
ABSTRACT
The mechanisms underlying spatial and temporal control of cortical neurogenesis of the brain are largely elusive. Long non-coding RNAs (lncRNAs) have emerged as essential cell fate regulators. Here we found LncKdm2b (also known as Kancr), a lncRNA divergently transcribed from a bidirectional promoter of Kdm2b, is transiently expressed during early differentiation of cortical projection neurons. Interestingly, Kdm2b’s transcription is positively regulated in cis by LncKdm2b, which has intrinsic-activating function and facilitates a permissive chromatin environment at the Kdm2b’s promoter by associating with hnRNPAB. Lineage tracing experiments and phenotypic analyses indicated LncKdm2b and Kdm2b are crucial in proper differentiation and migration of cortical projection neurons. These observations unveiled a lncRNA-dependent machinery in regulating cortical neuronal differentiation.
Similar content being viewed by others


Introduction
The mammalian cerebral cortex, also known as the neocortex, is a six-layered structure and responsible for performing the most sophisticated cognitive and perceptual functions such as sensory perception, generation of motor commands, conscious thought and language. The adult neocortex comprises a plethora of projection neurons, interneurons and glial cells. Projection neurons (PNs) are the main functional units, expressing excitatory neurotransmitters, with their long axons projecting into subcortical regions or contralateral cortex of the brain. In mice, cortical PNs are largely generated between embryonic (E) day 11.5 to E17.5 indirectly from radial glial progenitor cells (RGPCs), whose nuclei lie in the region close to the lateral ventricles, ventricular zone (VZ). RGPCs usually divide asymmetrically to self-renew and simultaneously give rise to intermediate progenitor cells (IPCs), which are multipolar and reside basally to RGPCs in the subventricular zone (SVZ). IPCs divide symmetrically to generate either two IPCs or two postmitotic PNs. PNs then migrate radially along the basal processes of RGPCs to propagate the cortical plate (CP) in the basal part of the cortex, which eventually forms cortical layers (Fietz and Huttner, 2011; Kwan et al., 2012). Many cellular and molecular aspects governing cortical neurogenesis have been extensively studied, including cell-autonomous and non-autonomous regulation of RGPCs’ asymmetric cell division, neuronal fate commitment, as well as PNs’ radial migration (Ayala et al., 2007; Greig et al., 2013; Imayoshi and Kageyama, 2014). However, mechanisms that control the initial numbers and proliferation rates of RGPCs, as well as the proliferative or neurogenic choices of IPCs, are largely elusive (Greig et al., 2013; Homem et al., 2015).
Recent studies indicate a few long non-coding RNAs could be essential cell fate regulators in development (Grote et al., 2013; Klattenhoff et al., 2013). Long non-coding RNAs (lncRNAs), defined as RNAs longer than 200 nucleotides but lacking protein-coding potentials, are abundant in brain and display cell-type-, and developmental stage-specific expression patterns compared to protein-coding transcripts (Mercer et al., 2010; Belgard et al., 2011; Aprea et al., 2013; Molyneaux et al., 2015). LncRNAs may regulate gene transcription by recruiting transcription factors, RNA-binding proteins and chromatin-remodeling machineries to the site of transcription and creating a locus-specific environment (Ng et al., 2013; Lin et al., 2014; Wang et al., 2015). LncRNAs are often derived from bidirectional promoters, such that initiating Pol II can generate divergently-oriented transcripts simultaneously, the sense (protein-coding mRNA) direction or the upstream-antisense (divergent non-coding) direction, with these mRNA/divergent lncRNA pairs having coordinated expression (Lepoivre et al., 2013; Sigova et al., 2013; Scruggs and Adelman, 2015). Moreover, the transcription of divergent lncRNAs could affect the expression of their neighboring protein-coding transcripts in cis (Orom et al., 2010; Luo et al., 2016). Anti-sense promoters could serve as platforms for transcription factor (TF) binding and facilitate establishment of proper chromatin architecture to regulate sense-strand mRNA expression (Scruggs and Adelman, 2015; Scruggs et al., 2015). Although divergent lncRNAs are prevalent in both embryonic and adult nervous system, only a few functional divergent lncRNAs have been characterized, including roles of Emx2os in regulating the expressions of their neighboring protein-coding transcripts Emx2, an essential cortical RGPC gene (Noonan et al., 2003; Spigoni et al., 2010). Furthermore, these are largely invitro studies and it’s still lack of in vivo evidence showing the significance of divergent lncRNAs in cortical neuronal differentiation (Wang et al., 2017).
Here we characterized LncKdm2b (also known as Kancr—Kdm2b upstream-antisense non-coding RNA), a divergent lncRNA that can positively regulate the transcription of Kdm2b in cis. Both LncKdm2b and Kdm2b are transiently expressed in committed neuronal precursors and newborn cortical PNs and essential for their proper differentiation. LncKdm2b cis-regulates Kdm2b’s expression and facilitates a permissive chromatin environment by binding to hnRNPAB. Our findings advance understandings of molecular events that govern cortical neuronal differentiation and might have general implications in regulation of cell differentiation.
Results
LncKdm2b and Kdm2b are transiently expressed in committed neuronal precursors and newborn cortical projection neurons
In an effort to identify pairs of divergent lncRNA/protein-coding transcript that exert roles in cortical neurogenesis of the mouse brain, we analyzed a database comprising both in-house and publicized transcriptome data of developing mouse cerebral cortex (dorsal forebrain). In-house data are RNA-seq data from embryonic (E) day 10.5 and E12.5 dorsal forebrain. We also included RNA-seq data of mouse embryonic stem cells (mESCs), mESCs derived neural progenitor cells (NPCs), and tissues from later stages of cortical development including E14.5 ventricular zone (VZ), subventricular and intermediate zone (SVZ/IZ) and cortical plate (CP), E17.5 and adult cortex (Guttman et al., 2010; Ayoub et al., 2011; Dillman et al., 2013; Ramos et al., 2013). Interestingly, protein-coding genes associated with divergent lncRNAs within 5 kilobase from their transcription start sites (TSS) are highly enriched for signatures including transcription, cell cycle progression and catabolic process (Fig. S1A and Table S1), indicating their related roles (Ponjavic et al., 2009). One of these pairs is Kdm2b and its divergent non-coding transcript LncKdm2b (also known as Kancr and A930024E05Rik) (Diez-Roux et al., 2011; Saba et al., 2015; Liu et al., 2017). LncKdm2b is transcribed at 262 base pair upstream of Kdm2b’s TSS, and is predicted to be a lncRNA according to its low score in coding potential and inability to translate proteins (Fig. S1B and S1C). The expression of LncKdm2b peaks in E14.5 SVZ/IZ, where IPCs and migrating PNs reside. Similarly, the expression of Kdm2b in E14.5 SVZ/IZ is slightly higher than that in E14.5 VZ and CP (Fig. S1D). Notably, LncKdm2b is expressed at higher levels than Kdm2b in E14.5 VZ and SVZ/IZ and at comparable levels in other stages (Fig. S1D), which is contradictory to the common notion that divergent lncRNAs are expressed at much lower levels than their neighboring protein-coding transcripts (Sigova et al., 2013). Consistently, quantitative RT-PCR and immunoblotting experiments showed expression levels of both KDM2B and LncKdm2b peak in E12.5 and E14.5 dorsal forebrains, with much lower levels in E10.5 and adult stages (Fig. S1E, S1F and S1M). This pattern is quite similar to those of Tbr2, Dcx, Unc5d and Neurod1, markers for IPCs and immature PNs (Fig. S1G–M). Northern blot detected a ~1.8 kb band in poly(A) RNAs extracted from E14.5 and E16.5 cortices (Fig. S1N). Through analyzing the ENCODE database (Yue et al., 2014), we found the genomic region spanning the promoter of Kdm2b and its immediate upstream region that transcribes LncKdm2b is evolutionarily conserved across mammals, and is associated with Pol II (RNA polymerase II) and H3K4me3 in E14.5 mouse brain, indicating active transcription at this condition (Fig. 1A). In situ hybridization (ISH) revealed that both LncKdm2b and Kdm2b are predominantly expressed in the upper SVZ of the E16.5 dorsal forebrain, with the apical side of ISH signals overlapping with TBR2, an SVZ marker labeling intermediate cortical neural precursors (IPCs) (Figs. 1B, S1O and S1P); and basal side overlapping with TUJ1, a marker for fate-determined pyramidal neurons (Fig. S1P). These data suggest both LncKdm2b and Kdm2b are transiently expressed in committed IPCs and freshly differentiated projection neurons during the peak of cortical neurogenesis.

