
Abstract
Electron transfer dissociation of peptide ions with the diazirine-containing residue photomethionine (M*) results in side-chain dissociations by loss of C3H7N2 radicals in addition to standard backbone cleavages. The side-chain dissociations are particularly prominent upon activation of long-lived, charge-reduced, cation radicals (GM*GGR + 2H)+●. Investigation of these cation radicals by near-UV photodissociation and collisional activation revealed different fragmentation products and mechanisms resulting from these ion activation modes. The dissociations observed for photomethionine were dramatically different from those previously reported for the lower homologue photoleucine; here, a difference by a single methylene group in the side chain had a large effect on the chemistries of the cation radicals upon ETD and further activation. ETD intermediates and products were probed by tandem 355-nm UV photodissociation-collision induced dissociation and found to contain chromophores that resulted from electron attachment to the diazirine ring. The nature of the newly formed chromophores and ion energetics and kinetics were investigated by electron structure calculations combining ab initio and density functional theory methods and Rice-Ramsperger-Kassel-Marcus (RRKM) theory. The dramatic difference between the dissociations of L* and M* containing peptide cation radicals is explained by electronic effects that play a role in stabilizing critical reaction intermediates and steer the dissociations into kinetically favored reaction channels. In addition, a new alternating UVPD-ETD-UVPD MS4 experiment is introduced and utilized for ion structure elucidation.

ᅟ
Similar content being viewed by others
Introduction
The diazirine-labeled amino acid residues photoleucine (L-2-amino-4,4-azipentanoic acid, L*) and photomethionine (L-2-amino-5,5-azihexanoic acid, M*) have been shown to be useful surrogates of the respective natural amino acids [1] that can be introduced into proteins and used for photochemical footprinting [2, 3]. Photodissociation of L* and M* is analogous to the general behavior of aliphatic diazirines in that it results in N2 elimination, creating highly reactive carbenes positioned in the amino acid side chains that undergo competing insertions into X–H bonds and isomerization to nonreactive olefins [4]. In our previous studies of photodissociation [5], collision-induced dissociation [6], and electron transfer dissociation [7, 8] of model peptide ions containing photoleucine, we discovered several unusual reactions involving the diazirine ring. In particular, ETD of L*-containing peptide ions (Scheme 1, n = 1) resulted in diazirine ring fission and losses of small molecules such as N2H4, N2H3, and [NH4O] that competed with backbone dissociations. The side-chain dissociations were interpreted as being triggered by electron attachment to the diazirine π*(N = N) orbital forming an anion radical. The diazirine ring was calculated to have a substantial adiabatic electron affinity. For diazirine-containing peptide ions in particular, electron attachment is accompanied by exothermic intramolecular proton transfer to the reduced diazirine anion-radical, forming a diaziridinyl radical, which triggers a cascade of radical-induced isomerizations resulting in a ring cleavage and dissociation (Scheme 1).

Reduction of diazirine ring in photolabeled peptide ions
Photomethionine (Scheme 1, n = 2) is a homologue of photoleucine from which it differs by a single methylene group in the side chain, placing the diazirine ring to a slightly remote position with respect to the peptide backbone. It was therefore surprising to find that ETD of an M*-containing peptide ion displayed completely different fragments than did ETD of its L*-containing analogues, most notable of which was the limited observation of N2H4, N2H3, and [NH4O] losses. We attribute these differences primarily to the presence of this extra methylene group in photomethionine. Here, we utilize computational and mass spectrometry methods to describe the effect of this subtle difference. Characteristically, these methods include MSn combinations of ETD with collision-induced dissociation (CID) and near-UV photodissociation (UVPD) to elucidate the electron-induced dissociations of the diazirine ring in a model peptide ion, (GM*GGR + 2H)2+.
Experimental
Materials
Photomethionine was purchased from Pierce Biotechnology (Rockford, IL, USA). All peptides were synthesized on Wang resin (Bachem Americas, Torrance, CA, USA) using the
Fmoc technology according to literature procedures [9]. Fmoc N-protected photomethionine were prepared according to the literature [10].
Methods
Electron transfer dissociation (ETD) mass spectra were measured on a modified LTQ-XL ETD linear ion trap (LIT) mass spectrometer (ThermoElectron Fisher, San Jose, CA, USA), which was equipped with an auxiliary chemical ionization (CI) source for the production of fluoranthene anion radicals. Peptide solutions (5–10 μM) in 50/50/1 methanol/water/acetic acid were electrosprayed at 2.2–2.3 kV from a pulled fused silica capillary into an open microspray ion source described previously [6, 7]. Doubly charged ions were selected according to their m/z, stored in the LIT, and then allowed to react with fluoranthene anions injected into the LIT. The ion–ion reaction times were typically varied between 100 and 300 ms. MSn experiments were carried out by isolating the fragment ions and exposing them to resonant collisional excitation or photoexcitation. High resolution ETD mass spectra were obtained on an LTQ-Orbitrap Fusion instrument at a 120,000 resolving power.
Photodissociation
To accomplish photodissociation of trapped ions in the LIT, the LTQ-XL ETD mass spectrometer was modified as described previously [5, 8]. The typical light intensity used in the photodissociation experiments was 15–20 mJ/pulse. The typical experimental time sequence of events consists of preparing by MSn ETD/CID an ion with a chromophore absorbing at the laser wavelength and storing it in the LIT for a chosen time period. The mass-selected and stored ions are photodissociated with a chosen number of laser pulses. For example, 400-ms storage time can accommodate up to seven laser pulses spaced by 50 ms. This allows one to vary the number of pulses and determine the photodissociation kinetics.
