-based microfluidics. Anal. Chem. 81, 5821–5826 (2009)
Cui, J., Lisak, G., Strzalkowska, S., Bobacka, J.: Potentiometric sensing utilizing paper-based microfluidic sampling. Analyst 139, 2133–2136 (2014)
Bruckenstein, S., Raogadde, R.: Use of a porous electrode for in-situ mass spectrometric determination of volatile electrode reaction products. J. Am. Chem. Soc. 93, 793–794 (1971)
Liu, P.Y., Lu, M., Zheng, Q.L., Zhang, Y., Dewald, H.D., Chen, H.: Recent advances of electrochemical mass spectrometry. Analyst 138, 5519–5539 (2013)
Faber, H., Vogel, M., Karst, U.: Electrochemistry/mass spectrometry as a tool in metabolism studies: a review. Anal. Chim. Acta 834, 9–21 (2014)
Permentier, H.P., Bruins, A.P.: Electrochemical oxidation and cleavage of proteins with on-line mass spectrometric detection: Development of an instrumental alternative to enzymatic protein digestion. J. Am. Soc. Mass Spectrom. 15, 1707–1716 (2004)
Shumyantseva, V.V., Suprun, E.V., Bulko, T.V., Archakov, A.I.: Electrochemical methods for detection of post-translational modifications of proteins. Biosens. Bioelectron. 61, 131–139 (2014)
Permentier, H.P., Bruins, A.P., Bischoff, R.: Electrochemistry-mass spectrometry in drug metabolism and protein research. Mini-Rev. Med. Chem. 8, 46–56 (2008)
Li, J., Dewald, H.D., Chen, H.: Online Coupling of electrochemical reactions with liquid sample desorption electrospray ionization-mass spectrometry. Anal. Chem. 81, 9716–9722 (2009)
Zhang, Y., Dewald, H.D., Chen, H.: Online mass spectrometric analysis of proteins/peptides following electrolytic cleavage of disulfide bonds. J. Proteome Res. 10, 1293–1304 (2011)
McClintock, C., Kertesz, V., Hettich, R.L.: Development of an electrochemical oxidation method for probing higher order protein structure with mass spectrometry. Anal. Chem. 80, 3304–3317 (2008)
Baumann, A., Lohmann, W., Jahn, S., Karst, U.: On-line electrochemistry/electrospray ionization mass spectrometry (EC/ESI-MS) for the Generation and identification of nucleotide oxidation products. Electroanalysis 22, 286–292 (2010)
Jahn, S., Karst, U.: Electrochemistry coupled to (liquid chromatography/) mass spectrometry-current state and future perspectives. J. Chromatogr. A 1259, 16–49 (2012)
Xu, K., Zhang, Y., Tang, B., Laskin, J., Roach, P. J., Chen, H.: Study of highly selective and efficient thiol derivatization using selenium reagents by mass spectrometry. Anal. Chem. 82, 6926–6932 (2010)
Wang, Z., Zhang, Y., Zhang, H., Harrington P., Chen, H.: Fast and selective modification of thiol proteins/peptides by N-(phenylseleno)phthalimide. J. Am. Soc. Mass Spectrom. 23, 520–529 (2012)
Modestov, A.D., Srebnik, S., Lev, O., Gun, J.: Scanning capillary microscopy/mass spectrometry for mapping spatial electrochemical activity of electrodes. Anal. Chem. 73, 4229–4240 (2001)
Momotenko, D., Qiao, L., Cortes-Salazar, F., Lesch, A., Wittstock, G., Girault, H.H.: Electrochemical push-pull scanner with mass spectrometry detection. Anal. Chem. 84, 6630–6637 (2012)
Gun, J., Bharathi, S., Gutkin, V., Rizkov, D., Voloshenko, A., Shelkov, R., Sladkevich, S., Kyi, N., Rona, M., Wolanov, Y., Rizkov, D., Koch, M., Mizrahi, S., Pridkhochenko, P.V., Modestov, A., Lev, O.: Highlights in coupled electrochemical flow cell-mass spectrometry, EC/MS. Isr. J. Chem. 50, 360–373 (2010)