LncKdm2bandKdm2bare transiently expressed in the developing mouse embryonic cortex. (A) Schematic illustration of the mouse LncKdm2b/Kdm2b locus. The top tracks depict ChIP-seq signals for Pol II, H3K4me3 and H3K36me3 in E14.5 mouse brain. Bottom tracks depict a parallel genomic alignment of 19 vertebrates to the mouse genome (UCSC mm9) at the LncKdm2b locus. Shaded lines indicate conserved sequences. (B) Top: In situ hybridization (ISH) of LncKdm2b (left) and Kdm2b (right) on coronal sections of E16.5 mouse dorsal forebrains. Bottom: Immunofluorescent staining for TBR2 (green) on ISH sections of LncKdm2b (left, red) and Kdm2b (right, red) on coronal sections of E16.5 mouse dorsal forebrains. (C) A schematic diagram illustrates the strategy for generating Kdm2bCreERT2 knock-in mice line. (D) Left: Immunofluorescent staining for EGFP (green), TBR2 (red), and TUJ1 (blue) on cortical sections of E16.5 heterozygous Kdm2bCreERT2 knock-in mice. Right: Immunofluorescent stainings for EGFP (green) and UNC5D (red) on cortical sections of E16.5 heterozygous Kdm2bCreERT2 knock-in mice. (E) A schematic diagram illustrates the strategy for lineage tracing of Kdm2b-expressing cortical cells using in utero electroporation. (F) E12.5 Kdm2bCreERT2/+ knock-in cortices were electroporated with conditional DsRed-expressing plasmids (pCALNL), followed by tamoxifen (TAM) injection at E12.75 and analyses for SATB2 (green) and CTIP2 (blue) expression at P0. Arrowheads indicate DsRed+, SATB2+ cells. Arrows indicate DsRed+, CTIP2+ cells. (G) Quantification of SATB2 or CTIP2 expression in DsRed+ recombined cells (F). A total of 711 cells from 3 animals were analyzed. Data shown are the mean + SD. Scale bars, 50 μm. Boxed areas are enlarged at the bottom-right corners in (B), (D) and (F). Ctx, cortex; LV, lateral ventricle; VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone. pA, polyA
Kdm2b-expressing cortical cells are fated to be cortical projection neurons
To further validate Kdm2b’s expression and the fate of Kdm2b-expressing cells during cortical neurogenesis, we generated a knock-in mouse line, Kdm2b-F2a-CreERT2-IRES-EGFP (referred to Kdm2bCreERT2), in which the F2a-CreERT2-IRES-EGFP cassette was inserted in frame into the third exon of Kdm2b (Fig. 1C). Southern blotting and genomic PCR validated the predicted genomic modification (Fig. S1Q). Expressions of CreERT2 and EGFP are driven by the endogenous Kdm2b promoter, which would allow us to perform detailed expression analyses and lineage tracing experiments for Kdm2b. Brain sections from embryos derived from mating of Kdm2bCreERT2/+ with wild-type (WT) C57/B6 were subjected to immunofluorescent staining. Consistent with ISH experiments, EGFP+ cells reside in upper SVZ and lower intermediate zone (IZ), overlapping with both TBR2+ IPCs and TUJ1+ projection neurons (Figs. 1D and S1R). Moreover, a large portion of EGFP+ cells also overlap with UNC5D, a marker for multipolar cells in embryonic SVZ/IZ and layer IV projection neurons (Fig. S1S). Notably, EGFP+ signals extend more basally than Kdm2b or LncKdm2b ISH signaling, probably because EGFP protein is more stable than transcripts of Kdm2b or LncKdm2b. We next bred Kdm2bCreERT2/+ with the Ai14 (Rosa-CAG-LoxP-STOP-LoxP-tdTomato-WPRE) reporter mice. Pregnant female mice were injected with tamoxifen at various stages to enable the excision of the STOP cassette, thus leading to tdTomato expression in the progenies of Kdm2b-expressing cells. Cortices were collected from E16.5 and newborn (P0) pups for immunofluorescent staining of SATB2 (a marker for layer II-IV callosal neurons) and CTIP2 (a marker for layer V subcortical neurons). Interestingly, most tdTomato-positive cells express either SATB2 (51.0% ± 2.5% at E16.5, 63.1% ± 2.5% at P0) or CTIP2 (20.7% ± 5.4% at E16.5, 7.0% ± 2.3% at P0), suggesting the progenies of Kdm2b-expressing cells are largely projection neurons (Fig. S2A–D). Of note, the Cre recombinase could be randomly activated in neural epithelial (NE) cells of Kdm2bCreERT2/+; Ai14 mice in the absence of tamoxifen, thus confounding the analysis of lineage-tracing data (Fig. S2E and S2F). Nonetheless, by P7, tdTomato-positive cells largely express SATB2 (63.6% ± 4.8%) and/or CTIP2 (44.3% ± 5.8%) (Fig. S2G and S2H). To overcome the issue, we electroporated the LoxP-STOP-LoxP-DsRed (pCALNL) reporter plasmid into the E12.5 Kdm2bCreERT2/+ cortices followed by tamoxifen injection six hours after electroporation. In line with genetic lineage-tracing data, the majority DsRed-positive cells express either SATB2 (71.9% ± 1.5%) or CTIP2 (18.2% ± 7.1%) (Fig. 1E–G). The above expression and lineage-tracing results suggest Kdm2b and LncKdm2b are transiently expressed in differentiating IPCs and freshly born PNs and might regulate neuronal differentiation during cortical neurogenesis.
LncKdm2b regulates Kdm2b’s expression in cis
The close proximity of Kdm2b and LncKdm2b’s TSS and their identical expression patterns in developing cortices prompted us to examine if LncKdm2b regulates Kdm2b’s expression. Since it’s impractical to maintain intermediate progenitor cells or immature projection neurons invitro, we utilized a few primary or immortalized cells that express both Kdm2b and LncKdm2b to address the issue. First, we transduced Neuro-2a neuroblastoma cells with LncKdm2b antisense oligonucleotides (ASOs), which mediate RNA degradation via the RNase H-dependent mechanism (Walder and Walder, 1988; Vickers et al., 2003). The levels of Kdm2b’s transcripts and protein were significantly decreased upon the ASO treatment (Fig. 2A and 2B). Consistently, knockdown of LncKdm2b by ASO or shRNAs in adherent cultured cortical cells leads to decreased Kdm2b expression (Figs. 2C, S3A and S3B). Next, we applied the CRISPR/Cas9 technique to delete the genomic region of LncKdm2b’s second exon in NE-4C mouse neural stem cells (LncKdm2bexon2-KO), which results in compromised expression of LncKdm2b and Kdm2b (Figs. 2D and S3D). Notably, there’re significant amounts of transcripts derived from LncKdm2b’s first and third exons in LncKdm2bexon2-KO cells (Fig. 2D). However, the expression levels of Zfp292, the downstream target of LncKdm2b in ILC3 cells (Liu et al., 2017), were not decreased upon LncKdm2b depletion, suggesting cell-type-specific effects by LncKdm2b (Fig. S3C and S3E). Therefore, LncKdm2b maintains Kdm2b’s expression in neural cells.

LncKdm2bmaintainsKdm2btranscription incis. (A) RT-qPCR analysis of LncKdm2b and Kdm2b RNA levels in Neuro-2a cells treated for two days with Scramble ASOs or ASOs targeting LncKdm2b. (B) Representative immunoblotting of Neuro-2a cells treated for four days with indicated ASOs using antibody against KDM2B and β-TUBULIN. (C) RT-qPCR analysis of LncKdm2b and Kdm2b RNA levels in adherent cultures derived from E12.5 cortices. The cultures were treated with indicated shRNAs. (D) RT-qPCR analysis of LncKdm2b and Kdm2b RNA levels in wild-type or LncKdm2b’s exon 2 knockout NE-4C cells. The expression levels of individual exons of LncKdm2b were examined. (E) Left: Schematic diagram showing the insertion of a pAS cassette at 1.8 kb downstream the TSS of LncKdm2b in mESCLncKdm2b-pAS/+. pAS, 3 × SV40 polyA and a BGH polyA signal. Right: RT-qPCR analysis of Kdm2b mRNA levels in mESCLncKdm2b-pAS/+ and wild-type mESCs. (F) Left: Schematic diagram showing the indels of Kdm2b’s second exon in two mESCLncKdm2b-pAS/+ clones, 1B1 and 2D5. Right: RT-qPCR analysis of Kdm2b’s expression levels from individual alleles of clone 1B1 and 2D5. The y-axis represents relative expression normalized to genomic DNA. (G) The effects of LncKdm2b knockdown on nascent transcripts in nuclear run-on assays. RT-qPCR analysis of LncKdm2b, and Kdm2b nascent transcripts in Neuro-2a cells treated for two days with scramble ASO or ASOs targeting LncKdm2b. The y-axis represents relative expression normalized to Gapdh nascent transcript. In (A), (C), and (D–G), quantification data are shown as mean + SD (n = 3 unless otherwise indicated). In (A), (C and D), and (G), statistical significance was determined using 2-way ANOVA followed by the Bonferroni’s post hoc test. In (E and F), statistical significance was determined using unpaired 2-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, “ns” indicates no significance. The y-axis represents relative expression normalized to Gapdh transcript unless otherwise indicated
Cross-talk among neighboring genes could involve trans- and/or cis-regulatory mechanisms, the latter including enhancer-like activity of gene promoters, the process of transcription, and the splicing of the transcript (Bassett et al., 2014; Yin et al., 2015; Engreitz et al., 2016). To discriminate these possibilities, four polyadenylation sequences (pAS) were inserted 1.8 kb downstream of LncKdm2b’s TSS to prematurely terminate its transcription in one allele of mouse C57/B6 embryonic stem cells (mESCLncKdm2b-pAS/+), but to keep undisturbed the essential promoter region for Kdm2b and LncKdm2b’s transcription, which is DNase I hypersensitive (HS) (Fig. 2E). Consistently, the expressions of LncKdm2b and Kdm2b were significantly decreased upon pAS insertion (Fig. 2E), suggesting LncKdm2b’s transcription process and/or transcripts themselves are required for Kdm2b’s expression. We next studied if LncKdm2b maintains Kdm2b’s transcription in cis. First, subcellular fractionation followed by RT-qPCR and RNA ISH assays revealed that most LncKdm2b resides in the cytosol with a fraction in the nuclei of cortical cells (Fig. S3F and S3G). Next, we genetically modified mESCLncKdm2b-pAS/+ cells so that indels were created in the second exon of Kdm2b in an allele-specific manner. Quantitative RT-PCR experiments of the two clones (1B1 and 2D5) showed it’s the allele with pAS insertion that has significantly lower Kdm2b transcription than the other allele (Fig. 2F). Lastly, nuclear extracts from LncKdm2b-depleted Neuro-2a cells were collected and subjected to nuclear run-on assay. Data showed depletion of LncKdm2b results in significantly lower yield of Kdm2b nascent transcripts (Fig. 2G). In contrast, overexpressing LncKdm2b in trans didn’t elevate Kdm2b transcripts’ levels in Neuro-2a and NE-4C cells (Fig. S3H–J). Depletion of Kdm2b has no effect on LncKdm2b’s expression, suggesting the linear cis-regulation of Kdm2b by LncKdm2b (Fig. S3K). In summary, LncKdm2b maintains Kdm2b transcription in cis.
