
Abstract
Hemoglobinopathies are the most common inherited disorders in humans and are thus the target of screening programs worldwide. Over the past decade, mass spectrometry (MS) has gained a more important role as a clinical means to diagnose variants, and a number of approaches have been proposed for characterization. Here we investigate the use of matrix-assisted laser desorption/ionization time-of-flight MS (MALDI-TOF MS) with sequencing using in-source decay (MALDI-ISD) for the characterization of Hb variants. We explored the effect of matrix selection using super DHB or 1,5-diaminonaphthalene on ISD fragment ion yield and distribution. MALDI-ISD MS of whole blood using super DHB simultaneously provided molecular weights for the alpha and beta chains, as well as extensive fragmentation in the form of sequence defining c-, (z + 2)-, and y-ion series. We observed sequence coverage on the first 70 amino acids positions from the N- and C-termini of the alpha and beta chains in a single experiment. An abundant beta chain N-terminal fragment ion corresponding to βc34 was determined to be a diagnostic marker ion for Hb S (β6 Glu→Val, sickle cell), Hb C (β6 Glu→Lys), and potentially for Hb E (β26 Glu→Lys). The MALDI-ISD analysis of Hb S and HbSC yielded mass shifts corresponding to the variants, demonstrating the potential for high-throughput screening. Characterization of an alpha chain variant, Hb Westmead (α122 His→Gln), generated fragments that established the location of the variant. This study is the first clinical application of MALDI-ISD MS for the determination and characterization of hemoglobin variants.

ᅟ
Similar content being viewed by others


Introduction
Hemoglobin mutations form one of the most common human genetic disorders world-wide and occur sporadically in all populations. Hemoglobinopathies are a diverse group of disorders caused by hemoglobin variants and are involved in disease states with broad physiological manifestations such as cyanosis, erythrocytosis, and hemolytic anemia. More than 1000 variants have been characterized and although the great majority of them are not clinically significant, there are close to 150 variant hemoglobins that are unstable, and these can cause hemolytic anemia of various severity [1–5]. The most common and clinically important variant hemoglobins are: Hb S (β6 Glu→Val), Hb C (β6 Glu→Lys), Hb E (β26 Glu→Lys), Hb D (β121 Glu→Gln) (Table 1). Hb S or sickle hemoglobin, caused by β-globin gene codon 6 GAG>GTG or glutamic acid residue replaced by valine. It is commonly found in sub-Sahara Africa, Middle East, and parts of Indian subcontinent. Patients with homozygous Hb S or sickle cell anemia suffer vaso-occlusions, multiple organ damage, and shortened lifespan. It is estimated that up to 330,000 neonates are born annually with this serious condition worldwide [6]. Hb C caused by β codon 6 GAG>AAG or glutamic acid replaced by lysine. It is the second most common variant hemoglobin worldwide found mostly in people of African descent. While homozygous Hb C is a mild condition, compound heterozygote with Hb S, known as Hb SC disease, can have severe clinical course similar to sickle cell anemia. Hb E caused by β codon 26 GAG>AAG or glutamic acid replaced by lysine, is widespread in southeast Asia, southern China, and eastern India. Homozygous Hb E is a mild condition. However, compound heterozygotes with β-thalassemia mutation, Hb E/β-thalassemia, can present as β-thalassemia major with severe anemia that requires monthly blood transfusions throughout life. While less common in the United States, D-Los Angeles (D-Punjab) is common in other regions of the world and thus may be included in screening programs. Of concern is the compound heterozygote Hb S/Hb D, where Hb D may co-polymerize with Hb S and cause severe sickling. Diseases caused by Hb S, Hb C, and Hb E are of public health importance in the US and in many countries where malaria was and may still be endemic. With population migrations, these diseases are now found throughout all parts of the world [3]. The detection and characterization of clinically relevant Hb variants is of paramount importance to generate a correct diagnosis. Proper identification of all variant hemoglobins is necessary in order to provide appropriate medical care, prognosis, and family counseling for those who have inherited these globin gene mutations.
Classical methods for the biochemical diagnosis of hemoglobinopathies such as electrophoresis and cation exchange chromatography rely on the detection of a charge difference induced by the mutation [7]. In the case of HPLC, variant identification is made on the basis of matching the elution profile with a library of known variants. Difficulties arise with co-eluting variants and components exhibiting unmatched retention times [8]. Therefore, in some cases the detection and characterization of hemoglobin variants present challenges to classical methods and require more sophisticated techniques such as DNA analysis or mass spectrometry. DNA analysis is a confirmatory technique not amenable to high throughput applications [9]. Mass spectrometry offers a rapid and accurate means for the detection and characterization of Hb variants as the speed, sensitivity, and selectivity of the method are compelling analytical assets, in addition to its capability for de novo determinations.
Detection and characterization of hemoglobin variants by mass spectrometry have a rich and diverse history [10]. These endeavors have utilized the full range of advances in biomolecular mass spectrometry. In the 1980s, field desorption [11] and fast atom bombardment (FAB) ionization modes [12] were used to analyze tryptic peptides of hemoglobin variants. With high quality FAB-MS and MS/MS data from a four-sector (EBEB) instrument, even novel hemoglobins could be unambiguously identified in clinical samples [13]. The detection of hemoglobin variants by molecular weight profiling of intact globin chains using electrospray ionization (ESI) on magnetic sector [14], triple quadrupole mass spectrometers [15], and matrix-assisted laser desorption/ionization (MALDI) [16], as well as peptide mass mapping of their tryptic digests by MALDI-TOF MS [17–19], were amongst the first. MALDI-TOF MS molecular weight profiling was recently used as a first tier screen in which newborn samples were analyzed to detect the beta chain mass shift characteristic of the sickle cell variant (β6 Glu→Val) [20]. A combination of ESI-MS molecular weight profiling of intact hemoglobins with MS/MS of peptides from the tryptic digest of whole blood for variant detection and characterization was reported [21]. Gatlin et al. demonstrated the feasibility of automatic detection and identification of point mutations in proteins by LC-MS/MS analysis of proteolytic digests [22]. This approach has been used for Hb variant analysis [23, 24]. Although bottom-up methods [25] remain the workhorse of proteomics, the characterization of point mutations by LC-MS/MS can be made difficult by the higher false positive rate that is observed when sequence variations are allowed during searches of very large databases and may necessitate the use of custom-built databases [24]. The requirements for chromatographic separation and proteolytic digestion prior to analysis render the bottom-up approach time-consuming, discouraging the use of this approach for high throughput screening. Targeted analysis for common hemoglobin variants through multiple reaction monitoring (MRM) acquisition mode MS of whole blood tryptic digests has been proposed as a high-throughput population screening methodology [26].