Calculations
All electron structure calculations were carried out with the Gaussian 09 suite of programs [11]. Structures of GM*GGR dication precursors were built by modifying the side chain in the lowest-energy conformers of GL*GGR homologues and then fully optimized by density functional theory B3LYP [12, 13] and M06-2X [14] calculations with the 6-31+G(d,p) basis set. The structures were characterized by harmonic frequency analysis. Additional energies were obtained by single-point B3LYP, M06-2X, ωB97XD [15, 16], and MP2 (frozen core) [17] calculations with the larger 6-311++G(2d,p) basis set. Cation-radical structures were obtained by gradient optimizations with B3LYP and M06-2X in the spin-unrestricted format (UB3LYP and UM06-2X) and the 6-31+G(d,p) basis set. Local minima were characterized by harmonic frequency analysis as having all real frequencies. Transition states were located by mapping the relevant parts of the potential energy surface and fully optimized to first-order saddle points that were characterized by frequency analysis as having one imaginary frequency. Single point energies were calculated for all cation-radical structures, as mentioned above. The UMP2 energies were corrected for contamination by higher spin states using the standard spin annihilation procedure [18, 19]. The spin-projected single-point MP2 energies (PMP2) were combined with B3LYP energies (B3-PMP2) to improve accuracy through cancellation of small errors inherent to both methods [20–22]. Excited state energies and oscillator strengths were calculated using time-dependent DFT theory [23] with the M06-2X, ωB97XD, and LC-BLYP [24] functionals and the 6-311++G(2d,p) basis set. Typically, the 15 lowest excited electronic states were analyzed. Unimolecular rate constants were calculated according to Rice-Ramsperger-Kassel-Marcus (RRKM) theory [25]. These RRKM calculations used a QCPE program [26] that was expanded to allow molecular systems with up to 1000 normal modes to be treated, and the QCPE Unix program was recompiled to be run under the Windows NT and Windows 7 operating systems [27]. Iterative analysis of complex kinetic systems was carried out by solving first order linear differential equations using the ode23 solver in Matlab version R2004b (Mathworks, Natick, MA, USA).
Results
Electron Transfer and Photodissociation Spectra
Electron transfer to (GM*GGR + 2H)2+ ions results in standard backbone as well as side-chain dissociations (Figure 1a). The backbone fragments are represented by the z 1 –z 3 , y 1 , and y 3 ions, as assigned in the Figure 1a mass spectrum and corroborated by accurate mass measurements (Table S1, Supplementary Data). In addition, the spectrum displayed an unusual side-chain fragment at m/z 415, which was due to loss of a C3H7N2 radical, as evidenced by accurate mass measurements (Δm = 71.0610 Da, C3H7N2 requires 71.0609 Da). The m/z 415 ion relative intensity amounted to 25% of combined relative intensities of z 1 –z 4 ions and the m/z 469 (loss of ammonia) fragment ion. The ETD spectrum of (GM*GGR + 2H)2+ displayed only very minor peaks of fragment ions at m/z 454 (loss of N2H4) and m/z 452 (loss of NH4O) that were both prominent in the ETD mass spectra of L*-containing peptide ions [7, 8]. Hence, a minor structure change of replacing L* with M* had a major effect on ETD. The origin of the C3H7N2 moiety was ascertained by a UVPD-ETD experiment. The diazirine ring is the only chromophore in (GM*GGR + 2H)2+ ions absorbing at 355 nm [5], so it could be selectively targeted by UVPD. Photon absorption resulted in elimination of N2 from the diazirine ring to form the m/z 229 photoproduct (Figure 1b), which partly dissociated to y 4 2+ and y 3 backbone fragment ions (Figure 1b). The stable m/z 229 ion, (G(M* − N2)GGR + 2H)2+, was mass-selected and subjected to ETD. The ETD mass spectrum of this photodissociation product (Figure 1c) showed a complete series of backbone z ions at m/z 159, 216, 273, and 384, along with the complementary c 4 fragment ion at m/z 300. In contrast, there was no m/z 387 fragment ion suggesting loss of 71 Da that could be assigned to C3H7N2. The fact that selective photodissociative removal of the diazirine ring resulted in a complete suppression of the C3H7N2 loss upon ETD provided convincing evidence that this dissociation was associated with the M* side chain.

(a) ETD mass spectrum of (GM*GGR + 2H)2+ at m/z 243. (b) UVPD mass spectrum of (GM*GGR + 2H)2+. Inset shows the enlarged part of the spectrum. (c) UVPD-ETD MS3 spectrum of [G(M* – N2)GGR + 2H]2+ at m/z 229
The Figure 1a and c ETD spectra further showed some differences attributable to the photochemical detachment of the diazirine ring. The Figure 1a spectrum showed a very minor charge-reduced (GM*GGR + 2H)+● ion at m/z 486, whereas the corresponding [G(M* − N2)GGR + 2H]+● ion at m/z 458 was quite abundant in the Figure 1c ETD spectrum. Other major differences were due to the fragment ion formed by loss of ammonia and the z 4 ion, which were weak in the Figure 1a spectrum (m/z 469 and m/z 412, respectively) while strong in the Figure 1c spectrum (m/z 441 and m/z 384, respectively).
The low abundance of the long-lived m/z 486 (GM*GGR + 2H)+● cation radical in the Figure 1a ETD spectrum was an impediment to further experimental studies of this ion’s structure and electronic properties. To further elucidate the nature of the m/z 486 ion, we employed our previously introduced method of selective cation radical preparation using noncovalent complexes with 18-crown-6-ether (CE) [7, 8]. ETD of the (GM*GGR + CE + 2H)2+ complex (m/z 375) resulted in charge reduction accompanied by an abundant loss of the CE molecule, forming the (GM*GGR + 2H)+● cation-radical at m/z 486 (Figure 2a), which was characterized by accurate mass measurements (Table S1, Supplementary Data). It should be noted that although ETD has sometimes been presented as a method that preserves noncovalent interactions [28], the ETD spectra of this and several other peptide-CE complexes [5, 8, 29, 30] attest to the opposite. The (GM*GGR + 2H)+● ion was selected by mass and subjected to CID (Figure 2b) and UVPD (Figure 2c).