LncKdm2b modulates the configuration of Kdm2b’s cis-regulatory elements
Specific gene expression is coordinated by cis-regulatory elements such as the promoter/enhancer, cell-type-specific transcription factors and chromatin states (Heintzman et al., 2009; Perino and Veenstra, 2016). To understand these mechanisms underlying Kdm2b transcription, we first analyzed the genomic region both upstream and downstream of the Kdm2b and LncKdm2b’s TSS. This genome region contains multiple active and/or repressive epigenetic modifications including DNase I HS, H3K27ac (indicative of active enhancers), H3K4me1 (active or poised enhancers), H3K27me3 (repressive or poised cis-elements), and CTCF-association (insulators) in developing mouse brain (Vierstra et al., 2014; Yue et al., 2014), suggesting it may contain putative cis-regulatory sequences (enhancers) for Kdm2b (T1 to T7,Fig. 3A). Since cis-regulatory elements/enhancers can be recruited spatially adjacent to promoters to control gene expression, we performed the chromosome conformation capture (3C) followed by qPCR experiments and identified a peak of high crosslinking frequency at the H3K4me1-enriched T5 locus (5.9 kb upstream of Kdm2b’s TSS) when using a constant EcoRI fragment located close to the Kdm2b’s promoter (Fig. 3B), indicating the T5 locus is significantly associated with Kdm2b’s promoter. Interestingly, depletion of LncKdm2b significantly attenuated the association between T5 and Kdm2b’s TSS, suggesting transcribed LncKdm2b maintains Kdm2b’s expression by inducing a local 3D chromatin structure to bring close Kdm2b’s enhancer and promoter (Figs. 3B and S4A). In addition, luciferase (Luc) assays revealed that the 1.67 kb-long DNA fragment containing the T5 locus has strong enhancer/promoter activities in Neuro-2a cells when it was reversely placed (opposite of Kdm2b’s transcription direction) at 5′ of the firefly Luc cassette (Fig. 3C). We further narrowed the T5 locus to an evolutionarily conserved 484 bp-long region (T5-mini) and revealed this fragment can also significantly drive Luc expression if reversely placed at 5′ of the firefly Luc cassette. In line with the T5 locus being an evolutionarily conserved cis-regulatory element, both mouse T5 and T5-mini sequences are able to drive reporter gene expression in human HEK293T cells (Fig. S4B).

LncKdm2bregulates the configuration ofKdm2b’scis-elements. (A) Schematic illustration of the LncKdm2b/Kdm2b locus. The top tracks show ChIP-seq signals of H3K4me3, H3K27ac, H3K4me1, H3K27me3 and CTCF; and DNase I hypersensitivity (HS) in E14.5 mouse brain, along with sequence conservation among mammals. Bottom tracks show a higher-magnification view of the genomic region covering the promoter for Kdm2b and its upstream region that transcribes LncKdm2b. Indigo box indicates the conservative region T5 that is also enriched with H3K4me1. KUS, Kdm2b upstream sequence. T1 to T7 marks putative regulatory cis-elements. (B) Relative crosslinking frequency measured in Neuro-2a cells by 3C-qPCR using a constant primer in an EcoRI fragment at the Kdm2b TSS. Cells were treated with Scramble ASO or a mix of ASOs targeting LncKdm2b (ASO 1, 2, 3, 4) for two days. Crosslinking frequency is relative to a negative region (the magenta arrow). (C) Luciferase activities in experiments where indicated vectors were transfected into Neuro-2a cells for 24 h. ‘Forward’ and ‘Reverse’ indicate directions same as or opposite to Kdm2b’s transcription orientation. (D) E13.5 mouse cortices were electroporated with KUS-d2EGFP or KUSR-d2EGFP, along with CAG-driving mCherry-expressing vectors. Embryos were sacrificed at E15.5, followed by TBR2 immunofluorescent stainings on coronal cortical sections. Scale bars, 50 μm. KUSR: Kdm2b upstream sequence, reversed. (E) RT-qPCR analysis of LncKdm2b and Kdm2b RNA levels in NE-4C cells with the T5 region knocked out. (F) RT-qPCR analysis of LncKdm2b and Kdm2b RNA levels in cortical cells with the T5 region knocked out. EGFP+ cells express gRNAs and the Cas9 protein. In (B), quantification data are shown as mean ± SD (n = 3). In (C), (E), and (F), quantification data are shown as mean + SD (n = 3). In (B), statistical significance was determined using 2-tailed Student’s t test. In (C), statistical significance was determined using 1-way ANOVA with Tukey’s post hoc tests. In (E) and (F), statistical significance was determined using 2-way ANOVA followed by the Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, “ns” indicates no significance. The y-axis represents relative expression normalized to Gapdh
To validate if the genomic region embedded with the T5 element is sufficient to initiate spatiotemporal transcription in cortices, we cloned a piece of 8.0 kb genomic DNA (KUS—Kdm2b upstream sequence, −0.6 kb to +7.3 kb relative to Kdm2b’s TSS) from the mouse genome. In utero electroporation assay revealed that this genomic region alone can efficiently drive the expression of short-lived d2EGFP (Corish and Tyler-Smith, 1999) in embryonic cortices at either orientation with a pattern reminiscent of endogenous Kdm2b or LncKdm2b (Fig. 3D). In addition, we found the T5 region is essential for Kdm2b’s expression, as genomic deletion of T5 leads to compromised Kdm2b’s expression in NE-4C cells and in cortical cells (Figs. 3E, 3F, S4C, S4D and S4E). Together, these data indicate the Kdm2b’s upstream region contains evolutionarily conserved cis-regulatory elements essential for expression of Kdm2b and LncKdm2b, and its configuration is modulated by LncKdm2b.
LncKdm2b facilitates a permissive chromatin environment for Kdm2b’s expression by associating with hnRNPAB
In order to test whether LncKdm2b displays intrinsic ability to promote gene expression, we used the Gal4-λN/BoxB system to tether this lncRNA to a heterologous reporter promoter (Fig. S5A) (Wang et al., 2011; Li et al., 2013; Trimarchi et al., 2014). The data showed the full-length LncKdm2b and its evolutionarily conserved 5′ part (1–908 nt, transcribed from LncKdm2b gene’s first and second exons) could enhance luciferase activities in a dosage-dependent manner, whereas its less conserved 3′ part (909–1,896 nt) couldn’t (Fig. S5B–D). Therefore, the 5′ conserved part of LncKdm2b’s transcript bears intrinsic-activating function. LncKdm2b’s intrinsic-activating capability could be due to its association with trans-factor(s). We carried out RNA pull-down experiments using biotinylated LncKdm2b and antisense-LncKdm2b RNAs. RNA pull-down assay was performed using nuclear protein extracts from cortical NPCs followed by mass spectrometry (MS). A number of RNA binding proteins were enriched in LncKdm2b-precipitating extracts compared to those precipitated by antisense LncKdm2b (Table S2). One of the most enriched protein is heterogeneous nuclear ribonucleoprotein A/B (hnRNPAB), which is validated by RNA pull-down followed by immunoblotting (Fig. 4A and 4B). Notably, SATB1, the protein partner of LncKdm2b in group 3 innate lymphoid cells (ILC3) cells (Liu et al., 2017), was not identified to be associated with LncKdm2b in this study, probably due to cellular specificity. hnRNPAB is dynamically expressed during brain development and has implications in neuronal differentiation (Sinnamon et al., 2012). Depletion of hnRNPAB in Neuro-2a cells significantly decreased Kdm2b’s expression (Fig. S5E). In control experiments, knockdown the expression of Dhx9, Satb1, Bptf, Hnrnpa2b1, Hnrnpa3, Dhx5, Ncl or Lmnb1, genes encoding other putative LncKdm2b-associated proteins, had no significant effect on Kdm2b’s expression (Fig. S5E). RNA in situ hybridization followed by immunofluorescent staining showed colocalization of LncKdm2b and hnRNPAB in cortical NPCs (Fig. S5F). RNA immunoprecipitation experiments (RIP) in either native or the formaldehyde-fixed condition confirmed association of hnRNPAB with LncKdm2b but not with Actb or Gapdh RNAs (Fig. 4C and 4D).