For the top-down approach, proteins are introduced directly into the mass spectrometer and individual components can be mass-selected and dissociated in the instrument, yielding product ions containing sequence information. A significant advantage of top-down [27–29] over bottom-up methods for variant characterization is that connectivity is maintained between the molecular weight profile information and the fragment ion mass spectrum. This connectivity is of vital importance in protein variant detection and characterization. The molecular weight profile provides the mass shift caused by the presence of a variant and limits the number of possible amino acid substitutions to be considered, facilitating the analysis of the fragmentation data by guiding the search of variant fragment ions. The avoidance of chromatographic separation and the minimal requirement for sample preparation represent a considerable saving in terms of time. As noted above, the potential of top-down mass spectrometry for the analysis of hemoglobin variants was recognized in the early 1990s using triple quadrupole instruments [30]. At that time, the efforts were impeded by charge state ambiguities, spectra complexity, and the lack of suitable software tools for spectra interpretation. Recent improvements in instrument capabilities and performance as well as software development have greatly increased the power of top-down mass spectrometry. We, and others, have recently proposed top-down analytical platforms for hemoglobin variant analysis based on high performance, high resolution mass spectrometers [31, 32].
Despite the scope of the efforts deployed to use mass spectrometry techniques to detect and characterize hemoglobin variants, the application of MALDI in-source decay (ISD) [33–35] mass spectrometry in this area has not yet been explored. MALDI-ISD MS was initially developed on linear MALDI-TOF instruments and later used on higher performance TOF/TOF instruments [36]. The capabilities of MALDI-ISD MS in terms of top-down protein sequencing were demonstrated by determining the primary sequence of a 13.6 kDa single heavy chain camelid antibody [37]. The closest application to Hb variant analysis described to date used MALDI-ISD MS to identify sequence variants in tubulin isoforms [38] originating from HeLa cells. In-source decay occurs prior to acceleration of the ions. The fragmentation gives rise mainly to c, y, and (z + 2) fragment ions. The MALDI-ISD MS process is analogous to ECD MS and ETD MS where the fragmentation is mediated by a hydrogen radical transfer [39]. In MALDI-ISD MS, the type and distribution of the fragment ions can be strongly influenced by the choice of matrix [40]. The technique offers a fast, convenient means of generating top-down data from proteins and has been reported to be an efficient method for C- and N-terminal sequencing [36] and a range of other applications including tissue imaging [41]. More recent instrumentation offers the possibility for performing MS/MS analysis of ISD fragment ions using T3 sequencing [42]. We note that hemoglobins are well suited for MALDI-ISD MS since sample purity and quantity are not an issue, given that simple dilution of whole blood virtually eliminates the contribution of other components while still providing a suitably high concentration of hemoglobin for analysis. The method can provide simultaneous molecular weight profile information and sequence-defining fragmentation. We present here a prospective clinical application of MALDI-ISD MS with the potential for high throughput screening and sequencing of clinically relevant Hb variants.
Experimental
Whole blood was obtained from patients, with their informed consent, as part of a screening program of the Hemoglobin Diagnostic Reference Laboratory at the Boston University School of Medicine. The blood was diluted approximately 1:250 in water. The “Super DHB” (sDHB) (Bruker Daltonics, Billerica, MA, USA) matrix solution was prepared to a concentration of 50 g/L in 50% acetonitrile/water/0.1% formic acid. Super DHB is a mixture of 2,5-dihydroxy-benzoic acid and 2-hydroxy-5-methoxy-benzoic acid 10:1 w/w [43]. A saturated solution of 1,5-DAN (Acros Organics, Fisher Scientific, Pittsburgh, PA, USA) matrix was prepared in 50% acetonitrile/water/0.1% formic acid. The sDHB samples were prepared by spotting 0.5 μL of the matrix solution and 0.5 μL of diluted whole blood on a ground stainless target. For 1,5-DAN samples, 1 μL of matrix solution was used.
All MALDI-TOF mass spectra were acquired on a Bruker UltrafleXtreme MALDI-TOF/TOF mass spectrometer (Bruker Daltonics) equipped with a Smartbeam II laser (wavelength 355 nm, 3 ns pulse width, and a power level of 100 μJ/pulse) that was operated at 1 kHz repetition rate. In-source decay (ISD) measurements were used for top-down sequencing [42] and were conducted in the reflectron mode. The acceleration voltage was set to 25.00 kV, extraction voltage was set to 22.60 kV, lens voltage was at 7.75 kV, and reflector voltage was at 26.45 kV. The pulsed ion extraction (PIE) time was set to 180 ns. ISD spectra consisted of 12,000 to 15,000 accumulated laser shots and were externally calibrated using c-type fragment ions generated from intact bovine serum albumin in the range m/z 1000–5000.
The T3 sequencing spectra of ISD fragment ions were acquired by preselecting the ISD fragment ions of interest with the precursor ion selector and fragmenting them further. The acceleration voltage was set to 7.50 kV, extraction voltage was set to 6.80 kV, lens voltage was at 3.50 kV, LIFT 1 voltage was at 19.00 kV, and reflector voltage was set to 29.50 kV. The PIE time was set to 90 ns.
Data acquisition and spectral processing were accomplished using Compass 1.4 software followed by top-down sequencing analysis in BioTools 3.2 software (Bruker Daltonics). The assignment of fragment ions was performed using BUPID Top Down (Boston University Protein Identifier Top Down), a custom-programmed software algorithm written in-house [44, 45], which can be used to assign sequence and mass information to top-down T3 sequencing data. Fragment ion isotope patterns were calculated using the Protein Prospector MS-isotope tool [46].
Safety Considerations
1,5-DAN is a known carcinogen and appropriate precautions should be taken to avoid accidental exposure.
Results and Discussion
The goal of this work is to explore the major practical considerations that could enable the application of MALDI-ISD MS to the screening and sequencing of hemoglobin variants. For this purpose, we investigated the effect of matrix selection and experimental conditions on the fragmentation of hemoglobin alpha and beta chains to evaluate the feasibility of MALDI-ISD MS for application to clinical diagnosis of hemoglobinopathies. After the methodology was developed using wild-type hemoglobin, representative alpha and beta chain variants were analyzed to demonstrate the applicability of the method. For the purpose of this feasibility study, we focused on two clinically relevant beta chain variants: Hb S (β6 Glu→Val), Hb C (β6 Glu→Lys), and a representative alpha chain variant Hb Westmead (α122His→Gln) [47]. MS characteristics of these samples including mass of intact protein and subsequent masses and assignments of tentative diagnostic ions are summarized in Table 2. We implemented an experimental scheme developed in our laboratory for data acquisition and interpretation based upon our experience using top-down MS/MS for the characterization of hemoglobin and transthyretin variants [31]; this is visualized in the Scheme 1. After measuring an intact protein mass and determining if a mass difference between normal and variant forms exists, initial sequence information can be obtained. Following a database search or pattern mass mapping of the peaks against known values for initial variant localization, select ions may be further sequenced to pinpoint the exact location of the amino acid substitution. In the case of ESI top-down we used nozzle skimmer dissociation followed by MS/MS. Here we explore MALDI ISD followed by T3 sequencing. In all instances of data analysis, it is possible to compare MS and MS/MS data with library spectra, or in the case of heterozygotes with ions in the same mass spectra, to match m/z values, peak intensities, and mass differences between normal and variant amino acids.