Fenn, J.B., Mann, M., Meng, C.K., Wong, S.F., Whitehouse, C.M.: Electrospray Ionization for mass-spectrometry of large biomolecules. Science 246, 64–71 (1989)
Yamashita, M., Fenn, J.B.: Electrospray ion-source—another variation on the free-jet theme. J. Phys. Chem. 88, 4451–4459 (1984)
Yamashita, M., Fenn, J.B.: Negative-ion production with the electrospray ion-source. J. Phys. Chem. 88, 4671–4675 (1984)
Blades, A.T., Ikonomou, M.G., Kebarle, P.: Mechanism of electrospray mass-spectrometry—electrospray as an electrolysis cell. Anal. Chem. 63, 2109–2114 (1991)
Zeleny, J.: The electrical discharge from liquid points, and a hydrostatic method of measuring the electric intensity at their surfaces. Phys. Rev. 3, 69–91 (1914)
Taylor, G.: Disintegration of water drops in electric field. Proc. Royal Soc. Lond. A-Mat. 280, 383–397 (1964)
Taylor, G.: Electrically driven jets. Proc. Royal Soc. Lond. A-Mat. 313, 453–475 (1969)
Rohner, T.C., Lion, N., Girault, H.H.: Electrochemical and theoretical aspects of electrospray ionization. Phys. Chem. Chem. Phys. 6, 3056–3068 (2004)
Van Berkel, G.J., McLuckey, S.A., Glish, G.L.: Electrochemical origin of radical cations observed in electrospray ionization mass spectra. Anal. Chem. 64, 1586–1593 (1992)
Van Berkel, G.J., Zhou, F.M.: Electrospray as a controlled current electrolytic cell—electrochemical ionization of neutral analytes for detection by electrospray mass-spectrometry. Anal. Chem. 67, 3958–3964 (1995)
Van Berkel, G.J., Zhou, F.M.: Characterization of an electrospray ion-source as a controlled-current electrolytic cell. Anal. Chem. 67, 2916–2923 (1995)
Van Berkel, G.J., Zhou, F.M., Aronson, J.T.: Changes in bulk solution pH caused by the inherent controlled-current electrolytic process of an electrospray ion source. Int. J. Mass Spectrom. 162, 55–67 (1997)
Bond, A.M., Colton, R., D'Agostino, A., Downaed, A.J., Traeger, J.C.: A role for electrospray mass spectrometry in electrochemical studies. Anal. Chem. 67, 1691–1695 (1995)
Xu, X., Lu, W., Cole, R.B.: On-line probe for fast electrochemistry/electrospray mass spectrometry. investigation of polycyclic aromatic hydrocarbons. Anal. Chem. 68, 4244–4253 (1996)
Zhou, F., Van Berkel, G.: Electrochemistry combined on-line with electrospray mass spectrometry. Anal. Chem. 67, 3643–3649 (1995)
Oberacher, H., Pitterl, F., Erb, R., Plattner, S.: Mass spectrometric methods for monitoring redox processes in electrochemical cells. Mass Spectrom. Rev. 34, 64–92 (2015)
Zhang, T.Y., Palii, S.P., Eyler, J.R., Brajter-Toth, A.: Enhancement of ionization efficiency by electrochemical reaction products in on-line electrochernistry/electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 74, 1097–1103 (2002)
Looi, D.W., Eyler, J.R., Brajter-Toth, A.: Electrochemistry-electrospray ionization FT ICR mass spectrometry (EC ESI MS) of guanine-tyrosine and guanine-glutathione crosslinks formed on-line. Electrochim. Acta 56, 2633–2640 (2011)