LncKdm2btranscripts can activate gene expression and modulate local chromatin state. (A) Identification of proteins associated with LncKdm2b. Protein extracts from E14.5 mouse cortices were incubated with the biotinylated LncKdm2b (sense) or control (antisense LncKdm2b) followed by SDS-PAGE and silver staining. (B) Immunoblots of hnRNPAB and SATB1 of protein extracts that are associated with sense or antisense LncKdm2b in Neuro-2a cells. (C and D) RNA immunoprecipitation (RIP) of anti-hnRNPAB and control IgG antibodies in native (N-RIP, C) and formaldehyde treated (F-RIP, D) E14.5 mouse embryos cortex. Extracted RNAs were subjected to RT-qPCR analysis of indicated transcripts. (E) Digoxigenin-labeled LncKdm2b truncations were incubated with Flag-hnRNPAB bound to anti-Flag-agarose beads. hnRNPAB associated RNAs were chemiluminescently detected. (F) Left: schematic diagram showing the putative stem-loop structure in LncKdm2b’s 455–908 nt region. Right: point mutations made to disrupt hairpin formation. (G) Digoxigenin-labeled LncKdm2b’s stem loops described in (F) were incubated with Flag-hnRNPAB bound to anti-Flag-agarose beads. hnRNPAB associated RNAs were chemiluminescently detected. (H) EMSA assays of digoxigenin-labeled LncKdm2b RNA (463–625 nt) incubated with purified hnRNPAB. (I and J) Schematic illustration of primer sets used in ChIP-qPCR experiments around the TSS of the LncKdm2b/Kdm2b locus (I) and the T5 region (J). Kdm2b’s promoter region (pKdm2b) and the putative hnRNPAB-binding CArG box for luciferase reporter assay in (M) was also shown. (K and L) ChIP-qPCR analysis of indicated primer sets showed in (I and J) enriched by anti-hnRNPAB antibody upon depletion of LncKdm2b. The y-axis shows fold enrichment normalized to scramble ASO control. (M) Relative luciferase activity of T5 region with or without the CArG box in Neuro-2a cells. Cells were treated for two days with siRNAs against hnRNPAB. (N) Relative crosslinking frequency between the T5 and Kdm2b’s TSS upon hnRNPAB depletion measured by 3C-qPCR in Neuro-2a cells. The y-axis shows fold enrichment normalized to the scramble control (siNC). (O and P) Neuro-2a cells were treated with Scramble ASO or a mix of ASOs targeting LncKdm2b (ASO 1, 2, 3, 4) for 48 h before ChIP-qPCR of H3K4me3 (O), and H3K27ac (P) at the Kdm2b promoter. The y-axis shows fold enrichment normalized to the input. Positions of promoter primers are shown on the bottom of Fig. 3A. Quantification data are shown as mean + SD (n = 3). In (C and D), (K and M), and (O and P), statistical significance was determined using 2-tailed Student’s t test. In (N), statistical significance was determined using 1-way ANOVA with Tukey’s post hoc tests. *P < 0.05, **P < 0.01, ***P < 0.001, “ns” indicates no significance
In line with the fact that the 5′ conserved part of LncKdm2b has intrinsic-activating function (Fig. S5B–D), in vitro binding experiments indicated the two 5′ conserved regions (1–454 nt and 455–905 nt) of LncKdm2b could interact with hnRNPAB, with the 455–905 nt region having stronger association with hnRNPAB than the 1–454 nt region. On the other hand, the 3′ non-conserved region (909–1,391 nt and 1,392–1,872 nt) couldn’t associate with hnRNPAB (Fig. 4E). RNA structure analysis using RNAfold predicts two stem-loops in the 455–905 nt region (Fig. S5G). Particularly, the stem-loop 1 (463–625 nt) has two hairpin arms, P1 and P2 (Fig. 4F). To ask if these hairpin arms are required for the interaction between LncKdm2b and hnRNPAB, we mutated a few nucleotides to disrupt the hairpin formation (Fig. 4F). In vitro binding experiments indeed showed disruption of the hairpin formation in P1 would greatly compromise the interaction (Fig. 4G). Moreover, restoration of the P1 hairpin (P1 rescue) would partially rescue the association, but the P2 hairpin or the stem-loop 2 (840–918 nt) is not required for the association of LncKdm2b with hnRNPAB. The EMSA (electrophoretic mobility shift assay) experiment further validated the binding of LncKdm2b’s stem-loop 1 region (463–625 nt) to hnRNPAB (Fig. 4H). Together, these analyses revealed that the hairpin P1 of LncKdm2b’s conserved 5′ part directly interacts with hnRNPAB, which might be responsible for LncKdm2b’s intrinsic-activating function (Fig. S5B–D).
hnRNPAB, also known as CArG box-binding factor-A (CBF-A), is an RNA binding protein with transcription activity (Venkov et al., 2007; Zhou et al., 2014). We went on to ask if hnRNPAB binds to genomic regions essential for Kdm2b expression, and if the binding is regulated by LncKdm2b. ChIP-qPCR showed hnRNPAB binds to multiples sites in the Kdm2b’s promoter and the T5 region, many of which were positively regulated by LncKdm2b (Figs. S5H and 4I–L). The reporter activity driven by Kdm2b’s promoter (pKdm2b) is mediated by the hnRNPAB-binding CArG box. Downregulating hnRNPAB would significantly lower pKdm2b’s reporter activity, whereas exert no effect on CArG box-deleted pKdm2b (Fig. 4M). Moreover, the association between the T5 and Kdm2b’s TSS was significantly compromised upon hnRNPAB depletion (Fig. 4N). Finally, the Kdm2b’s promoter (−78 bp to −20 bp relative to Kdm2b’s TSS) was less enriched for H3K4me3 and H3K27ac, two histone markers indicative of active transcription, in LncKdm2b-depleted Neuro-2a cells (Fig. 4O and 4P). Collectively, Kdm2b’s expression correlates positively with the association between Kdm2b’s promoter and an essential enhancer (T5), which is facilitated by LncKdm2b’s transcripts and its associated protein hnRNPAB. These findings point a role of LncKdm2b in regulating transcription locally.
KDM2B promotes cortical neuronal differentiation
Since LncKdm2b regulates the expression of Kdm2b, and Kdm2b is transiently expressed in freshly born projection neurons, we next explored roles and mechanisms of KDM2B in cortical neurogenesis. We first electroporated E13.5 embryonic cortices with plasmids overexpressing Kdm2b and collected brains at E15.5 (Fig. S6A). Significantly more Kdm2b transduced cells reside in the cortical plate (CP, future cortices) with fewer cells in the VZ/SVZ, indicating accelerated cortical neurogenesis and radially neuronal migration (Fig. 5A and 5B). In line with this, fewer mCherry+Kdm2b transduced cells express TBR2 and PAX6, markers for IPCs and RGPCs respectively (Fig. 5C–E). Embryonic brains of Kdm2bCreERT2/CreERT2 mice have significant amount of residual KDM2B protein probably due to inefficient transcriptional termination (Fig. S6B), which might lead to subsequent use of alternative start codons. We therefore performed Kdm2b loss-of-function studies by electroporating plasmids expressing short-hairpin RNAs (shRNAs) against Kdm2b into E13.5 embryonic cortices. To minimize non-specific effects, we chose the shmiRNA system to express long RNA hairpins with shRNAs embedded into endogenous miRNA loop and flanking sequences (Bauer et al., 2009; Baek et al., 2014). Significant more Kdm2b-shRNA electroporated cells reside in the VZ/SVZ at E16.5 (Fig. 5F and 5G). Next, E16.5 Kdm2b-shRNA transduced cortices were immuno-stained with TBR2 and NEUROD2, a transcriptional factor expressed in cortical PNs. Results showed more transduced cells are co-labeled with TBR2 but fewer cells express NEUROD2, with significantly more NEUROD2+ transduced cells localized in the VZ/SVZ (Figs. 5H, 5I, S6C and S6D). Moreover, more transduced cells are colocalized with PAX6-positive RGPCs (Fig. 5J and 5K). This phenotype can be fully rescued by simultaneously overexpressing Kdm2b (Figs. 5F, 5G, S6E and S6F). Furthermore, significantly more Kdm2b-depleted cells (EGFP+) are BrdU positive and in S-phase, as embryos were injected BrdU 30 min before sacrifice; and more PAX6+EGFP+ RGPCs are BrdU positive, suggesting depletion of Kdm2b promotes proliferation of RGPCs (Fig. 5L–N). We didn’t observed changes of programmed cell death (cleaved caspase-3+ cells) in Kdm2b-shRNA transduced cortices (Fig. S6G). All these data support the notion that KDM2B promotes cortical neuronal differentiation in vivo (Table S3).

Kdm2bpromotes cortical neurogenesis. (A–E) E13.5 mouse cortices were electroporated with empty or KDM2B-expressing vector, along with mCherry-expressing vector to label transduced cells. Embryos were sacrificed at E15.5 for immunofluorescent analysis. Coronal Sections were stained for DAPI (A), and the relative location of mCherry-positive cells was quantified (B). Ten embryos in control and nine embryos in KDM2B-overexpression. Representative VZ/SVZ images of control and KDM2B-expressing cortices immunostained for TBR2 (top) and PAX6 (bottom). Arrowheads denote double-labeled cells (C). Quantification of TBR2+ (D) or PAX6+ (E) cells in transduced cells. (F–N) E13.5 mouse cortices were electroporated with indicated combination of vectors, with transduced cells labeled with EGFP. Embryos were sacrificed at E16.5 for immunofluorescent analyses. The relative location of EGFP+ cells was quantified (F and G). Three embryos in scramble and shKDM2B, five embryos in shKDM2B plus KDM2B. Representative VZ/SVZ images of scramble or KDM2B shRNA electroporated sections immunostained for TBR2 (H) and quantification of TBR2+ transduced cells (I). Representative VZ/SVZ images of scramble or KDM2B shRNA electroporated sections immunostained for PAX6 (J) and quantification of PAX6+ transduced cells (K). Sections were co-immunostained with PAX6 and BrdU (30 min) (L). Percentiles of BrdU+ transduced cells (M) and of BrdU+PAX6+EGFP+/PAX6+EGFP+ cells (N). In (B), (D and E), (G), (I), (K), and (M and N), quantification data are shown as mean + SEM. In (B), (D and E), (I), (K), and (M and N), statistical significance was determined using 2-tailed Student’s t test. In (G), statistical significance was determined using 2-way ANOVA followed by the Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, “ns” indicates no significance. Scale bars, 50 μm. VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone; CP, cortical plate
LncKdm2b promotes cortical neuronal differentiation via KDM2B
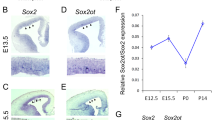
As we have shown that LncKdm2b is transiently expressed in freshly born projection neurons and LncKdm2b cis-activates Kdm2b expression, we expected that LncKdm2b and Kdm2b may have similar function on cortical neuronal differentiation. To this end, we first knocked down the expression of Kdm2b or LncKdm2b by transfecting adherent-cultured cortical progenitor cells (NPCs) with low titer lentiviral shRNAs to study cell fate changes at the clonal level. NPCs depleted with Kdm2b or LncKdm2b showed enhanced self-renewal but decreases neuronal differentiation: significantly more Kdm2b or LncKdm2b-depleted cortical cells expressing SOX2 with fewer cells expressing TUJ1 compared to scramble shRNA-transfected cells (Fig. 6A and 6B); more precursor-containing clones with fewer neuron-containing clones and fewer TUJ1-only neuronal clones (Fig. 6C–E); and more SOX2+ cells per clone upon Kdm2b or LncKdm2b depletion (Fig. 6F and 6G). Thus, LncKdm2b and Kdm2b are required for proper neuronal differentiation of cortical NPCs invitro (Table S3).