Generalized scheme for analysis of variant hemoglobins by MS. Screening may be performed with MALDI-ISD. T3 sequencing provides the opportunity for more detailed confirmatory analysis
MALDI-ISD MS requires 10–50 pmol of relatively pure sample. This requirement makes hemoglobins ideal candidates for MALDI-ISD MS analysis, given the approximately 8 mM concentration of the proteins in blood, and the required concentration for MALDI-ISD MS analysis can be achieved by simple dilution of a few microliters of whole blood. A dilution step also has the advantage of diminishing the concentration of less abundant proteins, further decreasing the possibility that proteins other than hemoglobin will interfere with the analysis. MALDI-ISD MS is tolerant of contaminants and offers the advantages of MALDI, such as speed and sensitivity, as well as ease of sample preparation. Furthermore, MALDI ISD MS offers rapid and extensive sequence coverage of proteins and peptides in a single experiment [48].
MALDI-ISD MS of Wild-Type Human Hemoglobin
The MALDI-ISD MS data obtained from normal hemoglobin with sDHB matrix exhibited rich fragmentation (m/z 600–8500), as well as some abundant higher mass peaks corresponding to the doubly and triply charged ions of the intact alpha [M + 2H]2+ (m/z 7564.0) and beta [M + 2H]2+ (m/z 7934.5) chains (Figure 1) that provided very helpful molecular weight profile information in addition to the N- and C-terminal sequence information. The m/z values of the doubly and triply charged ions of the intact alpha and beta chains are given as average mass values as these peaks were not isotopically resolved. The measured molecular weight of the alpha chain (MW 15126.0, calc. MW 15126.4) and the beta chain (MW 15867.0, calc. MW 15867.2) obtained from the observed average mass of the doubly charged ions of the intact protein are in agreement with their calculated values. The triply charged ions of the intact alpha chain [M + 3H]3+ (m/z 5043.5) and beta chain [M + 3H]3+ (m/z 5290.0) were also observed. The molecular weight profile offers valuable information for the detection and characterization of Hb variants since the value of the mass shift limits the number of possible amino acid substitutions to be considered and also guides the analysis of the fragmentation data. It is important to note that no interference from other whole blood components was observed.

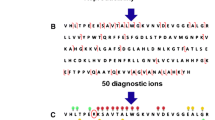
MALDI-ISD mass spectrum of wild-type hemoglobin obtained from diluted whole blood in sDHB matrix labeled with c-ion assignments for the beta chain. Full ion assignments are shown in Supplementary Figures 1 and 2. Inset is an expanded view of m/z 3550–3620 showing the βc34 fragment ion at m/z 3584.98. Greater than 80% sequence coverage was obtained using BioTools 3.2 and BUPID Top-Down software. Sequence coverage is illustrated for the alpha and beta chains shown below. Assignments correspond to values shown in Supplementary Table 1. An asterisk (*) indicates matrix adducts
The MALDI-ISD MS fragment ion spectrum obtained from whole blood in sDHB featured abundant product ions as presented in Figure 1; for clarity, only c-ions are shown for the beta chain. Complete ion series assignments for the alpha and beta chains are provided in Supplementary Figures 1 and 2 and in Supplementary Table 1. We applied a combination of methods for the initial ion assignments using BioTools 3.2 peak fitting software, BUPID Top-Down software, and manual confirmation with theoretical ions. In general, ions could be assigned with confidence with an average absolute mass error of 0.05 Da ± 0.05 Da (range 0 ± 0.35 Da). Although both y- and (z + 2)-ion series were present in the MALDI-ISD mass spectrum of normal hemoglobin obtained in sDHB, the signals from the y-ion series were generally more intense and the coverage was more extensive. The c-ion series were sometimes accompanied by weaker series of a-ions. The a- and (z + 2)-ions can help distinguish N- and C-terminal related fragmentation. The N-terminal c- and a-ions are 45 Da apart and the C-terminal y- and (z + 2)-ions are separated by15 Da. Assignment of the fragment ions observed for the alpha and beta chains provided extensive sequence coverage for both the alpha and beta chains (Figure 1). The coverage extends approximately 70 residues from the N- and C-termini for both proteins. A total of 389 fragment ions were assigned to both chains: 180 for the beta chain (Supplementary Figure 1) and 209 for the alpha chain (Supplementary Figure 2). In a manner analogous to ECD, some gaps appear because of the presence of proline, whose cyclic structure directs the type of fragmentation that gives rise to c- and (z + 2) ions [49, 50]. The expected complexity resulting from the presence of two different proteins did not appear to significantly hinder data interpretation, although some alpha and beta chain fragment ion mass values overlapped because of the presence of isobaric peaks (Supplementary Table 2). From the 389 peak assignments, a total of 13 potential isobaric peaks were observed: four pairs of fragment ion assignments could be assigned to the beta chain, six could be assigned to the alpha chain, and three could be co-assigned to either the alpha or beta chain. The occurrence of overlap can be recognized by paying close attention to the isotope pattern expected from the fragment ion of interest. Although the coverage of the hemoglobin chains is not complete, this is not necessarily a limitation, as the majority of clinically significant hemoglobin variants have the modified amino acid within the readily accessible regions of the sequence [26].
Consistently, the relative abundances of the fragment ions of the beta chain appeared to be lower than those of the alpha chain. This is in agreement with the results of MALDI-ISD MS as applied to mixtures of proteins in the context of MALDI-MS tissue imaging al [41]. Using protein standards of varying molecular weights, this study revealed a high sensitivity to protein ionization efficiency. In the case of hemoglobins, the alpha chain exhibits higher ionization efficiency than the beta chain. For hemoglobins, this behavior is particularly marked in electrospray/nanospray ionization where the beta chain’s ionization efficiency can be less than a third that of the alpha chain. For the MALDI-MS experiments reported herein, the relative intensity of signals from the intact beta chain relative to the alpha chain was about 50%. This difference did not affect the quality of the fragment ion series or the number of assignments obtained from the beta chain.