Mautjana, N.A., Estes, J., Eyler, J.R., Brajter-Toth, A.: One-electron oxidation and sensitivity of uric acid in on-line electrochemistry and in electrospray ionization mass spectrometry. Electroanalysis 20, 2501–2508 (2008)
Mautjana, N.A., Estes, J., Eyler, J.R., Brajter-Toth, A.: Antioxidant pathways and one-electron oxidation of dopamine and cysteine in electrospray and on-line electrochemistry electrospray ionization mass spectrometry. Electroanalysis 20, 1959–1967 (2008)
Mautjana, N.A., Looi, D.W., Eyler, J.R., Brajter-Toth, A.: Sensitivity of Positive Ion Mode Electrospray Ionization Mass Spectrometry (Esi Ms) in the Analysis of Purine Bases in Esi Ms and on-Line Electrochemistry Esi Ms (Ec/Esi Ms). Electrochim. Acta 55, 52–58 (2009)
Mautjana, N.A., Looi, D.W., Eyler, J.R., Brajter-Toth, A.: Sensitivity of Positive Ion Mode Electrospray Ionization Mass Spectrometry in the Analysis of Thiol Metabolites. Electroanalysis 22, 79–89 (2010)
Lu, W., Xu, X., Cole, R.B.: On-line linear sweep voltammetry electrospray mass spectrometry. Anal. Chem. 69, 2478–2484 (1997)
Kertesz, V., Van Berkel, G.J.: Study and application of a controlled-potential electrochemistry-electrospray emitter for electrospray mass spectrometry. Anal. Chem. 77, 4366–4373 (2005)
Van Berkel, G.J., Asano, K.G.: Controlling analyte electrochemistry in an electrospray ion source with a three-electrode emitter cell. Anal. Chem. 76, 1493–1499 (2004)
Albert, A., Shelley, J.T., Engelhard, C.: Plasma-based ambient desorption/ionization mass spectrometry: state-of-the-art in qualitative and quantitative analysis. Anal. Bioanal. Chem. 406, 6111–6127 (2014)
Awad, H., Khamis, M.M., El-Aneed, A.: Mass spectrometry, review of the basics: ionization. Appl. Spectrosc. 50, 158–175 (2015)
Cooks, R.G., Ouyang, Z., Takats, Z., Wiseman, J.M.: Ambient mass spectrometry. Science 311, 1566–1570 (2006)
Harris, G.A., Galhena, A.S., Fernandez, F.M.: Ambient Sampling/ionization mass spectrometry: applications and current trends. Anal. Chem. 83, 4508–4538 (2011)
Ifa, D.R., Wu, C.P., Ouyang, Z., Cooks, R.G.: Desorption electrospray ionization and other ambient ionization methods: current progress and preview. Analyst 135, 669–681 (2010)
Li, L.P., Feng, B.S., Yang, J.W., Chang, C.L., Bai, Y., Liu, H.: Applications of ambient mass spectrometry in high-throughput screening. Analyst 138, 3097–3103 (2013)
Monge, M.E., Harris, G.A., Dwivedi, P., Fernandez, F.M.: Mass spectrometry: recent advances in direct open air surface sampling/ionization. Chem. Rev. 113, 2269–2308 (2013)
Takats, Z., Wiseman, J.M., Gologan, B., Cooks, R.G.: Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306, 471–473 (2004)
Venter, A., Nefliu, M., Cooks, R.G.: Ambient desorption ionization mass spectrometry. TRAC-Trend. Anal. Chem. 27, 284–290 (2008)
Miao, Z., Chen, H.: Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS). J. Am. Soc. Mass Spectrom. 20, 10–19 (2009)
Liu, P.Y., Zhang, J., Ferguson, C.N., Chen, H., Loo, J.A.: Measuring protein-ligand interactions using liquid sample desorption electrospray ionization mass spectrometry. Anal. Chem. 85, 11966–11972 (2013)
Ferguson, C.N., Benchaar, S.A., Miao, Z.X., Loo, J.A., Chen, H.: Direct ionization of large proteins and protein complexes by desorption electrospray ionization-mass spectrometry. Anal. Chem. 83, 6468–6473 (2011)