(a) ETD mass spectrum of the (GM*GGR + CE + 2H)2+ complex at m/z 375. Inset shows the peak of the (GM*GGR + 2H)+● ion at m/z 486. (b) CID-MS3 spectrum of the (GM*GGR + 2H)+● at m/z 486. Inset shows the x 3 peak profile. (c) UVPD-MS3 spectrum of the (GM*GGR + 2H)+● m/z 486
The CID-MS3 mass spectrum of (GM*GGR + 2H)+● in Figure 2b showed a major dissociation forming the z 3 ion at m/z 273 along with the (y 3 – H) and x 3 backbone fragment ions at m/z 288 and 316, respectively. The position of the x 3 ion in the spectrum, (Figure 2b inset), which was 0.2 m/z units lower than the expected value and tailed toward low m/z, indicated that it was metastable [31, 32] and readily dissociated by loss of HNCO to form the z 3 ion [33]. The UVPD-MS3 spectrum of the (GM*GGR + 2H)+● ion (Figure 2c) indicated that it contained a chromophore absorbing at 355 nm. The major UVPD fragments were at m/z 415 (loss of C3H7N2), 428 (loss of CH4N3), and 470 (loss of NH2). The spectrum also contained ions at m/z 485 and 457, as discussed below.
The course of photodissociation was followed by recording the relative intensities of the major ion species as a function of the number of laser pulses (Figure S1, Supplementary Data). The m/z 486 and 485 ions that were present in the mass-selected ion population showed photodepletion albeit at very different rates. This allowed us to distinguish their dissociations as shown in Figure S2a and b (Supplementary Data) and analyze them separately. The photodepletion data for the (GM*GGR + 2H)+● ions (Figure S2a, Supplementary Data) were fitted with an exponential decay function, I[m/z 486] = e −0.182n+0.0095, where I is the ion relative intensity and n is the number of laser pulses. The fit showed a correlation coefficient of r 2 = 0.9993 and gave a 0.6% root-mean square deviation (RMSD) from the experimental data. The photodepletion data of the (GM*GGR + H)+ ions (Figure S2b, Supplementary Data) were also fitted with an exponential function, I[m/z 485] = e −0.0728n+0.0486, which showed r 2 = 0.996 and 1.7% RMSD from the experimental data. The nature of the chromophore group that was generated by ETD in the charge reduced m/z 486 cation radical will be addressed below.
Light absorption by the m/z 485 even-electron ion can only be due to the diazirine ring chromophore [5] because amide and other side-chain groups in the peptide ion are transparent at 355 nm. This was probed by two separate experiments described in the Supplementary Data (Figure S3), and the ion structure was unambiguously assigned to (GM*GGR + H)+ with an intact M* residue.
Tandem UVPD-ETD-UVPD Spectra
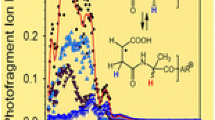
Photodissociation was also used to probe charge reduced [G(M* − N2)GGR + 2H]+● cation radicals. This was accomplished in a novel tandem MS4 experiment, called UVPD-ETD-UVPD, in which the [G(M* − N2)GGR + 2H]2+ precursor dications were prepared by UVPD of the diazirine-tagged peptide ions, mass-selected, and allowed to react by electron transfer from fluoranthene anions, as shown in Figure 1b, c. The surviving charge-reduced [G(M* − N2)GGR + 2H]+● ion (m/z 458) was selected by mass and interrogated by photodissociation; a CID spectrum was also taken for reference (Figure 3a, b). The CID spectrum (Figure 3a) showed a combination of proton and radical-driven dissociations, the former leading to loss of ammonia and water, m/z 441 and 440, respectively. The radical dissociations resulted in loss of 43 Da (m/z 415), presumably H2NC●=NH from the Arg side chain, and loss of 56 Da (m/z 402), presumably C4H8 from the M* side chain or HN=CHCO● radical from the Gly1 residue. In contrast, the UVPD spectrum (Figure 3b) was dominated by loss of H (m/z 457), which was completely absent in the CID spectrum. The loss of 58 Da upon UVPD can be ascribed to the H2NCH2CO● radical from the Gly1 residue. The UVPD spectrum confirmed that the charge-reduced [G(M* − N2)GGR + 2H]+● ion contained a chromophore absorbing at 355 nm. Furthermore, the photofragment relative ion intensities obtained for photodissociation with laser pulses ranging from one (Figure 3b inset) to nine (Figure 3b) showed no major changes. This strongly indicated that these fragments were photon nonabsorbing even-electron ions and therefore their formation must have involved losses of neutral radicals. Monitoring fragment ion intensities as a function of laser pulses is a useful tool for distinguishing even-electron and odd-electron ions based on their susceptibility to photodissociation [5].

(a) CID-MS4 spectrum of [G(M* − N2)GGR + 2H]+●. (b) UVPD-MS4 spectrum of [G(M* − N2)GGR + 2H]+● obtained with nine laser pulses. Inset shows the high mass region of the single-pulse spectrum
The formation of photofragment ions from [G(M* − N2)GGR + 2H]+● can be visualized by standard radical-induced reactions [34, 35] depicted in Scheme S1 and described by an accompanying text in the Supplementary Data. In short, electron transfer can trigger hydrogen atom migration from the N-terminal ammonium to the olefin side chain [36], which is followed by a rearrangement to a photoactive M* Cα radical whose optimized structure and absorption spectrum are shown in Figure S4 (Supplementary Data) [5, 37].
Ion and Radical Structures and Energetics
To interpret the experimental data concerning GM*GGR ions, we undertook extensive theoretical analysis of local energy minima, interconnecting transition states, and excited electronic states of pertinent cation radicals at several levels of ab initio and density functional theory. Conformers of the precursor (GM*GGR + 2H)2+ dications were modeled on their GL*GGR homologues, for which an exhaustive conformational search was reported previously [6]. The relative enthalpies and free energies of conformers with the most stable backbone folding motif are summarized in Table S2 (Supplementary Data), and the structures (1a 2+–1c 2+) are visualized in Scheme 2. These M* side-chain conformers had very similar free energies and were expected to be populated at thermal equilibrium in the gas phase. The lowest free-energy conformer (1a 2+) had the diazirine ring oriented towards the Gly1 amide, forming a weak hydrogen bond. Conformer 1b 2+ had the diazirine ring oriented towards the Gly3 amide, forming a weak hydrogen bond there, whereas conformer 1c 2+ had an all-anti M* side chain with an uncoordinated diazirine ring. Regardless, between all three structures, the folding of the backbone was mostly the same.