MALDI-ISD MS with T3 Sequencing
A key feature in the MALDI-ISD mass spectra obtained from diluted whole blood is a fragment ion observed at m/z 3584.98. This fragment was assigned as βc34 (calc. m/z 3584.98). The importance of this fragment ion lies in that it occurs in a region of the MALDI-ISD mass spectrum (Figure 1 inset) that is relatively free of other fragment ions (m/z 3560–3620) and describes the N-terminus of the beta chain where the sequence variations of the most common clinically relevant hemoglobin variants reside, as noted above. This suggests that the βc34 fragment ion could be useful as a diagnostic ion in the detection of these beta chain N-terminal variants. It should be noted that the experimental conditions do not preclude the possibility that an isobaric ion from β(z + 2)33 (calc. m/z 3584.89) or from αc34 (calc. m/z 3585.86) could contribute to the signal from the βc34 fragment ion. We observed that the calculated isotope distribution of the molecular composition of βc34 is slightly different from that observed in the MALDI-ISD mass spectrum shown in Figure 1 inset; a direct comparison is shown in Supplementary Figure 3. The difference in peak intensities suggests that αc34 is present as an overlapping isotopic cluster. However, this contribution of overlapping fragment ions to βc34 is minimal. In order to further probe the composition of βc34, T3 sequencing was performed to obtain sequence information on this primary fragment. The resulting secondary fragment ion mass spectrum (Figure 2) exhibits extensive product ions consistent with the sequence of βc34, representing sequence coverage of >65%. Therefore, whilst the fragment ion cluster assigned as βc34 in the MALDI-ISD mass spectrum of hemoglobin overlaps with a low abundance signal from αc34, the βc34 fragment is still a useful diagnostic ion for the detection of clinically relevant hemoglobin variants residing in the N-terminal region of the beta chain.

T3 sequencing fragment ion mass spectrum obtained from the m/z 3584.98 precursor generated from MALDI-ISD MS of hemoglobin obtained from diluted whole blood in 1,5-DAN matrix describing the fragmentation and sequencing of βc34. Numbering of the y-ion series considers residue 34 of the intact protein sequence as the peptide C-terminus. Greater than 64% sequence coverage was obtained. Ions were assigned using BioTools 3.2 software
Use of 1,5-DAN as a Matrix for MALDI-ISD MS
We also explored MALDI-ISD MS analysis for hemoglobin using 1,5-DAN as the matrix. The use of 1,5-DAN as the preferred matrix for MALDI-ISD MS has been advocated based on its ability to reduce disulfide bonds and to generate more intense c-ion and (z + 2)-ion signals than does DHB [40, 48, 51]. We noted a few significant differences between the MALDI-ISD mass spectra of hemoglobins recorded with 1,5-DAN and sDHB matrices. First, the MALDI-ISD mass spectra obtained with 1,5-DAN do not exhibit doubly charged intact protein ions and, hence, lack the molecular weight information that might reveal the presence of a variant. Second, since 1,5-DAN generally favors N-terminal fragmentation, it generates somewhat simpler ISD spectra than does sDHB (Figure 3). Consequently, we found that the abundances of fragment ions representing the C-terminus of both the alpha and beta chains were significantly diminished in comparison to their N-terminal counterparts compared with the ratio of these fragment types observed with sDHB. MALDI-ISD mass spectra obtained in sDHB thus had a more complex appearance, with abundant c-ions, y-ions, and (z + 2)-ions, and thereby more complete sequence coverage for both the alpha and beta chains. In the mass spectra obtained with 1,5-DAN, (z + 2)-ions tended to be the dominant type of C-terminal fragments, whereas y-ions were observed to be more abundant when using sDHB as the matrix. These observations are in agreement with previously reported results concerning the comparison of MALDI-ISD mass spectra of proteins in 1,5-DAN to spectra recorded for samples in DHB [40], but the literature relevant to these phenomena is still very limited. It should be noted that 1,5-DAN is an aminonapthalene derivative and a potent carcinogen. A further drawback is its instability in solution and its unfavorable sublimation properties that can cause significant ion source contamination. The latter consideration must be taken into account given the large number of laser shots necessary to generate MALDI-ISD mass spectra, and is especially relevant to selection of proper safety precautions during ion source cleaning. One of the potential advantages of using 1,5-DAN instead of sDHB is that the potential for higher fragment ion yields may facilitate the process of obtaining T3 sequencing mass spectra from ISD fragment ions (e.g., βc34 fragment ion), which can yield sequence information through T3 sequencing [42]. The option of performing T3 sequencing on the variant-containing fragment ion is potentially very useful. Although this process is not yet straightforward enough that it may be considered as part of a high throughput clinical screening assay, it may have an application for confirmation of putative variants following first pass MALDI-ISD MS screening. As the results obtained in Figure 2 suggest, the sequence coverage may not be sufficient to confirm unequivocally the presence of common variants such as Hb S, Hb C, and Hb E. Future improvements in instrumentation, matrices, and methodology could facilitate implementation of routine T3 sequencing analysis of variant-containing ISD fragment ions.

Expanded view of m/z 3100–3750 from the MALDI-ISD mass spectrum of wild-type hemoglobin obtained from diluted whole blood in (a) sDHB and (b) 1,5-DAN matrix. Dominant N-terminal defining c-ions can be observed by MALDI-ISD MS obtained with 1,5-DAN. The MALSI-ISD mass spectrum obtained with sDHB displays abundant fragmentation from both termini
Analyses of Variant Hemoglobins from a Select Group of Patient Samples
The combination of intact protein molecular weight and extensive sequence information obtained in a single MALDI-ISD MS experiment could be tremendously advantageous for high throughput screening for hemoglobin variant determination. In order to test the suitability of using MALDI-ISD MS for screening and determination of hemoglobin variants, we analyzed a pilot set of patient samples obtained from the Hemoglobin Diagnostic Reference Laboratory at the Boston University School of Medicine. These included normal hemoglobin and known clinically relevant variants. The MALDI-ISD MS analysis of known Hb variants affords the opportunity to test the methodology and validate the ion assignments made on the basis of their calculated monoisotopic mass values. As noted above, assignment of ion identities strictly on the basis of mass values can be difficult since some alpha and beta chain fragment ion masses may be isobaric or may have overlapping isotopic clusters that create complex patterns whereby it becomes difficult to extract the monoisotopic masses of the contributing fragments. Additionally, higher mass fragment ions may not exhibit well resolved isotope patterns because of the presence of isotopomers with low signal-to-noise values and, thus, the determination of monoisotopic mass becomes more difficult. In the case of a heterozygous variant, the presence of a variant fragment ion exhibiting the mass shift consistent with the amino acid substitution represents a secondary and important validation of the fragment ion assignments.