LncKdm2bmaintains mouse cortical neurogenesis through KDM2B. (A–G) E12.5 cortical neural precursors were infected with lentivirus expressing indicated shRNAs for three days followed by immunostaining of SOX2 and TUJ1. Transfected cells were labeled with ZsGreen. Quantification analyses were performed to calculate percentiles of SOX2+ (A) or TUJ1+ (B) ZsGreen+ transduced cells; percentiles of clones with at least one SOX2+ precursor (C), clones with at least one TUJ1+ neuron (D), neuron only clones (E); and the average number of SOX2+ cells in SOX2+ clones (F and G). (H–J) E13.5 mouse cortices were electroporated with indicated siRNAs and vectors, with transduced cells labeled with EGFP. Embryos were sacrificed at E16.5 for PAX6 immunofluorescent staining (H). The relative location of EGFP+ cells (I) and percentiles of PAX6+ transduced cells (J) were quantified. Three embryos in control (siNC), five embryos in siKDM2B, LncKdm2b ASO, and LncKdm2b ASO plus KDM2B. (K–N) E13.5 mouse cortices were electroporated with indicated siRNAs, with transduced cells labeled with EGFP. Embryos were sacrificed at E16.5 for PAX6 immunofluorescent staining (M). The relative location of EGFP+ cells (L) and percentiles of PAX6+ transduced cells (N) were quantified. Three embryos each. In (A–G), (I and J), (L), and (N), quantification data are shown as mean + SEM. (L) and (N) * indicates P value < 0.05, ** indicates P value < 0.01, *** indicates P value < 0.001, “ns” indicates no significance. Scale bars, 50 μm. VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone; CP, cortical plate
Next, we explored whether LncKdm2b regulates cortical neurogenesis in vivo through Kdm2b. E13.5 embryonic cortices were electroporated with siRNAs or antisense oligonucleotides (ASO) targeting Kdm2b or LncKdm2b respectively followed by phenotypic analyses at E16.5. Transcripts of LncKdm2b and Kdm2b were efficiently down-regulated in cortical cells electroporated with ASOs against LncKdm2b (Fig. S7A). Significantly fewer siKDM2B- or LncKdm2b ASO- transduced cells reside in the CP with more cells in the VZ/SVZ, indicating delayed neuronal differentiation. In line with this, more transduced cells express PAX6. LncKdm2b depletion doesn’t lead to enhanced apoptosis (Fig. S7B). Most importantly, overexpressing Kdm2b can mostly rescue the phenotypes caused by LncKdm2b knockdown (Fig. 6H–J). Finally, we ask if hnRNPAB, the LncKdm2b-associated protein, also regulates neuronal differentiation in developing neocortex. To this end, we electroporated E13.5 cortices with siRNAs against hnRNPAB and indeed found hnRNPAB-depleted cells showed delayed neuronal migration to the CP at E16.5 and hampered differentiation of NSPCs (neural stem/progenitor cells)—more sihnRNPAB-transduced cells localized in the VZ/SVZ and co-localized with PAX6 (Fig. 6K–N). On the other hand, depletion of hnRNPA2B1 didn’t cause such defects (Fig. S7C–E). Moreover, overexpression of LncKdm2b has no effect on neuronal migration and differentiation (Fig. S7F and S7G), which is in line with aforementioned data showing LncKdm2b couldn’t trans-activate Kdm2b expression (Fig. S3H–J). Together, LncKdm2b promotes cortical neuronal differentiation via KDM2B (Table S3).
We finally analyzed fates of LncKdm2b-depleted cells at postnatal day 10 (P10), when cortical development is largely complete. Electroporated cortical cells and their progenies were labeled with stably-expressed EGFP mediated by the piggyBac transposon (Fig. S8A). More LncKdm2b-depleted cortical cells reside in deep layers, the white matter and the SVZ with fewer cells in upper layers (Fig. 7A and 7B). Neuronal differentiation was hampered upon LncKdm2b depletion, but gliogenesis was not altered (Figs. 7C, 7D and S8B), which is in accordance with LncKdm2b’s transient expression in cortical neurogenesis. More LncKdm2b-depleted NeuN+ and SATB2+ neurons reside in deep layers, with more LncKdm2b-depleted cells in SVZ expressing SOX2 (Figs. 7E–H and S8C). Moreover, a good portion of LncKdm2b-depleted cells would form ectopic aggregates (periventricular heterotopias, PH) beneath the white matter and express SATB2 (Fig. 7I), a phenotype indicating defects of neuronal differentiation and migration (Lu and Sheen, 2005; Sarkisian et al., 2008). No increased apoptosis was observed upon LncKdm2b depletion (Fig. S8D). These data support LncKdm2b’s role in maintaining normal neuronal differentiation and migration.

LncKdm2bregulates cortical neuronal differentiation and migration. E13.5 mouse cortices were electroporated with piggyBac-CRISPR/Cas9 vectors and brain sections were analyzed at P10. (A–C) Representative images showing distribution of EGFP+ cells in cortices at P10 (A). The relative locations of EGFP+ cells were quantified (B). Four brains each. Examples of EGFP+ cells with neuronal and glial morphology were positive for SATB2, GFAP and OLIG2 respectively (C). (D) Quantification of EGFP+NeuN+ neurons and EGFP+ glial cells at P10. Four brains each. (E and F) Neuronal migration was analyzed at P10 by quantifying percentiles of NeuN+EGFP+ neurons in each bin. Arrowheads indicate delayed projection neurons. Four brains each. (G and H) Representative images of SOX2 immunofluorescent staining in the SVZ (G). SOX2+EGFP+ cells were quantified (H). Three brains each. (I) The periventricular heterotopias (PH) are evident in sgLncKdm2b-electroporated cortices. Enlarged boxed area at the right shows SATB2+ projection neurons in the PH. (J) A model for LncKdm2b promoting cortical neurogenesis by cis-activating Kdm2b. LncKdm2b and Kdm2b are transiently expressed in freshly born projection neurons. LncKdm2b RNA facilitates an open chromatin configuration locally by bringing together the upstream regulatory cis-element T5, Kdm2b promoter and hnRNPAB to maintain Kdm2b’s transcription. In (B), (D), (F), and (H), quantification data are shown as mean + SEM. In (B), (D), (F), and (H), statistical significance was determined using 2-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, “ns” indicates no significance. In (A), (E), and (I, left), scale bars, 100 μm. In (C), (G), and (I, right), scale bars, 20 μm. WM, white matter; SVZ, subventricular zone; PH, periventricular heterotopias; RGC, radial glial cells; IPC, intermediate progenitor cells; imPN, immature projection neurons; mPN, mature projection neurons; LOF, loss-of-function; GOF, gain-of-function
In summary, we found the precise balance of self-renewal and neuronal differentiation of NSPCs during cortical neurogenesis is modulated by KDM2B. Moreover, the expression of Kdm2b is positively regulated by its divergent lncRNA LncKdm2b, which facilitates a permissive chromatin configuration locally by bringing together the upstream regulatory cis-element T5, Kdm2b’s promoter and hnRNPAB (Fig. 7J).
Discussion
The generation of layer-specific PNs over developmental time is precisely controlled and largely attributed to cell-intrinsic properties of NSPCs (Shen et al., 2006; Gaspard et al., 2008). Cell fates choices are mostly the results of specific transcriptional events, which are coordinated by cis-regulatory elements, cell-specific transcription factors, and epigenetic states including DNA methylation, histone modification and chromatin accessibility (Heintzman et al., 2009; Perino and Veenstra, 2016). Some lncRNAs can regulate gene transcription locally (cis) and/or distally (trans) by modifying epigenetic states (Rinn and Chang, 2012; Berghoff et al., 2013; Grote et al., 2013; Fu, 2014). Here we found lncRNA gene LncKdm2b shares the same promoter with its bidirectional protein-coding gene Kdm2b, and both of them are transiently expressed in committed IPCs and freshly-born PNs during cortical neurogenesis. Unlike most bidirectional coding-noncoding transcripts, LncKdm2b’s expression level is comparable with that of Kdm2b at the peak of cortical neurogenesis, strongly indicating LncKdm2b’s regulatory roles. Indeed, LncKdm2b maintains Kdm2b’s expression in cis to control neuronal differentiation and migration. Mechanistically, the LncKdm2b transcripts enhances physical association of Kdm2b’s promoter and a key enhancer T5 via binding to hnRNPAB. LncKdm2b’s transcript, especially its evolutionarily conserved 5′ part, bears intrinsic-activating function and interacts with hnRNPAB via one of its putative stem-loop structures (Figs. 4A–H and S5A–D). Similarly, a 5′ fragment of LncKdm2b (450–700 nt) is necessary for its binding to SATB1 or SRCAP in ILC3 and ES cells respectively (Liu et al., 2017; Ye et al., 2018). hnRNPAB, an RNA binding protein with transcription activity (Venkov et al., 2007; Zhou et al., 2014), was shown to be associated with Kdm2b’s TSS and the T5 region in neural cells, and the strength of the association depends on the presence of LncKdm2b (Figs. S5H, 4K and 4L). Moreover, the cis-activity of Kdm2b’s promoter also relies on hnRNPAB’s binding (Fig. 4M). The core T5-region (T5-mini), a conserved cis-regulatory element embedded in LncKdm2b’s second intron, can drive gene expression in both mouse and human cells when reversely placed upstream of reporters, and its deletion results in decreased expression of Kdm2b. In summary, this study indicates a role of lncRNA in coordinating the association of cis-regulatory elements (Kdm2b’s TSS and T5) and trans-factor(s) (hnRNPAB) in transcriptional regulation, which probably relies on RNA’s specific secondary structures.