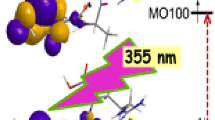
M06-2X/6-31 + G(d,p) optimized structures of (GM*GGR + 2H)2+ cations and cation radicals
Loss of N2 from ions 1a 2+–1c 2+ was calculated to be substantially exothermic when forming side-chain olefin isomers of the [G(M* – N2)GGR + 2H]2+ ions (Scheme S1, Supplementary Data). Formation of both the terminal CH2=CHCH2CH2- and internal trans-CH3CH=CHCH2- olefin groups in the M* residue was >200 kJ mol−1 exothermic, with the internal olefin being 15 kJ mol−1 more stable than the terminal one. The energetic loss of N2 upon photodissociation results in vibrational excitation in the [G(M* – N2)GGR + 2H]2+ ions, which explains their concomitant backbone dissociation forming y 3 fragment ions (Figure 1b). Note that a substantial part of this vibrational excitation originates from the highly exothermic rearrangement of the primary carbene (M* − N2) intermediate to the more stable olefins [6].
The course of electron attachment to 1a 2+–1c 2+ depended on the diazirine ring orientation (Scheme 2). Electron capture by conformers 1a 2+and 1b 2+ first formed transient anion-radical zwitterions consisting of a reduced diaziridine anion radical and the protonated Arg and N-terminal groups. These intermediates were not local potential energy minima and, upon gradient optimization of the ground electronic state, they underwent a spontaneous transfer of conformationally available peptide protons to the diaziridine anion radical. Starting from 1a 2+, this produced cis-diazirinyl radical 1a +● that can readily isomerize via a low-energy transition state (TS1, ~20 kJ mol−1) to its nearly isoenergetic trans-isomer 1d +●. Note that the proton transfer in the charge-reduced ion proceeds in two tandem steps, the first one being the transfer of the Gly1 amide proton on the diazirine ring forming a transient amidyl anion. This is followed by transfer of an N-terminal ammonium proton onto the Gly1 amidyl anion forming stable ion 1a +● (Scheme 2). Interestingly, the amidyl anion was found as a very shallow potential energy minimum (74 kJ mol−1 vs 1a +●) by B3LYP gradient optimization, whereas M06-2X optimization resulted in a spontaneous collapse to 1a +●.
Electron attachment to 1b 2+ involved a single step in which the electronic state of the diazirine anion radical was quenched by transfer of the M* amide proton forming zwitterion radical 1b +●. In contrast, electron attachment to conformer 1c 2+ avoided the diazirine ring while proceeding to the Gly1 amide group and forming the zwitterionic structure 1c +● as an intermediate. Ion 1c +● can be expected to readily eliminate ammonia or undergo N−Cα bond cleavage forming the z 4 fragment ion. The fact that neither of these dissociations is prominent in the ETD mass spectrum indicates that ions of type 1c +● are not substantially populated by electron transfer. The rather unusual reaction sequences and structure development upon electron attachment to GM*GGR ions 1a 2+–1c 2+ are analogous to those reported for GL*GGR conformers [8] and further point to the nature of electronic structure development as something that is very sensitive to the precursor peptide ion conformation.
Cation radicals formed by electron attachment to GM*GGR ions 1a 2+–1c 2+ differed very substantially in potential energy (Table 1). Cation radicals 1a +● and 1d +● represent the lowest energy structures that are nearly isoenergetic as judged on the basis of several DFT and ab initio calculations (Table 1). Ions 1b +● and 1c +● can be viewed as high-energy intermediates with the respective potential energies at 109 and 162 kJ mol−1 relative to 1a +●. Ion 1c +● can undergo a process towards the highly exothermic elimination of ammonia yielding an ion–molecule complex (1e +●), which is 148 kJ mol−1 more stable (Scheme S2, Supplementary Data). In contrast, the M* amidyl anion in 1b +● does not have a facile pathway to stabilization because there is no proximate proton for transfer to reform the M* amide group. An isomerization to 1a +● or 1d +●, albeit energetically very favorable, requires peptide refolding to bring the N-terminal ammonium close to the M* amidyl group to allow proton transfer.
Structure 1d +● is a crucial intermediate for the backbone and side-chain dissociations induced by collisions and UV absorption of long-lived (GM*GGR + 2H)+●. These were investigated for two branches of consecutive and competitive reactions. In the first one (Scheme 3), the N-diaziridinyl radical in 1d +● undergoes competitive transfers of the M* α (TS3) and β (TS4) hydrogen atoms forming the respective C-centered α and β radicals 3 +● and 4 +●. The respective transition state energies for TS3 and TS4 favor the former, although both transfers are endothermic and thus reversible. This is further underscored by the high energy for the Cβ–Cγ bond dissociation resulting in loss of 3-methyldiaziridinyl-3-methyl radical (5) from 3 +● (TS5, ETS5 = 161 kJ mol−1 relative to 1a +●) and formation of the peptide fragment ion 6 + (m/z 415) with a threshold energy of 140 kJ mol−1 relative to 1a +●.