As a first example, we performed MALDI-ISD MS on a heterozygous sickle cell sample, Hb S (β6 Glu→Val). The variant type had already been determined by gene sequencing. The MALDI-ISD mass spectrum (sDHB matrix) showed the doubly charged ion typically observed for the beta chain wild-type ([M + 2H]2+ m/z 7934.5) as well as the variant protein ([M + 2H]2+ m/z 7919.6) (Figure 4). The observed mass difference (–29.9 Da) is consistent with the –30.0 Da calculated mass shift resulting from the Glu→Val amino acid substitution. For the resultant fragment ions, the observation of a monoisotopic mass shift of –29.97 Da in the sickle cell MALDI ISD mass spectrum, relative to the peak assigned to a normal beta chain ion, would confirm the validity of the assignment. Furthermore, a decrease in the relative abundance of the assigned “normal” beta chain ions should be observed, given the heterozygous nature of the sample.

MALDI-ISD mass spectra of diluted whole blood. Expanded view of the range m/z 7890–7970 for (a) wild-type hemoglobin and (b) sickle cell hemoglobin, prepared with sDHB matrix. The doubly charged ions of the intact wild-type beta chain (m/z 7934.5) and sickle cell (β6 Glu→Val) variant (m/z 7919.6) can be observed. The average mass difference of –29.9 Da is consistent with a Val→Glu substitution (asterisks indicate peaks corresponding to prompt water loss and/or ammonia loss from the protein). Expanded view of the range m/z 3550–3600 for (c) wild-type hemoglobin in sDHB matrix, (d) sickle cell (β6 Glu→Val) heterozygote hemoglobin in sDHB matrix, and (e) sickle cell (β6 Glu→Val) heterozygote hemoglobin in 1,5-DAN matrix. The mass differences observed (29.94 Da and 29.96 Da) between the wild-type and sickle cell (β6 Glu→Val) βc34 fragment ions in (c) and (d), respectively, are consistent with a Val→Glu substitution (–29.97 Da)
The analysis of MALDI-ISD MS data obtained with a known beta chain hemoglobin variant affords the opportunity of confirmng the assignment of the fragment ion originally assigned as βc34 in the MALDI-ISD mass spectrum of wild-type hemoglobin as well as confirming the potential of using this precursor as a diagnostic fragment ion. The presence of a fragment ion corresponding to the Glu→Val variant at m/z 3555.04 (calc. m/z 3555.01) (Figure 4d) is accompanied by a decrease in the abundance of the peak at m/z 3584.98 relative to a wild-type sample. The observed –29.94 Da mass difference is consistent with the Glu→Val substitution. The ratio of the abundance of the sickle cell βc34 fragment ion to that of the wild-type βc34 is lower than that observed for the doubly charged ions corresponding to the intact beta chains. This further confirms our original observation that other fragment ions overlap with the wild-type βc34 ion. Inspection of the isotope patterns of the variant and wild-type βc34 reveals a difference in isotope distribution. A comparison of these isotopic distributions with those calculated on the basis of the molecular formulas of the respective fragment ions shows a close match for the sickle cell βc34. This was not the case for the normal βc34 fragment ion (m/z 3584.98) as shown before (Figure 1 inset). Beta chain c-ions with a –29.97 Da mass shift not present in the normal hemoglobin MALDI-ISD mass spectrum include a series of peaks indicating the presence of a sickle cell variant can be observed down to βc9 (m/z 1038.56).
A potential challenge is that data interpretation can be complicated by the overlap of variant-related fragment ions with other ions. Also, nonlinear calibration of low m/z values and interference due to the presence of matrix ions may add to the difficulty of observing informative fragment ions below m/z 1000. The data shown here demonstrate the characterization of the sickle cell variant given that no other known beta chain amino acid substitution can give rise to a –30 Da nominal mass shift in the first nine positions relative to the N-terminus. We show that the c-ion series assignments of the beta chain can be validated by looking for a variant-characteristic mass shift in relationship to the wild-type fragment ion. This is very useful in cases where isobaric fragment ions cannot be distinguished. For example, a peak was observed at m/z 6948.65 in the MALDI-ISD mass spectrum of normal hemoglobin. The peak has low abundance and was not well resolved isotopically. It could reasonably be assigned as a βc64 (calc. m/z 6948.60) fragment ion or a β(z + 2)63 (calc. m/z 6948.59) fragment ion (Supplementary Figure 4). The presence of a –30.04 Da peak in the mass spectrum obtained from the sickle cell sample, along with an observed decrease in the relative abundance of m/z 6948.65, confirmed the assignment of this fragment ion as βc64. This does not rule out the presence of some contribution by β(z + 2)63 but firmly establishes the βc64 assignment. A similar example may be observed in the MALDI-ISD mass spectrum of wild-type hemoglobin for the peak at m/z 4710.48, where the assignment may be αa44 (calc. m/z 4710.42) or βc42 (calc. m/z 4710.52) (Supplementary Figure 5). The presence of a –29.89 Da peak in the sickle cell sample, along with an observed decrease in the relative abundance of the peak at m/z 4710.48, confirmed the assignment of this fragment ion as βc42. Again, the possibility that αa44 is present cannot be discounted but the main component at m/z 4710.48 may confidently be assigned as βc42.
A significant challenge of using mass spectrometry for variant characterization involves the class of variants that differ by mass by only ± 1 Da. Under the conditions used in the present study, identification of ± 1 Da mass changes in the intact hemoglobin molecular weight profile could be considered difficult. However, the MALDI-ISD MS experiments performed here readily allow the detection of variants such as Hb C (β6 Glu→Lys) and Hb E (β26 Glu→Lys). The fragment ions resulting from the ISD process yield information permitting the assignment of these variants, since ISD fragment ions exhibiting a –0.95 Da mass shift can be observed with higher mass accuracy and sensitivity than the [M + 2H]2+ precursors. Here we demonstrate this with the example of an analysis of hemoglobin from a compound heterozygous patient expressing Hb C (β6 Glu→Lys) and Hb S (β6 Glu→Val) (HbSC). In the higher mass region of the MALDI-ISD mass spectrum (not shown), a peak corresponding to the intact beta chain sickle cell variant can be observed, [M + 2H]2+ m/z 7920.0, as well as a doubly charged ion, [M + 2H] 2+ m/z 7934.5, consistent with the molecular weight of wild-type beta chain. However, the measured mass difference is –29.0 Da, suggesting the possibility of Hb SC since no single amino acid substitution may result in a –29.0 Da mass shift. Examination of the beta chain ISD mass spectrum starting with the diagnostic fragment ion βc34 reveals that the peak cluster is indeed shifted by –0.92 Da (calculated Δm Glu→Lys = 0.95 Da), with the monoisotopic peak being located at m/z 3584.06 (Figure 5b). This observation, in conjunction with the presence of a βc34-29.94 Da fragment ion at m/z 3555.05, strongly indicates the possibility of Hb SC. Again, as in the case of the heterozygous sickle cell sample discussed above, the –0.95 Da mass difference can be observed down to βc9. Although there are several known variants in the first nine positions of the beta chain N-terminus that can give rise to a –0.95 Da mass shift, their occurrences are very rare. Thus, MALDI-ISD MS of diluted whole blood could be used to detect Hb S, Hb C, and Hb E on the basis of the βc34 fragment ion’s indication of the presence of these clinically important variants.