A recent study by Liu et al. showed LncKdm2b activates expression of Zfp292 in trans via recruiting the chromatin organizer SATB1 and the nuclear remodeling factor (NURF) complex onto the Zfp292 promoter in innate lymphoid cells (ILCs) (Liu et al., 2017). Similarly, LncKdm2b activates the expression of Zbtb3 by promoting the assembly and ATPase activity of the SRCAP complex in mESCs (Ye et al., 2018). Surprisingly, these studies didn’t detect expression alterations of Kdm2b in LncKdm2b-null ILC3s and mESCs. In our study, LncKdm2b was not found to be associated with SATB1. Furthermore, the expression levels of Zfp292 were not decreased in neural cells depleted with LncKdm2b (Fig. S3C–E). These discrepancies could be due to different cellular context and/or distinct inactivation approaches. LncKdm2b’s second exon was deleted in previous studies to abolish its transcripts, which might not hamper LncKdm2b’s transcription process per se and/or LncKdm2b’s conserved region with intrinsic-activating function could still exist. In fact, a good fraction of LncKdm2b’s transcripts derived from the first and third exons could be detected in NE-4C cells with LncKdm2b’s second exon deleted (Fig. 2D). In contrast, our study also used siRNAs and ASOs to target LncKdm2b, and inserted pAS sites into LncKdm2b’s first intron in mESCs, thus impeding LncKdm2b transcription, ultimately leading to attenuation of Kdm2b transcription (Fig. 2). Interestingly, although LncKdm2b controls Kdm2b’s expression at the transcriptional level in cell nuclei, a good fraction of LncKdm2b transcripts resides in the cytoplasm. Previous studies also indicated LncKdm2b localizes in both nuclei and cytosol in mESCs and innate lymphoid cells (Liu et al., 2017; Ye et al., 2018). It remains to be elucidated if LncKdm2b functions in cytosol, and if LncKdm2b’s cytosolic translocation would facilitate its decay to ensure Kdm2b’s transient expression during neuronal differentiation. This finding is just the beginning to understand how lncRNAs regulate cortical neuronal differentiation by controlling local transcription and might have general implications in cell fate determinations.
It will be also worthy of exploring how the transient expressions of Kdm2b and LncKdm2b are initiated and maintained in cortical IPCs and freshly-born PNs. A report showed KDM2B’s expression in primary MEFs and cancer cells is induced by FGF-2 via CREB phosphorylation and activation, downstream of DYRK1A kinase (Kottakis et al., 2011). Since both FGF-2 and DYRK1A have essential roles in cortical development, it remains to be studied if they regulate KDM2B’s expression in this context (Vescovi et al., 1993; Ghosh and Greenberg, 1995; Fotaki et al., 2002; Benavides-Piccione et al., 2005; Arron et al., 2006). Since normal cortical development is key to neurological functions such as cognition, KDM2B may have implications in neuropsychiatric disorders. In line with this, KDM2B is among the most frequently deleted genes in the 12q24.31 microdeletion syndrome, which is characterized by principal clinical features including autism, intellectual disability, epilepsy, and craniofacial anomalies (Labonne et al., 2016). Intriguingly, human LncKDM2B is also transcribed divergently from the promoter of KDM2B with high sequence homology with LncKdm2b (Ye et al., 2018). It remains to be investigated if LncKDM2B’s cis-regulating roles and KDM2B’s function in promoting neuronal differentiation are conserved in human.
Materials and methods
Mouse
All animal procedures were approved by the Animal Care and Ethical Committee of College of Life Sciences at Wuhan University. CD-1 and C57BL/6 mice were purchased from HNSJA. Mice were housed in a certified specific-pathogen-free (SPF) facility. The noon of the day on which the vaginal plug is found is counted as embryonic (E) day 0.5.
Generation of Kdm2b CreERT2/+ knock-in reporter mice
Kdm2bCreERT2/+ knock-in reporter mice were generated by Biocytogen (Beijing, China). A sequence encoding the self-cleaving T2A peptide was fused in frame with exon 3 of the Kdm2b followed by the CreERT2-IRES-EGFP cassette. To generate the Kdm2b targeting vector, a 1 kb 5′ homology (LR), a 1 kb 3′ homology arm (RR), F2a-iCreERT2, or IRES-EGFP were amplified by PCR. Fragment LR and F2a-iCreERT2 were overlapped to form fragment LR-F2a-iCreERT2 (SalI to BamHI). Fragment IRES-EGFP and RR were overlapped to form IRES-EGFP-RR (BamHI to SacI). Then fragment LR-F2a-iCreERT2 and IRES-EGFP-RR were cloned into the TV-2G vector. For cloning the sgRNA-expression cassette, annealed DNA was ligated with pT7-sgRNA. sgRNAs were transcribed in vitro by T7 RNA Synthesis Kit (NEB). Targeting vector, Cas9 vector, and sgRNAs were microinjected into mouse zygotes. After injection, zygotes were immediately transferred into pseudo-pregnant female mice to generate founders, which were genotyped by PCR and sequencing. Positively founders were crossed with C57BL/6 wild-type mice to generate F1 mice. F1 mice were screened by PCR, and positive mice were confirmed by Southern blot using the iCre internal probe and 3′ external probe. The genders of embryos were not determined for analyses conducted in this study. See Table S4 for sgRNA sequences and genotyping primers.
Genetic lineage-tracing
All animals used for analyses in Figs. 1 and S1 were heterozygous for the Cre allele (Kdm2bCreERT2/+). In Fig. S2, data were generated by crossing Kdm2bCreERT2/+ with Ai14fl/fl animals, both with congenic C57BL/6J backgrounds. Tamoxifen was dissolved in corn oil as previously described (Guo et al., 2013). To perform lineage-tracing analyses using the Kdm2bCreERT2/+;Ai14 mice, tamoxifen was injected into pregnant dams at indicated stages with a concentration of 100 mg/kg body weight.
Generation of LncKdm2b polyA knock-in (mESCsLncKdm2b-pAS/+) and Kdm2b indels mouse ES cells
LncKdm2b polyA knock-in mouse ES cells (mESCsLncKdm2b-pAS/+) were generated by Biocytogen (Beijing, China). The targeting vector contains two homology arms (1 kb each), the 3× SV40 polyA signal sequence and a BGH polyA signal (a total of 4× polyA signals), followed by an expression cassette of ΔTK and Neo flanked by two loxP sites. The targeting vector was electroporated into mouse ES cells with Cas9-expressing vectors and sgRNAs that target the genomic site 1.8 kb downstream of the LncKdm2b TSS. Out of 200 neomycin resistant clones, one heterozygous knock-in ESC clone was obtained through PCR and sequencing analyses. To generate mESCsLncKdm2b-pAS/+ with Kdm2b indels, sgRNAs that target the second exon of Kdm2b were electroporated into mESCsLncKdm2b-pAS/+. mESC clones with distinguishable indel mutations between two alleles were selected by PCR and sequencing analyses. See Table S4 for sgRNA sequences, genotyping and qPCR primers.
Cell lines
HEK293T cells were gifts from Dr. Hongbing Shu (Wuhan University). Neuro-2a cells and NE-4C cells were purchased from the Cell Bank of Chinese Academy of Sciences Cells were maintained in indicated culture media (DMEM or MEM) containing 10% fetal bovine serum (Life Technologies or Hyclone) and used within ten passages since arrival.
Plasmids construction
For constructing eukaryotic expression vectors, full-length mouse Kdm2b was PCR amplified from the pMXs-Kdm2b-Flag vector, a gift from Dr. Baoming Qin (Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences), then subcloned into the pCAGGS vector using EcoRI/MluI. KDM2B shRNA-resistant mutants were constructed using site-directed mutagenesis. Full-length LncKdm2b was PCR amplified from the cDNA of the E16.5 C57BL/6 embryonic cortex, and the PCR product was cloned into pCAGGS using EcoRI/NotI. All putative ORFs of LncKdm2b were fused in frame at their 3′ with sequence encoding 3× Flag tag and cloned into the eukaryotic expression vector pFLAG-N3 (a gift from Dr. Zhiyin Song, Wuhan University) using EcoRI/SacII. The CDS sequence of mouse hnRNPAB was PCR amplified from the cDNA of the E16.5 C57BL/6 embryonic cortex, was cloned into the eukaryotic expression vector pFLAG-N3 using XhoI/EcoRI in frame with sequence encoding C-terminal 3× Flag tag. pCALNL was a gift from Dr. Xiaoqun Wang (Institute of Biophysics, Chinese Academy of Sciences). Luciferase reporter vector was constructed according to the previous study (Li et al., 2017). Briefly, the T5 Forward, T5 Reverse, T5-mini Forward, or T5-mini Reverse were PCR amplified from the genomic DNA of C57BL/6 mice and cloned into pGL3-Basic Vector using MluI and XhoI. Kdm2b promoter and Kdm2b promoter with the CArG box deletion were cloned into pGL3-Basic Vector using SacI and XhoI. For constructing RNA tethering vectors, the LacZ sequence from pcDNA3-BoxB-LacZ was removed by XhoI and XbaI digestion, and LncKdm2b was amplified from the pCAGGS-LncKdm2b Vector and cloned into the same sites with XhoI and XbaI. 5× UAS-TK-Luc, pcDNA3-Gal4-λN, and pcDNA3-BoxB-LacZ were gifts from Dr. Xiang Lv (CAMS & PUMC). For constructing KUS-d2EGFP or KUSR-d2EGFP, EGFP and ODC (422–461 aa) were amplified by PCR, overlapped to form fragment d2EGFP (EcoRI to BglII) (Corish and Tyler-Smith, 1999). Then d2EGFP was cloned into the pCAGGS vector using EcoRI and BglII. pCAGGS-d2EGFP was digested by ApaI, followed by Mung Bean Nuclease (Takara Bio) modification and removal of the CAG promoter by SalI digestion. KUS or KUSR were PCR amplified from the genomic DNA of C57BL/6 mice and cloned into the same site with XhoI. For construction short-hairpin RNA (shRNA) vectors, the oligonucleotides for shRNA targeting Kdm2b or LncKdm2b were cloned into pLKO.1-zsGreen or pCAG-mir30 vectors using AgeI/EcoRI or XhoI/EcoRI. A scramble shRNA plasmid was used as a negative control. Primer sequences for all constructs were listed in Table S4.