Proposed mechanism for the loss of C3H7N2 ● from diaziridinyl radicals
In the second branched pathway, radical 1d +● can competitively undergo diaziridine ring opening, which requires 108 kJ mol−1 in the pertinent transition state (TS2) forming diazene radical 2 +● (Scheme 4). The ring opening is substantially exothermic and therefore practically irreversible, as follows from the energetics of the further reactions of 2 +●. The N-centered diazene radical 2 +● can undergo competitive transfers of the M* α (TS6) or β (TS7) hydrogen atoms forming the respective C-centered α- and β-radicals 7 +● and 9 +●, which are hydrazones with potential energies above 2 +●. Dissociation of the Cβ–Cγ bond in 7 +● results in loss of acetonyl hydrazone radical (8) to form 6 +, which can proceed via TS8 at 91 kJ mol−1 relative to 7 +●. The β-radical 9 +● is a reactant for the cleavage of the backbone Cα−CO bond resulting in a complex (10 +●) of the incipient x 3 +● (11 +●) and a 2 (12) fragments [33]. After fragment separation, the x 3 +● ion can readily eliminate HNCO to form the z 3 +● fragment ion found in the ETD spectrum. Alternatively, HNCO elimination can occur in complex 10 +●, where it requires relatively low energy in the transition state. The potential energy profiles for both branches of reactions starting from 1a +● are plotted in Figure 4 to visualize the following discussion of the dissociation kinetics. The fully optimized structures are shown in Schemes S3–S7 (Supplementary Data).

Proposed mechanism for the loss of C3H7N2 ● from diazene radicals

Dissociation Kinetics
Having a detailed description of the relevant transition states and their energies, we attempted to calculate the branching ratios for the formation of major products upon CID and UVPD. Because of the exothermic isomerization of 1a +● to 2 +● (Table 1) and the ensuing irreversible diazirine ring opening in 1a +●, it was possible to separate the top and bottom reaction schemes in Figure 4 and treat them independently. The RRKM rate constants for the isomerizations of ions with an intact diazirine ring (1a +●, 1d +●, 3 +●, 4 +●) are shown in Figure S5, Supplementary Data. Because of the low energy for TS1, the cis and trans diazirinyl radicals 1a +●and 1d +● were expected to rapidly interconvert. The rate constant for the Hα migration (k 1, TS3) was substantially larger than that for the Hβ migration (k 2, TS4), indicating that the former reaction will be preferred in 1a +● and 1d +●. However, because of their relative energies, intermediates 3 +● and 4 +● are depleted by fast reverse hydrogen migrations reforming 1a +● and 1d +●. This is indicated by the rate constant for the reverse H atom migration to the M* Cα position (k 1r, Figure S5, Supplementary Data). Further, the high TS5 and threshold energy for the loss of C3H7N2 hampers this reaction from efficiently competing with the diazirine ring opening through TS2, which is characterized by rate constant k 3. From this kinetic analysis it follows that α and β radicals 3 +●and 4 +● are expected to be at low equilibrium concentrations among the non-dissociating (GM*GGR + 2H)+● cations.

Top panel (a): RRKM rate constants for dissociations and isomerizations of 1d +● on the combined B3LYP-PMP2/6-311++G(2d,p) potential energy surface; bottom panel (b): calculated molar fractions from iterative solution of equations S1–S6. Reaction times were 50 ms (full lines) and 200 ms (broken lines)
The reaction scheme for the isomerizations and dissociations of ion 2 +● involves a system of several reversible competitive and consecutive reactions (Scheme S8) described by equations S1–S6 in the Supplementary Data. The pertinent RRKM rate constants (k 3, k 4, k 4r, k 5, k 5r, k 6, and k 7) were calculated for TS energies obtained at various levels of theory and used for iterative solution of differential equations S1–S6. These calculations were carried out to yield molar fractions of the involved species at reaction times of 50, 100, and 200 ms that cover the UVPD and CID time scales. Note that transfer of the β-H in 2 +● (TS7, Scheme 4) can involve either of the diastereotopic hydrogen atoms. These reactions were treated under the assumption that the pertinent rate constants were similar and, therefore, the RRKM rate constant for the hydrogen transfer (k 5) was doubled.
The kinetic analysis showed a substantial kinetic shift for the ring opening in 1d +●. This is indicated by the slowly rising curve for the pertinent rate constant (k 3, Figure 5a) and the delayed depletion of 1d +● , which shows <1% change up to ion internal energies of 300 kJ mol−1 (Figure 5b). The kinetic shift depends on the dissociation time scale, as shown for dissociations taking place at 50 ms (solid line curve in Figure 5b) and 200 ms (broken line curve). RRKM rate constants obtained for the B3-PMP2, M06-2X, and ωB97XD potential energy surfaces from Table 1 gave qualitatively similar results. Noteworthy is the low molar fraction of intermediate 2 +● (<0.4%), which is efficiently depleted by consecutive isomerizations to 9 +● and 7 +● and their further dissociations. Radicals 3 +● and 4 +● also showed very low intermittent molar fractions (<0.01%, not plotted in Figure 5b). The overall kinetics was dominated by the rate determining ring opening via TS2. When TS2 was passed, the intermediates of consecutive reversible isomerizations underwent competitive dissociations forming ion 6 + and complex 10 +● , which further dissociated to the x 3 and z 3 fragment ions. The branching ratio between 6 + and 10 +● consistently favored the former fragment ion when based on RRKM rate constants pertinent to the calculated TS energies. This result was contrary to experiment, where slow heating of the (GM*GGR + 2H)+● ion upon CID preferentially produced the z 3 fragment ion (Figure 2b), implying that this dissociation was preferred at excitation energies near the dissociation threshold.