Expanded view of m/z 3550–3600 from the MALDI-ISD mass spectra of hemoglobin obtained from diluted whole blood prepared with sDHB matrix (a) wild-type hemoglobin and (b) heterozygote variant hemoglobin SC (β6 Glu→Val and β6 Glu→Lys). The observed –0.92 Da mass shift in the isotope pattern of βc34 is consistent with the presence of a Glu→Lys substitution (–0.945 Da). The mass of the fragment ion m/z 3555.04 corresponds to the sickle cell (β6 Glu→Val) βc34 ion
Lastly, we extended our approach to the characterization of an alpha chain variant, Hb Westmead (α122His→Gln)47, a variant that is common in Guangxi province in southern China. The region of the MALDI-ISD mass spectrum containing the doubly charged ion corresponding to the intact alpha chain did not clearly indicate the presence of a variant (Figure 6a and b). This is due to the low abundance of the variant, which has been reported to be mildly unstable [47]. We observed this decrease in signal intensity of the variant chain in a previous ESI-MS analysis performed using ESI-MS on a Bruker Solarix FTMS (Supplementary Figure 6). Comparison of the variant spectrum with that of a wild-type sample indicates the presence of an unknown low abundance component between the doubly charged ion of normal alpha chain and the peak corresponding to prompt water or ammonia loss from the protein. Inspection of the ISD fragmentation pertaining to the alpha chain led to the observation of a low abundance series of peaks exhibiting a shift of –9.02 Da at m/z 2481.38 (versus wt αy23 m/z 2490.42, Figures 6c and 6d) and m/z 2929.59 (versus wt αy27 m/z 2938.62). These data are sufficient to characterize the variant as (α122 His→Gln) given that the –9.00 Da mass shift is unique to this amino acid substitution and there is only one histidine in the alpha chain C-terminal sequence defined by αy23 and αy27. Therefore, alpha chain variants can also be detected and, as in this case, characterized in a rapid and facile experiment.

MALDI-ISD mass spectra of hemoglobin obtained from diluted whole blood prepared using sDHB matrix. Expanded view of the region m/z 7540–7590 for (a) wild-type hemoglobin and (b) Hb Westmead (α122 His→Gln) heterozygote variant hemoglobin: comparison of the variant spectrum with that of the wild-type suggests the presence of an unknown low abundance component between the doubly charged ion of wild-type alpha chain and the peak corresponding to prompt water or ammonia loss from the protein. Expanded view of the region m/z 2477–2500 for (c) wild-type hemoglobin and (d) Hb Westmead: the wild-type and variant αy23 fragment ions were observed at m/z 2490.40 and m/z 2481.38 and show a mass shift of 9.02 Da consistent with a His→Gln substitution (9.00 Da)
Although we did not yet analyze C-terminal beta chain variants, the MALDI-ISD MS data accumulated in our work strongly indicate the possibility of finding a diagnostic fragment ion for such variants. In a manner similar to using the fragment ion βc34 to describe the beta chain N-terminus, MALDI-ISD MS analysis could be applied to the detection of Hb D-Los Angeles (β121 Glu→Gln) beta chain mutation by using the βy28 and βy34 fragment ions to describe the beta chain C-terminus. The βy28 (m/z 2994.63) and βy34 (m/z 3699.02) fragment ions cover positions 119-146 and 113-146, respectively (Supplementary Figure 7).
Conclusion
Significant advances have been made over the past decade in the application of mass spectrometry to the identification and characterization of hemoglobin variants. Approaches using chromatography and bottom-up peptide mass mapping/sequencing are powerful, yet require additional sample processing that reduces their suitability for high-throughput analyses. The top-down approach obviates these requirements and shows great promise as the next step in this area of research. While top-down ESI-MS and MS/MS may afford accurate mass and sequence information, significantly more stringency is still required in terms of sample preparation and handling, and the analysis time for each sample in the order of minutes. Here we demonstrate that MALDI-ISD MS of diluted whole blood readily provides molecular weight and ISD sequence information for both alpha and beta hemoglobin chains, enabling the precise and confident detection of clinically relevant variants. MALDI-ISD MS of hemoglobins provides results as informative as those obtained from a combination of molecular weight profiling and peptide mass mapping through tryptic digestion, but requires far less time and can easily be adapted to high-throughput protocols. Direct analyses of hemoglobins from diluted blood samples are well suited for MALDI-ISD as preparation of samples requires only simple dilution of whole blood; the method is tolerant to contaminants that greatly hinder the application of electrospray-based top-down techniques, and interpretation of the results is not hampered by the presence of signals from other blood components. While not highlighted, the speed of acquisition can be less than a minute per sample for MS and ISD and could be reduced to a few seconds per sample: a speed that would be ideal for screening.
Our results show that extensive sequence coverage may be obtained using MALDI-ISD MS in a single rapid experiment. Sequence coverage is greater than 80% for the alpha and beta chains and comprises approximately 70 residues that constitute the N-termini and C-termini of both the alpha and beta chains. When sDHB is used as the matrix, intact globin masses and fragment ion series are obtained simultaneously in a rapid and facile experiment. Mass shifts and changes in peak intensities relative to the peaks assigned as a normal globin chain confirm and validate peak assignments. We have applied MALDI-ISD MS successfully to the identification of clinically relevant hemoglobin variants present in patient samples. These included heterozygote Hb S (β6 Glu→Val); compound heterozygote Hb C (β6 Glu→Lys)/Hb S (β6 Glu→Val) (Hb SC); and heterozygote Hb Westmead (α122His→Gln). In all cases, the variant could be assigned by MALDI ISD-MS. Using T3 sequencing, we demonstrate that it is possible to obtain significant additional information and sequence coverage, complementing the initial MS and ISD data. Althugh the routine use of T3 sequencing is demanding at this time and might preclude its application to high throughput initial screening, if a putative variant is identified first by MALDI-ISD MS, T3 sequencing would offer a powerful means for further sequence analysis and confirmation. We note that future improvements in instrumentation and methodology should increase the yield of ISD fragment ions, provide greater sequence coverage, allow faster acquisition, and facilitate more routine application of T3 sequencing analysis to variant-containing ISD fragment ions.