Protein expression and purification for hnRNPAB
Plasmids expressing Flag-tagged hnRNPAB were transfected into HEK293T cells. Cells were harvested after 2 days to achieve optimal expression. 2 × 108 HEK293T cells were resuspended in lysis buffer [20 mmol/L Tris pH 8.0, 100 mmol/L NaCl, 1 mmol/L PMSF, protease inhibitor cocktail (Biotool)] followed by sonication with 30% power output (3 min, 0.5 s on, 0.5 s off) on ice. After centrifugation at 12,000 rpm for 10 min at 4 °C, supernatants were incubated with 50 μL anti-Flag agarose beads (Biotool) for 2 h at 4 °C. The agarose beads were washed 4 × 5 min with TBS buffer, and bound protein was eluted with 200 ng/μL 3× FLAG peptide (Sigma-Aldrich, F4799) in TBS buffer at 4 °C for 30 min. the eluted sample was ultrafiltrated and concentrated with 0.5 mL Amicon Ultra-centrifugal filters (Millipore, UFC501024). The concentration of purified protein was determined using Bicinchoninic Acid Protein Assay Kit (Beyotime Biotechnology) and by Western blot.
Lentivirus production and cell infection
To obtain lentiviral particles, HEK293T cells (5 × 106 cells in a 10-cm dish) were transiently transfected with 12 μg pLKO.1 shRNA constructs, 6 μg of psPAX2 and 6 μg pMD2.G. The supernatant containing lentivirus particles was harvested at 48 h after transfection, and filtered through Millex-GP Filter Unit (0.22 μm pore size, Millipore). Viral particles were then stored at −80 °C ultra-cold freezer until use. Cortical NPCs infected by lentivirus at a low viral titer. Knockdown efficiency was evaluated by RT-qPCR analysis three days post-infection.
Antisense oligonucleotide (ASO) treatment
Phosphorothioate-modified antisense oligodeoxynucleotides (ASOs) were synthesized at BioSune (Shanghai, China), and transduced into Neuro-2a cells using Lipofectamine® 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocol at 100 nmol/L. For transfecting primary NPCs, ASOs were introduced into tertiary cortical NPCs derived from E14.5 CD-1 mouse cortex by nucleofection (Lonza) according to the manufacturer’s protocol at 1 μmol/L for 1 × 106 cells. Optimal programs and solutions of the Lonza Cell Line Nucleofector Kit for the ASO delivery were tested. Total RNAs were collected for RT-qPCR analysis two days post-transfection. Total protein was collected for immunoblotting analysis four days post-transfection. See Table S4 for ASO sequences.
Knockout of Lnckdm2b and T5 by CRISPR/Cas9
CRISPR/Cas9-mediated genomic knockout was performed essentially as described previously (Cheng et al., 2016). Briefly, annealed oligonucleotides for sgRNAs targeting T5 or Lnckdm2b’s exon2 were cloned into a piggyBac-based vector (pPB-sgRNA-Cas9). pPB-sgRNA-Cas9 and the transposase-expressing vector were mixed in a 1:1 ratio and co-transfected into NE-4C cells using Lipofectamine 3000 or electroporated into E13.5 CD-1 mouse embryonic cortices. NE-4C cells that stably express Cas9 and sgRNAs through transposon-mediated random insertion were selected by flow cytometry and maintained as mono-clones for two weeks. Individual NE-4C clones (23–26 clones) were picked, expanded and analyzed by PCR genotyping. E15.5 cortical cells were isolated from embryonic cortices two days after electroporation and maintained in neurosphere culture medium for a week. Cortical cells that stably express Cas9 and sgRNAs were selected by flow cytometry and an aliquot was subjected to genomic DNA or RNA isolation followed by PCR genotyping and qPCR. See Table S4 for sgRNA sequences, genotyping and qPCR primers.
Northern blot
Dorsal forebrain tissues were resected from E14.5 and E16.5 mouse embryos under dissecting microscopes. Total RNAs were extracted twice using Trizol (Thermo Fisher). The polyA+ RNA fractions were enriched using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB). About 1 μg of polyA+ RNA from each sample was subjected to formaldehyde denaturing agarose electrophoresis followed by transferring to positively charged nylon membrane with 20× SSC buffer (3.0 mol/L NaCl and 0.3 mol/L sodium citrate, pH 7.0). Membrane was UV-cross-linked and incubated with DIG-labeled RNA probes (LncKdm2b, 217–1307 nt) generated by in vitro transcription with the DIG-RNA Labeling Mix (Roche). Hybridization was done overnight at 65 °C in DIG Easy Hyb Hybridization solution (Roche). Membranes were stringently washed three times in wash buffer 1 (0.1× SSC and 0.1% SDS) for 15 min at 65 °C, then rinsed in wash buffer 2 [0.1 mol/L maleic acid, 0.15 mol/L NaCl, 0.3% Tween 20 (pH 7.5)] and incubated in blocking reagent (Roche) for 1 h at room temperature. Subsequently, membranes were incubated with a 60,000-fold dilution of anti-DIG-AP Fab fragment (Roche) in blocking reagent for 30 min at room temperature, washed three times in wash buffer 2 for 10 min at room temperature, and immersed in detection buffer [0.1 mol/L Tris·HCl, 0.1 mol/L NaCl (pH 9.5)] for 5 min. Anti-DIG-AP was detected using CDP-star chemiluminescent substrate for alkaline phosphatase (Roche) and X-ray film exposure. See Table S4 for primers used in generating Northern blot probes.
In situ hybridization (ISH)
To make ISH probes, the 5′-overhang of forward primer was modified with a T7 promoter (See Table S4 for the primers used in ISH probes). Digoxigenin labeled riboprobes were transcribed using the DIG-RNA Labeling Mix (Roche). In situ Hybridization was performed as described (Li et al., 2017). In brief, all solutions were prepared properly to avoid RNase contamination. Digoxigenin-labeled LncKdm2b and Kdm2b riboprobes were transcribed in vitro using NTP mix containing digoxigenin-labeled UTP (Roche). E12.5 CD-1 mouse embryos and E16.5 mouse brains were fixed in chilled 4% paraformaldehyde (Sigma) in 1× PBS overnight followed by treatment of 20% sucrose in 1× PBS overnight. Tissues were embedded in OCT, and 14 μm sections were cut onto slides using a Leica CM1950 cryostat. Sections were permeabilized with 2 μg/mL proteinase K (Sigma) for 10 min followed by acetylation in 0.1 mol/L TEA (triethanolamine) solutions (10 mL 1 mol/L TEA solution and 250 μL acetic anhydride in 90 mL DEPC treated ddH2O) for 10 min. Slides were blocked in hybridization buffer (50% deionized formamide; 5× SSC, 5× Denharts; 250 μg/mL yeast RNA; and 500 μg/mL herring sperm DNA) at room temperature (R/T) for 3 h followed by incubating with 0.1–0.2 ng/μL digoxigenin-labeled riboprobe in hybridization buffer overnight at 60 °C in humidified boxes. Slides were washed with 65 °C 0.1× SSC for three times (20 min each) followed by blocking with 10% heat-inactivated sheep serum in buffer B1 (0.1 mol/L Tris-HCl, pH 7.4; 150 mmol/L NaCl) at room temperature for 1 h. Sections were incubated with an alkaline phosphatase-conjugated anti-digoxigenin antibody (1:5000, Roche) overnight at 4 °C. After washing three times in buffer B1, sections were equilibrated twice in buffer B3 (0.1 mol/L Tris-HCl; 0.1 mol/L NaCl; 50 mmol/L MgCl2; 0.1% Tween-20, pH 9.5) for 10 min. Colorization was performed using NBT/BCIP (Roche) containing B3 solutions at R/T overnight in the dark. Slides were dehydrated, cleared and mounted using gradient ethanol, xylene, and neutral balsam sequentially. Images were collected using a Nikon 80i microscope equipped with Nikon DS-FI1C-U3 camera system.