Miao, Z.X., Chen, H., Liu, P.Y., Liu, Y.: Development of Submillisecond time-resolved mass spectrometry using desorption electrospray ionization. Anal. Chem. 83, 3994–3997 (2011)
Miao, Z.X., Wu, S.Y., Chen, H.: The study of protein conformation in solution via direct sampling by desorption electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 1730–1736 (2010)
Cai, Y., Liu, Y., Helmy, R., Chen, H.: Coupling of Ultrafast Lc with Mass Spectrometry by DESI. J. Am. Soc. Mass Spectrom. 25, 1820–1823 (2014)
Cai, Y., Adams, D., Chen, H.: A new splitting method for both analytical and preparative LC/MS. J. Am. Soc. Mass Spectrom. 25, 286–292 (2014)
Zhang, Y., Yuan, Z.Q., Dewald, H.D., Chen, H.: Coupling of liquid chromatography with mass spectrometry by desorption electrospray ionization (DESI). Chem. Commun. 47, 4171–4173 (2011)
Zheng, Q.L., Zhang, H., Chen, H.: Integration of online digestion and electrolytic reduction with mass spectrometry for rapid disulfide-containing protein structural analysis. Int. J. Mass Spectrom. 353, 84–92 (2013)
Zhang, Y., Cui, W.D., Zhang, H., Dewald, H.D., Chen, H.: Electrochemistry-assisted top-down characterization of disulfide-containing proteins. Anal. Chem. 84, 3838–3842 (2012)
Lu, M., Wolff, C., Cui, W.D., Chen, H.: Investigation of some biologically relevant redox reactions using electrochemical mass spectrometry interfaced by desorption electrospray ionization. Anal. Bioanal. Chem. 403, 355–365 (2012)
Brown, T.A., Chen, H., Zare, R.N.: Identification of fleeting electrochemical reaction intermediates using desorption electrospray ionization mass spectrometry. J. Am. Chem. Soc. 137, 7274–7277 (2015)
Wang, H., Liu, J.J., Cooks, R.G., Ouyang, Z.: Paper spray for direct analysis of complex mixtures using mass spectrometry. Angew. Chem.-Int. Ed. 49, 877–880 (2010)
Liu, J.J., Wang, H., Manicke, N.E., Lin, J.M., Cooks, R.G., Ouyang, Z.: Development, characterization, and application of paper spray ionization. Anal. Chem. 82, 2463–2471 (2010)
Cooks, R.G., Manicke, N.E., Dill, A.L., Ifa, D.R., Eberlin, L.S., Costa, A.B., Wang, H., Huang, G.M., Zheng, O.Y.: New ionization methods and miniature mass spectrometers for biomedicine: desi imaging for cancer diagnostics and paper spray ionization for therapeutic drug monitoring. Faraday Discussions 149, 247–267 (2011)
Pulliam, C.J., Bain, R.M., Wiley, J.S., Ouyang, Z., Cooks, R.G.: Mass spectrometry in the home and garden. J. Am. Soc. Mass Spectrom. 26, 224–230 (2015)
Espy, R.D., Teunissen, S.F., Manicke, N.E., Ren, Y., Ouyang, Z., van Asten, A., Cooks, R.G.: Paper spray and extraction spray mass spectrometry for the direct and simultaneous quantification of eight drugs of abuse in whole blood. Anal. Chem. 86, 7712–7718 (2014)
Bag, S., Hendricks, P.I., Reynolds, J.C., Cooks, R.G.: Biogenic aldehyde determination by reactive paper spray ionization mass spectrometry. Anal. Chim. Acta. 860, 37–42 (2015)
Bain, R.M., Pulliam, C.J., Yan, X., Moore, K.F., Muller, T., Cooks, R.G.: Mass spectrometry in organic synthesis: Claisen-Schmidt base-catalyzed condensation and Hammett correlation of substituent effects. J. Chem. Educ. 91, 1985–1989 (2014)