Although MALDI-ISD mass spectral analysis of hemoglobins results in spectra with the potential for isobaric overlap of fragment ions and may yield incomplete sequence coverage, we propose that the combination of the molecular weight information provided by the intact alpha and beta chain doubly charged ions, primary sequence information provided by MALDI ISD-MS, comparative analysis of variants with respect to normal control samples and subsequent T3 sequence information obtained from select precursors (e.g., the βc34 fragment ion) provide straightforward diagnostic indicators of the presence of a variant and, thus, could be used as a screening method for clinically relevant Hb variants: Hb S, Hb C, and Hb E.
References
Available at: http://globin.cse.psu.edu. Accessed Apr 1 2015
Watson, M.S., Mann, M.Y., Lloyd-Puryear, M.A., Rinaldo, P., Howell, R.R.: Newborn screening: toward a uniform screening panel and system—executive summary. Pediatrics 117, S296–S307 (2006)
Weatherall, D.J., Clegg, J.B.: Inherited haemoglobin disorders: an increasing global health problem. Bull. World Health Organ. 79, 704–712 (2001)
Giardine, B., Borg, J., Viennas, E., Pavlidis, C., Moradkhani, K., Joly, P., Bartsakoulia, M., Riemer, C., Miller, W., Tzimas, G., Wajcman, H., Hardison, R.C., Patrinos, G.P.: Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 42, D1063–D1069 (2014)
Hoppe, C.C.: Newborn screening for hemoglobin disorders. Hemoglobin 35, 556–564 (2011)
Piel, F.B., Patil, A.P., Howes, R.E., Nyangiri, O.A., Gething, P.W., Dewi, M., Temperley, W.H., Williams, T.N., Weatherall, D.J., Hay, S.I.: Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381, 142–151 (2013)
Wild, B.J., Bain, B.J.: Detection and quantitation of normal and variant haemoglobins: an analytical review. Ann. Clin. Biochem. 41, 355–369 (2004)
Joutovsky, A., Hadzi-Nesic, J., Nardi, M.A.: HPLC retention time as a diagnostic tool for hemoglobin variants and hemoglobinopathies: a study of 60,000 samples in a clinical diagnostic laboratory. Clin. Chem. 50, 1736–1747 (2004)
Trent, R.J.A.: Diagnosis of the haemoglobinopathies. Clin. Biochem. Rev. 27, 27–38 (2006)
Zanella-Cleon, I., Joly, P., Becchi, M., Francina, A.: Phenotype determination of hemoglobinopathies by mass spectrometry. Clin. Biochem. 42, 1807–1817 (2009)
Wada, Y., Hayashi, A., Fujita, T., Matsuo, T., Katakuse, I., Matsuda, H.: Structural analysis of human hemoglobin variants with field desorption mass spectrometry. Biochim. Biophys. Acta 667, 233–241 (1981)
Pucci, P., Carestia, C., Fioretti, G., Mastrobuoni, A.M., Pagano, L.: Protein fingerprinting by fast atom bombardment mass spectrometry-characterization of normal and variant human hemoglobins. Biochem. Biophys. Res. Commun. 130, 84–90 (1985)
Wada, Y., Matsuo, T., Papayannopoulos, I.A., Costello, C.E., Biemann, K.: Fast-atom-bombardment and tandem mass spectrometry for the characterizarion of hemoglobin variants including a new variant. Int. J. Mass Spectrom. Ion Process 122, 219–229 (1992)
Shackleton, C.H.L., Falick, A.M., Green, B.N., Witkowska, H.E.: Electrospray mass spectrometry in the clinical diagnosis of variant hemoglobins. J. Chromatogr. B Biomed. Appl. 562, 175–190 (1991)
Lightwahl, K.J., Loo, J.A., Edmonds, C.G., Smith, R.D., Witkowska, H.E., Shackleton, C.H.L., Wu, C.S.C.: Collisonally activated dissociation and tandem mass spectrometry of intact hemoglobin beta chain variant proteins with electrospray ionization. Biol. Mass Spectrom. 22, 112–120 (1993)
Houston, C.T., Reilly, J.P.: Rapid analysis of hemoglobin from whole human blood by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1435–1439 (1997)
Houston, C.T., Reilly, J.P.: Toward a simple, expedient, and complete analysis of human hemoglobin by MALDI-TOFMS. Anal. Chem. 71, 3397–3404 (1999)
McComb, M.E., Oleschuk, R.D., Chow, A., Ens, W., Standing, K.G., Perreault, H., Smith, M.: Characterization of hemoglobin variants by MALDI-TOF MS using a polyurethane membrane as the sample support. Anal. Chem. 70, 5142–5149 (1998)
Kiernan, U.A., Black, J.A., Williams, P., Nelson, R.W.: High-throughput analysis of hemoglobin from neonates using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chem. 48, 947–949 (2002)
Hachani, J., Duban-Deweer, S., Pottiez, G., Renom, G., Flahaut, C., Perini, J.M.: MALDI-TOF MS profiling as the first-tier screen for sickle cell disease in neonates: matching throughput to objectives. Proteomics Clin. Appl. 5, 405–414 (2011)
Wild, B.J., Green, B.N., Cooper, E.K., Lalloz, M.R.A., Erten, S., Stephens, A.D., Layton, D.M.: Rapid identification of hemoglobin variants by electrospray ionization mass spectrometry. Blood Cells Mol. Dis. 27, 691–704 (2001)
Gatlin, C.L., Eng, J.K., Cross, S.T., Detter, J.C., Yates, J.R.: Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Anal. Chem. 72, 757–763 (2000)
Basilico, F., Di Silvestre, D., Sedini, S., Petretto, A., Levreri, I., Melioli, G., Farina, C., Mori, F., Mauri, P.L.: New approach for rapid detection of known hemoglobin variants using LC-MS/MS combined with a peptide database. J. Mass Spectrom. 42, 288–292 (2007)
Das, R., Mitra, G., Mathew, B., Ross, C., Bhat, V., Mandal, A.K.: Automated analysis of hemoglobin variants using nanolc-ms and customized databases. J. Proteome Res. 12, 3215–3322 (2013)
Aebersold, R., Mann, M.: Mass spectrometry-based proteomics. Nature 422, 198–207 (2003)
Daniel, Y.A., Turner, C., Haynes, R.M., Hunt, B.J., Neil Dalton, R.: Rapid and specific detection of clinically significant haemoglobinopathies using electrospray mass spectrometry-mass spectrometry. Br. J. Haematol. 130, 635–643 (2005)
Siuti, N., Kelleher, N.L.: Decoding protein modifications using top-down mass spectrometry. Nat. Methods 4, 817–821 (2007)