The rate constants are known to be very sensitive to the TS energies which, for the computational methods used here, have an expected accuracy of ±10 kJ mol−1. For example, lowering the TS2 energy by 8 kJ mol−1 from the Table 1 datum resulted in a 25- to 110-fold increase of the RRKM rate constant (k 3) in the kinetically relevant energy region of 270–380 kJ mol−1 (Figure 5). Likewise, stepwise adjustments by 4 kJ mol−1 of energies of TS6, TS7, 7 +●, and 9 +● resulted in sets of RRKM rate constants that produced increasing molar fractions of 10 +● to improve agreement with experiment, although the formation of 6 + still prevailed. Because the molar fractions had to be calculated by iterative solution of differential equations S1–S6, it was not possible to analyze the effect of multiple combinations of adjusted TS energies to obtain an optimized set of RRKM rate constants that would produce a [10 +●]/[6 +] ion intensity ratio matching the experimental datum. Nonetheless, the calculations with the TS energies that we used indicated that the [10 +●]/[6 +] ratio changed by less than 2% over a 250–450 kJ mol−1 range of 1d +● internal energies. This implied that the differences in the formation of fragment ions 6 + and z 3 in the CID and UVPD spectra cannot be accounted for by different internal energies in the precursor ions. If vibronic energy equilibration preceded dissociation on UVPD, the photon energy (3.49 eV, 337 kJ mol−1) combined with the thermal enthalpy of the precursor ion (95 kJ mol–1 at 310 K) would produce 1d +● with an internal energy of 432 kJ mol−1. According to the RRKM kinetic analysis (Figure 5b), at these internal energies the [10 +●]/[6 +] ratio stays flat and would be expected to be close to that from CID, which is contrary to the experimental data. This apparent discrepancy could be resolved by assuming that the dissociation in UVPD proceeds from an excited electronic state.
Excited Electronic States
The UVPD spectra of the (GM*GGR + 2H)+● ions clearly indicated that the long-lived species contained a chromophore group absorbing at 355 nm. Since the peptide ions were stored in the ion trap at ambient temperature (30 ○C), the UV absorption bands were likely to be broadened to 30–40 nm by excitation involving multiple vibrational states [38, 39], implying that the laser excitation line and the absorption band maximum do not have to match in absolute precision. The other constraint for observing UVPD is that the absorbing species must be significantly populated in the ensemble of (GM*GGR + 2H)+● ion structures produced by ETD to account for the observed photodissociation. The electronic transitions for photon absorption by 1d +●, 2 +●, 7 +●, and 9 +● were calculated by TD-DFT and are plotted as theoretical absorption spectra in Figure 6a–d. The spectra are plotted on the same oscillator strength scale for facile comparison.

UV absorption spectra from TD-DFT calculations. Blue bars and lines: ωB97XD/6-311++G(2d,p) energies; brown bars and lines: M06-2X/6-311++G(2d,p) energies
The accuracy of the TD-DFT-calculated excitation energies and transition intensities was checked by benchmarking to the equation-of-motion coupled cluster calculations (EOM-CCSD) [40, 41] all with the 6-311++G(2d,p) basis set, as described in detail in the Supplementary Data. The ωB97XD, M062X, and LC-BLYP methods have been evaluated previously with different molecular systems [42]. The calculated absorption maximum of the diazirinyl radical 1d +● was at 370 nm (Figure 6) which, when corrected for a ca. 18-nm red shift, could readily account for UVPD at 355 nm. The analogy between the electronic structure of 1d +● and the 3,3-dimethyldiazirin-1-yl radical is obvious from the comparison of the donor and acceptor molecular orbitals (MO) involved in the electron transition (Figure S6, Supplementary Data). In both cases, the component of largest transition density corresponds to a weakly allowed electron excitation from a diazirine doubly occupied in-plane πxy orbital to the singly occupied out-of-plane πz MO, and the excitation is localized in the diazirine ring. Interestingly, this is analogous to the πxy → πz * excitation in diazirine that occurs at a similar wavelength [4]. The diazene radical 2 +●, M*-α radical 7 +● and M* β-radical 9 +● were calculated to have weak absorption maxima at 320–330 nm (Figure 6), which would be close to 300 nm when a red shift was accounted for. Hence, the Figure 6 calculated spectra indicated the best match for photon absorption by the diazirinyl radical 1d +●. This conclusion is consistent with the analysis of the above-mentioned Figure 5 kinetic data that indicated a substantial fraction of non-dissociating 1d +●, whereas those of 2 +●,7 +●, and 9 +● were vanishingly low.
Discussion
Aside of the details of the dissociation kinetics, there are two salient features of the ETD spectra of the L* and M* tagged peptides. One is the competition between the backbone and side-chain dissociations. The other is the dramatically different courses of side-chain dissociations for L* and M* because of a minor structure difference by a single methylene group. Regarding the first feature, analysis of electronic states that develop in the course of electron attachment to both L* and M* containing peptide ions indicated that the ground doublet state in each case involved electron capture by the diazirine ring. This was discussed previously for L* where it was pointed out that the diazirine ring has a substantial intrinsic electron affinity, which is augmented by the Coulomb effect of the peptide charged groups [7, 8]. An important step in the development of the ground electronic state of the charge-reduced ion is the concomitant proton transfer that stabilizes the diazirine anion radical by transforming it to a neutral radical. In the absence of a suitable proton, as in GM*GGR conformer 1c 2+, the incoming electron reduces another functional group in the vicinity of the charge site. The calculated flow of electron density to the diazirine ring in L* and M* radicals may be viewed as being in partial conflict with ETD spectra, where the majority of electron-triggered dissociations (75% for GM*GGR) involve backbone N−Cα bond cleavages and only 25% affects the M* side chain. Note that the backbone N−Cα bond dissociations cannot be initiated from intermediates with reduced diazirine rings because of energy reasons [8]. This apparent conflict can be resolved by considering that the N−Cα bond cleavages do not proceed from the doublet ground state but involve excited electronic states in which the electron enters amide π* orbitals, as postulated by the Utah-Washington model [28, 43, 44]. As discussed for GL*GGR, intermediate structures produced by electron attachment to the dication show a number of low-lying electronic states in which the spin density is delocalized over several amide groups where it can trigger proton transfer and N−Cα bond cleavage. These processes have been analyzed in detail for a dipeptide ion using Ehrenfest dynamics [45]; however, a detailed analysis of excited state dynamics is currently lacking for larger peptide ions.