Hamid, A.M., Wei, P., Jarmusch, A.K., Pirro, V., Cooks, R.G.: Discrimination of candida species by paper spray mass spectrometry. Int. J. Mass Spectrom. 378, 288–293 (2015)
Hamid, A.M., Jarmusch, A.K., Pirro, V., Pincus, D.H., Clay, B.G., Gervasi, G., Cooks, R.G.: Rapid discrimination of bacteria by paper spray mass spectrometry. Anal. Chem. 86, 7500–7507 (2014)
Hirabayashi, A., Sakairi, M., Koizumi, H.: Sonic spray ionization method for atmospheric-pressure ionization mass spectrometry. Anal. Chem. 66, 4557–4559 (1994)
Hardman, M., Makarov, A.A.: Interfacing the Orbitrap mass analyzer to an electrospray ion source. Anal. Chem. 75, 1699–1705 (2003)
Hu, Q.Z., Noll, R.J., Li, H.Y., Makarov, A., Hardman, M., Cooks, R.G.: The Orbitrap: a new mass spectrometer. J. Mass Spectrom. 40, 430–443 (2005)
Makarov, A.: Electrostatic axially harmonic Orbital trapping: a high-performance technique of mass analysis. Anal. Chem. 72, 1156–1162 (2000)
Makarov, A., Denisov, E., Kholomeev, A., Baischun, W., Lange, O., Strupat, K., Horning, S.: Performance evaluation of a hybrid linear ion trap/Orbitrap mass spectrometer. Anal. Chem. 78, 2113–2120 (2006)
Makarov, A., Denisov, E., Lange, O., Horning, S.: Dynamic range of mass accuracy in LTQ Orbitrap hybrid mass spectrometer. J. Am. Soc. Mass Spectrom. 17, 977–982 (2006)
Perry, R.H., Cooks, R.G., Noll, R.J.: Orbitrap mass spectrometry: instrumentation, ion motion, and applications. Mass Spectrom. Rev. 27, 661–699 (2008)
Wijeratne, A.B.., Yang, S.H., Armstrong, D.W., Schug, K.A.: Solvent molecules undergo homolytic cleavage and radical recombination processes during negative-mode electrospray ionization: adduct formation with antimony(III)-tartrate dianion. Anal. Chem. 82, 5141–5146 (2010)
Dodd, E.E.: The statistics of liquid spray and dust electrification by the hopper and laby method. J. Appl. Phys. 24, 73–80 (1953)
Guricza, L.M., Schrader, W.: Electrospray ionization for determination of non-polar polyaromatic hydrocarbons and polyaromatic heterocycles in heavy crude oil asphaltenes. J. Mass Spectrom. 50, 549–557 (2015)
Miyabayashi, K., Naito, Y., Tsujimoto, K., Miyake, M.: Structure characterization of polyaromatic hydrocarbons in arabian mix vacuum residue by electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. Int. J. Mass Spectrom. 235, 49–57 (2004)
Magno, F., Bontempe, G.: Electrochemical behavior of diphenyl sulfide in acetonitrile medium at a platinum-electrode. J. Electroanal. Chem. 36, 389–397 (1972)
Venter, A., Sojka, P.E., Cooks, R.G.: Droplet dynamics and ionization mechanisms in desorption electrospray ionization mass spectrometry. Anal. Chem. 78, 8549–8555 (2006)
Acknowledgments
The Perry Research Laboratory gratefully acknowledges financial support from the University of Illinois at Urbana-Champaign (UIUC).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1.63 MB)
Rights and permissions
About this article

Cite this article
Liu, YM., Perry, R.H. Paper-Based Electrochemical Cell Coupled to Mass Spectrometry. J. Am. Soc. Mass Spectrom. 26, 1702–1712 (2015). https://doi.org/10.1007/s13361-015-1224-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-015-1224-9