Catherman, A.D., Skinner, O.S., Kelleher, N.L.: Top down proteomics: facts and perspectives. Biochem. Biophys. Res. Commun. 445, 683–693 (2014)
Kelleher, N.L., Lin, H.Y., Valaskovic, G.A., Aaserud, D.J., Fridriksson, E.K., McLafferty, F.W.: Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 121, 806–812 (1999)
Witkowska, H.E., Green, B.N., Morris, M., Shackleton, C.H.: Intact protein electrospray ionization tandem mass spectrometry can be the sole technique used for confirming the structure of a variant hemoglobin. Rapid Commun. Mass Spectrom. Spec No, S111–S115 (1995)
Theberge, R., Infusini, G., Tong, W.W., McComb, M.E., Costello, C.E.: Top-down analysis of small plasma proteins using an LTQ-Orbitrap. Potential for mass spectrometry-based clinical. assays for transthyretin and hemoglobin. Int. J. Mass Spectrom. 300, 130–142 (2011)
Edwards, R.L., Griffiths, P., Bunch, J., Cooper, H.J.: Top-down proteomics and direct surface sampling of neonatal dried blood spots: diagnosis of unknown hemoglobin variants. J. Am. Soc. Mass Spectrom. 23, 1921–1930 (2012)
Brown, R.S., Lennon, J.J.: Sequence-specific fragmentation of matrix assisted laser-desorbed protein peptide ions. Anal. Chem. 67, 3990–3999 (1995)
Hardouin, J.: Protein sequence information by matrix-assisted laser desorption/ionization in-source decay mass spectrometry. Mass Spectrom. Rev. 26, 672–682 (2007)
Ng, E.W.Y., Wong, M.Y.M., Poon, T.C.W.: Advances in MALDI mass spectrometry in clinical diagnostic applications. Top. Curr. Chem. 336, 139–175 (2014)
Suckau, D., Resemann, A., Schuerenberg, M., Hufnagel, P., Franzen, J., Holle, A.: A novel MALDI LIFT-TOF/TOF mass spectrometer for proteomics. Anal. Bioanal. Chem. 376, 952–965 (2003)
Resemann, A., Wunderlich, D., Rothbauer, U., Warscheid, B., Leonhardt, H., Fuchser, J., Kuhlmann, K., Suckau, D.: Top-down de novo protein sequencing of a 13.6 kDa camelid single heavy chain antibody by matrix-assisted laser desorption ionization-time-of-flight/time-of-flight mass spectrometry. Anal. Chem. 82, 3283–3292 (2010)
Calligaris, D., Villard, C., Terras, L., Braguer, D., Verdier-Pinard, P., Lafitte, D.: MALDI in-source decay of high mass protein lsoforms: application to alpha- and beta-tubulin variants. Anal. Chem. 82, 6176–6184 (2010)
Kocher, T., Engstrom, A., Zubarev, R.A.: Fragmentation of peptides in MALDI in-source decay mediated by hydrogen radicals. Anal. Chem. 77, 172–177 (2005)
Demeure, K., Quinton, L., Gabelica, V., De Pauw, E.: Rational selection of the optimum MALDI matrix for top-down proteomics by in-source decay. Anal. Chem. 79, 8678–8685 (2007)
Zimmerman, T.A., Debois, D., Mazzucchelli, G., Bertrand, V., De Pauw-Gillet, M.C., De Pauw, E.: An analytical pipeline for MALDI in-source decay mass spectrometry imaging. Anal. Chem. 83, 6090–6097 (2011)
Suckau, D., Resemann, A.: T(3)-sequencing: targeted characterization of the N- and C-termini of undigested proteins by mass spectrometry. Anal. Chem. 75, 5817–5824 (2003)
Tsarbopoulos, A., Karas, M., Strupat, K., Pramanlk, B.N., Nagabhushan, T.L., Hillenkamp, F.: Comparative mapping of recombinant proteins and glycoproteins by plasma desorption and matrix assisted laser desorption/ionization mass spectrometry. Anal. Chem. 66, 2062–2070 (1994)
Tong, R.T.W., Infusini, G., Cui, W., Perlman, D.H., Lin, C., McComb, M.E., Costello, C.E.: BUPID-Top-Down: Database Search and Assignment of Top-Down MS/MS Data. Proceedings of the 57th American Society Conference on Mass Spectrometry and Allied Topics, Philadelphia, PA, 31 May–4 June 2009
Heckendorf, C., Theberge, R., Spencer, J.L., Costello, C.E., McComb, M.E.: Algorithm for Identification and Sequencing of Protein Variants Using Top-Down MS Data, Proceedings of the 61st American Society Conference on Mass Spectrometry and Allied Topics, Minneapolis, MN, 9–13 June 2013
Available at: http://prospector.ucsf.edu/prospector/mshome.htm. Accessed Apr 1 2015
Fleming, P.J., Hughes, W.G., Farmilo, R.K., Wyatt, K., Cooper, W.N.: Hemoglobin Westmead alpha -2 122 (H5)His-Gln beta-2 new hemoglobin variant with the substitution in the alpha-1-beta-1 contact area. Hemoglobin 4, 39–52 (1980)
Asakawa, D.: Principles of hydrogen radical mediated peptide/protein fragmentation during matrix-assisted laser desorption/ionization mass spectrometry. Mass. Spectrom. Rev. (2014). doi:10.1002/mas.21444
Zubarev, R.A., Kelleher, N.L., McLafferty, F.W.: Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120, 3265–3266 (1998)
Asakawa, D., Smargiasso, N., Quinton, L., De Pauw, E.: Influences of proline and cysteine residues on fragment yield in matrix-assisted laser desorption/ionization in-source decay mass spectrometry. J. Am. Soc. Mass Spectrom. 25, 1040–1048 (2014)
Fukuyama, Y., Iwamoto, S., Tanaka, K.: Rapid sequencing and disulfide mapping of peptides containing disulfide bonds by using 1,5-diaminonaphthalene as a reductive matrix. J. Mass Spectrom. 41, 191–201 (2006)
Acknowledgment
The authors acknowledge funding for this project by NIH-NHLBI Contract HHSN268201000031C and NIH grants P41 RR10888/GM104603 and S10 OD010724. The authors thank the members of the Center for Biomedical Mass Spectrometry for their contributions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Roger Théberge and Sergei Dikler contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 831 kb)
Supplemental Table 1
(PDF 75 kb)
Supplemental Table 2
(PDF 37.4 kb)
Rights and permissions
About this article

Cite this article
Théberge, R., Dikler, S., Heckendorf, C. et al. MALDI-ISD Mass Spectrometry Analysis of Hemoglobin Variants: a Top-Down Approach to the Characterization of Hemoglobinopathies. J. Am. Soc. Mass Spectrom. 26, 1299–1310 (2015). https://doi.org/10.1007/s13361-015-1164-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-015-1164-4