The dramatic difference in the diazirine-affecting dissociations of L* and M* can be ascribed to subtle structure effects. Both reduced L* and M* are thought to involve diaziridinyl radicals as reactive intermediates that undergo exothermic ring opening to diazene radicals. Diazene radicals from both L* and M* can undergo hydrogen atom transfers forming α and β carbon-centered radicals, e.g., 7 +● and 9 +● (Scheme 4). The difference between L* and M* arises because the β- radical from L* is conjugated with the side-chain hydrazone π-system, which lowers its energy and allows for further α-H migration and cyclization in a cascade of thermoneutral or exothermic rearrangements (Scheme 5) [8]. In contrast, GM*GGR radical 7 +● is not stabilized by conjugation and undergoes simple Cβ–Cγ bond fission resulting in the loss of C3H7N2 radical.

Cyclization pathway leading to loss of NH2 and H2O from (GL*GGR + 2H)+● [8]
Conclusions
The dramatic differences in the fragmentation patterns upon electron transfer dissociation of photoleucine and photomethionine are accounted for by using experimental and theoretical methods. In both amino acid analogues, the high electron affinity of the diazirine tag contributes greatly to the radical forming processes pursued following electron transfer. Two major products predominate the ETD spectrum of the studied photomethionine containing peptide. Density functional calculations show that these two products can arise through two initial separate but congruent steps that compete through isomerization of various radical intermediates. Kinetic modeling was used to distinguish the relative branching ratios of the reactions following divergence via the initial step. The resulting radical intermediates were probed by ETD-UVPD MS3 and ETD-CID MS3 to identify photoactive radical centers that can account for the unique fragments. Divergence between CID and UVPD results and pulse dependence were used as a basis to suggest radical centers that were then characterized by electron structure theory calculations. These experiments included the first use of an alternating UVPD-ETD-UVPD MS4 experiment. This allowed the incipient liberation of the diazo species, allowing for identification of the minor fragmentation products as having occurred from backbone fragmentations.
References
Suchanek, M., Radzikowska, A., Thiele, C.: Photo-leucine and photo-methionine allow identification of protein–protein interactions in living cells. Nat. Methods 2, 261–268 (2005)
Hashimoto, M., Hatanaka, Y.: Recent progress in diazirine-based photoaffinity labeling. Eur. J. Org. Chem. 15, 2513–2523 (2008)
Das, J.: Aliphatic diazirines as photoaffinity probes for proteins: recent developments. Chem. Rev. 111, 4405–4417 (2011)
Liu, M.T.H.: Chemistry of diazirines Vol. I and II. CRC Press, Boca Raton (1987)
Shaffer, C.J., Marek, A., Pepin, R., Slováková, K., Tureček, F.: Combining UV photodissociation with electron transfer for peptide structure analysis. J. Mass Spectrom. 50, 470–475 (2015)
Marek, A., Turecek, F.: Collision-induced dissociation of diazirine-labeled peptide ions. evidence for Brønsted-acid assisted elimination of nitrogen. J. Am. Soc. Mass Spectrom. 25, 778–789 (2014)
Marek, A., Pepin, R., Peng, B., Laszlo, K.J., Bush, M.F., Tureček, F.: Electron transfer dissociation of photolabeled peptides. backbone cleavages compete with diazirine ring rearrangements. J. Am. Soc. Mass Spectrom. 24, 1641–1653 (2013)
Marek, A., Shaffer, C.J., Pepin, R., Slováková, K., Laszlo, K.J., Bush, M.F., Tureček, F.: Electron transfer reduction of the diazirine ring in peptide gas-phase ions. on the peculiar loss of [NH4O] from photoleucine. J. Am. Soc. Mass Spectrom. 26, 415–431 (2015)
Janz, J.M., Ren, Y., Looby, R., Kazmi, M.A., Sachdev, P., Grunbeck, A., Haggis, L., Chinnapen, D., Lin, A.Y., Seibert, C., McMurry, T., Carlson, K.E., Muir, T.W., Hunt, S., Sakmar, T.P.: Direct interaction between an allosteric agonist pepducin and the chemokine receptor CXCR4. J. Am. Chem. Soc. 133, 15878–15881 (2011)
Coste, J., LeNguyen, D., Castro, B.: PyBOP: a new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 31, 205–208 (1990)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford (2009)
Becke, A.D.: New mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377 (1993)
Becke, A.D.: Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993)
Zhao, Y., Truhlar, D.G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 120, 215–241 (2008)
Chai, J.D., Head-Gordon, M.: Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 128, 084106/1–084106/15 (2008)
Chai, J.D., Head-Gordon, M.: Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008)
Møller, C., Plesset, M.S.: A note on an approximation treatment for many-electron systems. Phys. Rev. 46, 618–622 (1934)
Schlegel, H.B.: Potential energy curves using unrestricted Moller-Plesset perturbation theory with spin annihilation. J. Chem. Phys. 84, 4530 (1986)
Mayer, I.: Spin-projected UHF method. IV. Comparison of potential curves given by different one-electron methods. Adv. Quantum Chem. 12, 189–262 (1980)
Tureček, F.: Proton affinity of dimethyl sulfoxide and relative stabilities of C2H6OS molecules and C2H7OS+ Ions. A comparative G2(MP2) ab initio and density functional theory study. J. Phys. Chem. A 102, 4703 (1998)
Polášek, M., Tureček, F.: Hydrogen atom adducts to nitrobenzene. Formation of the phenylnitronic radical in the gas phase and energetics of Wheland intermediates. J. Am. Chem. Soc. 122, 9511–9524 (2000)
Wolken, J.K., Yao, C., Tureček, F., Polce, M.J., Wesdemiotis, C.: Cytosine neutral molecules and cation-radicals in the gas-phase. Structures, energetics, ion chemistry, and neutralization-reionization mass spectrometry. Int. J. Mass Spectrom. 267, 30–42 (2007)
Furche, F., Ahlrichs, A.: Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 117, 7433–7447 (2002)
Iikura, H., Tsuneda, T., Yanai, T., Hirao, K.: A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 115, 3540–3544 (2001)
Gilbert, R.G., Smith, S.C.: Theory of unimolecular and recombination reactions, pp. 52–132. Blackwell Scientific Publications, Oxford (1990)
Zhu, L., Hase, W.L.: Quantum chemistry program exchange. Indiana University: Bloomington; Program No. QCPE 644 (1994)
Frank, A.J., Sadílek, M., Ferrier, J.G., Tureček, F.: Sulfur oxoacids and radicals in the gas phase. A variable-time neutralization-photoexcitation-reionization mass spectrometric and ab initio/RRKM study. J. Am. Chem. Soc. 119, 12343–12353 (1997)
Turecek, F., Julian, R.R.: Peptide cation radicals. Chem. Rev. 113, 6691–6733 (2013)
Maclot, S., Rangama, J., Nielsen, S.B., Poully, J.-C.: Nanosolvation by acetonitrile and 18-crown-6 ether induce strongly different effects on the electron-capture induced dissociation of aromatic tripeptide cations in the gas phase. Int. J. Mass Spectrom. 337, 1–11 (2013)
Bernigaud, V., Cederquist, H., Haag, N., Holm, A.I.S., Huber, B.A., Hvelplund, P., Kadhane, U., Larsen, M.K., Manil, B., Nielsen, S.B., Panja, S., Ptasinska, S., Rangama, J., Reinhed, P., Schmidt, H.T., Streletskii, A.V., Støchkel, K., Worm, E.S., Zettergren, H.: Electron capture-induced dissociation of AK dipeptide dications:i of ion velocity, crown-ether complexation and collision gas. Int. J. Mass Spectrom. 276, 77–81 (2008)
McClellan, J.E., Murphy III, J.P., Mulholland, J.J., Yost, R.A.: Effects of fragile ions on mass resolution and on isolation for tandem mass spectrometry in the quadrupole ion trap mass spectrometer. Anal. Chem. 74, 402–412 (2002)
Chung, T.W., Hui, R., Ledvina, A.R., Coon, J.J., Tureček, F.: Cascade dissociations of peptide cation-radicals. Part 1. Scope and effects of amino acid residues in penta-, nona-, and decapeptides. J. Am. Soc. Mass Spectrom. 23, 1336–1350 (2012)
Ledvina, A.R., Chung, T.W., Hui, R., Coon, J.J., Tureček, F.: Cascade dissociations of peptide cation-radicals. Part 2. Infrared multiphoton dissociation and mechanistic studies of z-ions from pentapeptides. J. Am. Soc. Mass Spectrom. 23, 1351–1363 (2012)
Sun, Q., Nelson, H., Stolz, B.M., Julian, R.R.: Side chain chemistry mediates backbone fragmentation in hydrogen deficient peptide radicals. J. Proteome Res. 8, 958–966 (2009)
Chung, T.W., Tureček, F.: Backbone and side-chain specific dissociations of z ions from non-tryptic peptides. J. Am. Soc. Mass Spectrom. 21, 1279–1295 (2010)
Shaffer, S.A., Wolken, J.K., Tureček, F.: Neutralization-reionization of alkenylammonium cations: an experimental and ab initio study of intramolecular N–H…C=C interactions in cations and hypervalent ammonium radicals. J. Am. Soc. Mass Spectrom. 8, 1111–1123 (1997)
Nguyen, H.T.H., Shaffer, C.J., Ledvina, A.R., Coon, J.J., Tureček, F.: Serine effects on collision-induced dissociation and photodissociation of peptide cation radicals of the z+● -type. Int. J. Mass Spectrom. 378, 20-30 (2015)
Kelly, O., Calvert, C.R., Greenwood, J.B., Zettergren, H., Nielsen, S.B., Wyer, J.A.: Effects of charge location on the absorptions and lifetimes of protonated tyrosine peptides in vacuo. J. Phys. Chem. A 116, 1701–1709 (2012)
Byskov, C.S., Jensen, F., Joergensen, T.J.D., Nielsen, S.B.: On the photostability of peptides after selective photoexcitation of the backbone: prompt versus slow dissociation. Phys. Chem. Chem. Phys. 16, 15831–15838 (2014)
Koch, H., Jørgensen, P.: Coupled cluster response functions. J. Chem. Phys. 93, 3333–3344 (1990)
Comeau, D.C., Bartlett, R.J.: The equation-of-motion coupled-cluster method. Applications to open- and closed-shell reference states. Chem. Phys. Lett. 207, 414–423 (1993)
Sonk, J.A., Schlegel, H.B.: TD-CI simulation of the electronic optical response of molecules in intense fields II: comparison of DFT functionals and EOM-CCSD. J. Phys. Chem. A 115, 11832–11840 (2011)
Sobczyk, M., Anusiewicz, I., Berdys-Kochanska, J., Sawicka, A., Skurski, P., Simons, J.: Coulomb-assisted dissociative electron attachment: application to a model peptide. J. Phys. Chem. A 109, 250–258 (2005)
Syrstad, E.A., Tureček, F.: Toward a general mechanism of electron-capture dissociation. J. Am. Soc. Mass Spectrom. 16, 208–224 (2005)
Moss, C.L., Liang, W., Li, X., Tureček, F.: The early life of a peptide cation radical. Ground and excited-state trajectories of electron-based peptide dissociations during the first 330 femtoseconds. J. Am. Soc. Mass Spectrom. 23, 446–459 (2012)
Acknowledgments
The authors gratefully acknowledge support of this research by the National Science Foundation Division of Chemistry (Grants CHE-1055132 and CHE-1359810). F.T. thanks the Klaus and Mary Ann Saegebarth Endowment for support. Technical advice was kindly provided by James Gladden (University of Washington), Drs. Joshua J. Coon (University of Wisconsin), Graeme McAlister, and John E. P. Syka (both ThermoElectron Fisher).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1931 kb)
Rights and permissions
About this article

Cite this article
Shaffer, C.J., Marek, A., Nguyen, H.T.H. et al. Combining Near-UV Photodissociation with Electron Transfer. Reduction of the Diazirine Ring in a Photomethionine-Labeled Peptide Ion. J. Am. Soc. Mass Spectrom. 26, 1367–1381 (2015). https://doi.org/10.1007/s13361-015-1139-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-015-1139-5