Immunofluorescence (IF) and immunoblotting
IF and immunoblotting were performed as described (Li et al., 2017). For immunofluorescent staining, 4% paraformaldehyde (PFA) fixed 14 μm sections or cells were permeabilized and blocked with blocking buffer (3% heat-inactivated normal goat serum, 0.1% bovine serum albumin and 0.1% Triton-X 100 in 10 mmol/L Tris-HCl, pH 7.4; 100 mmol/L NaCl) for one hour at R/T. Sections were then incubated with primary antibodies diluted in blocking buffer overnight at 4 °C or R/T. The next day, slides were washed three times for 10 min with 1× PBS and incubated with second antibodies in blocking buffer at R/T for an hour. Slides were mounted with anti-fade solution with DAPI after PBS wash. For triple IF labeling of EGFP/PAX6/BrdU, sections were stained for EGFP/PAX6 antibodies first, then treated with 20 μg/mL proteinase K (Sigma) for 5 min followed by 2 mol/L HCl for 30 min before BrdU staining. All immunofluorescence comparing expression levels were acquired at equal exposure times. Immunoblotting assays were carried out according to standard procedures.
RNA-seq transcriptome analysis
Dorsal forebrain (cortex) tissues were resected from E10.5 or E12.5 mouse embryos under dissecting microscopes. Total RNAs were extracted twice using Trizol (Thermo Fisher) and were treated with DNase I (NEB Biolabs). The integrity of RNAs was analyzed using Agilent Bioanalyzer 2100. Removal of ribosomal RNAs (rRNAs) and construction of libraries for standard strand-specific RNA-seq were performed using Illumina HiSeq 2000 in BGI Tech. Quality control reads alignment, and gene-expression analysis were also carried out in BGI Tech. Some low-quality RNA reads were present in original data. Thus, four kinds of reads were removed before mapping to the mouse genome. 1) Adaptor sequences; 2) Poor quality reads that Q5 or less mass value bases account for more than 50% of the entire reads; 3) Reads that have a proportion of “N” greater than 10%. 4) Reads that align with mouse rRNA. Next, the resulting clean reads were mapped to mouse genome (NCBI37/mm9) by TopHat (Trapnell et al., 2009) and an ab initio transcriptome reconstruction approach was performed by Cufflinks (Trapnell et al., 2012). To explore the expression patterns of coding and non-coding gene across embryonic cortical development, we used the Galaxy platform (Goecks et al., 2010) to integrate RNA-seq data from four other studies (mESCs and NPCs: GSE20851; mouse E14.5 VZ, IZ, and CP: GSE30765; E17.5 cortex: GSE39866; adult mouse cortex: GSE39866, GSE45282) (Guttman et al., 2010; Ayoub et al., 2011; Dillman et al., 2013; Ramos et al., 2013). Finally, we used Cuffnorm to calculate FPKM (fragments per kilobase of exon per million fragments mapped). GO analysis was performed using the DAVID Functional Annotation Bioinformatics Microarray Analysis tool (da Huang et al., 2009). The RNA-seq data of E10.5 or E12.5 mouse cortice were deposited in the Gene Expression Omnibus with accession no. GSE55600.
RNA fractionation
RNA fractionation was performed as previously described (Cabianca et al., 2012). In brief, neural progenitor cells from E14.5 mouse cortices were detached by treating with 1× trypsin, counted and centrifuged at 168 ×g for 5 min. The pellet was lysed with 175 μL/106 cells of cold RLN1 solution [50 mmol/L Tris-HCl, pH 8.0; 140 mmol/L NaCl; 1.5 mmol/L MgCl2; 0.5% NP-40; 2 mmol/L vanadyl ribonucleoside complex (Sangon Biotech)] for 5 min. The suspension was centrifuged at 4 °C and 300 ×g for 2 min. The supernatant, corresponding to the cytoplasmic fraction, was transferred into a new tube and stored on ice. The pellet containing nuclei was corresponding to nuclear fractions. Total RNA was extracted from the cytoplasmic and nuclear fractions using TRIzol solution. The samples were treated with DNase I, washed with 75% ethanol and then resolved in 30 μL RNase-free water. 1 μg of RNA was used for the first-strand synthesis with the PrimerScriptTM Reverse Transcriptase (Takara Bio) using oligo-dT and random primers. cDNA was used for qPCR with iTaqTM Universal SYBR® Green Supermix (Bio-rad) and analyzed by a CFX ConnectTM Real-Time PCR Detection System (Bio-rad). See Table S4 for qPCR primers.
Fluorescent RNA in situ hybridization (FISH) and immunofluorescence microscopy
FISH probes were designed using the Stellaris Probe Designer (Biosearch Technologies) (See Table S4 for primers used in FISH probes). A total of 38 probes with 20 nucleotides in length were used (Tsingke Biotech). Probes were biotinylated using terminal transferase (NEB, M0315S) with Bio-N6-ddATP (ENZO, ENZ-42809) as substrates. To detect LncKdm2b RNA, adherently cultured primary NPCs derived from E13.5 mouse cortex were rinsed in PBS and then fixed with 3.7% formaldehyde in PBS at room temperature for 10 min. Cells were permeabilized with 70% ethanol at 4 °C overnight. Cells were treated with RNase A (100 µg/mL) or with PBS (in the control group) at 37 °C for 1 h. After washing in Wash Buffer A (Biosearch Technologies, SMF-WA1-60) for 5 min, cells were incubated with DNA probes in hybridization buffer (Biosearch Technologies, SMF-HB1-10) at 37 °C overnight. After hybridization, cells were incubated with Alexa Fluor 555 conjugated streptavidin (1:1500 diluted in 1% BSA in PBS) at RT for 1 h. Cells were washed twice with Wash Buffer A at 37 °C for 30 min followed by nuclear counterstaining with DAPI. For colocalization studies, cells were co-stained with mouse anti-hnRNPAB (Santa Cruz Biotechnology).
Luciferase reporter assays
To perform luciferase assays, Neuro-2a and HEK293T cells at ~60% confluency in each well of 24-well plates were transfected with 500 ng of pGL3-T5 Forward, pGL3-T5 Reverse, pGL3-T5-mini Forward, or pGL3-T5-mini Reverse plus 5 ng of pTK-Ren vectors using Lipofectamine® 3000 (Thermo Fisher Scientific). For the RNA tethering experiment, Neuro-2a cells were grown in 24-well plates until 60% confluent and transfected with 150 ng of 5× UAS-TK-Luc, pcDNA3-Gal4-λN, and pcDNA3-BoxB-LacZ or pcDNA3-BoxB-LncKdm2b, plus 5 ng of pTK-Ren vectors. For the detection of dose-dependent repression effect of BoxB-LncKdm2b on reporter activity, different doses of the pcDNA3-BoxB-LncKdm2b plasmid at 50 ng, 100 ng or 200 ng were used. As the amount of BoxB-lncRNA plasmid was increased, an equal amount of BoxB-LacZ was reduced accordingly. Twenty-four hour after transfection, cells were harvested and assayed for reporter activity using the Dual-Glo Luciferase Assay System and the GloMax multidectection system according to manufacturer’s instructions (Promega). Each data point was taken as the average Luc/Ren ratio of triplicate wells. To test the effects of hnRNPAB knockdown on Kdm2b’s promoter activity (CArG Box-containing), Neuro-2a cells at ~50% confluency in each well of 24-well plates were transfected with 50 nmol/L siRNA targeting hnRNPAB. The second transfections with 500 ng pGL3-pKdm2b or pGL3-pKdm2bΔCArG plus 5 ng of pTK-Ren vectors were done twenty-four hour later. Cells were harvested 24 h later and assayed for reporter activity.
Nuclear run-on (NRO)
Nuclear run-on was performed as previously described (Roberts et al., 2015). About 1–4 × 106 Neuro-2a cells were harvested and washed with PBS for one run-on experiment. Cell pellets were added 1 mL of lysis buffer (10 mmol/L Tris-HCl, pH 7.4; 10 mmol/L NaCl; 3 mmol/L MgCl2; 0.5% NP-40) and incubated on ice for 5 min. After centrifugation at 300 ×g for 4 min at 4 °C, the pellet was washed with lysis buffer without NP-40 and re-suspended with 40 μL nuclear storage buffer (50 mmol/L Tris-HCl, pH 8.3; 40% glycerol; 5 mmol/L MgCl2; 0.1 mmol/L EDTA). Equal volume of 2× transcription buffer [20 mmol/L Tris-HCl, pH 8.3; 300 mmol/L KCl; 5 mmol/L MgCl2; 4 mmol/L DTT; 1 mmol/L each of ATP, GTP and CTP, 0.5 mmol/L UTP, 100 U RNase inhibitor (Takara Bio)] was added into nuclei and then supplied with 0.5 mmol/L BrUTP (Sigma). After incubation at 30 °C for 30 min, RNA was extracted by TRIzol, and digested by 6 U DNase I (Thermo Fisher). About 30 μL of protein G agarose beads were washed with PBST, resuspended in 30 μL PBST. 2 μg of anti-BrdU monoclonal antibody (Santa Cruz) was added and incubated on a rotating platform for 10 min at room temperature. 150 μL of blocking buffer (0.1% PVP and 0.1% BSA in PBST) was added and incubated for 30 min at room temperature. After four times wash with 300 μL PBSTR (80 U RNase Inhibitor per 10 mL of PBST), pellets were resuspended in 100 μL of PBSTR. NRO-RNA samples were treated at 65 °C for 5 min to denature RNA secondary structures and then incubated with the BrdU antibody-bound agarose for 60 min at room temperature on a rotating platform. After four times wash with 300 μL PBSTR, RNA was extracted by resuspending the pellet in 500 μL TRIzol. RNAs were reverse-transcribed, and detected by qPCR. RT-qPCR primers used to detect the pre-mRNAs of LncKdm2b, or Kdm2b were designed to cover one exon-intron junction, that is, one primer locates in the intron and the other in the adjacent exon. See Table S4 for qPCR primers.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed essentially as described previously (Wu et al., 2008). Briefly, 1 × 107 Neuro-2a cells per experiment were crosslinked with 1% formaldehyde in the medium for 10 min at room temperature and quenched by adding 0.125 mol/L glycine for 5 min. Cells were then washed twice with ice-cold PBS. Cells were then harvested in 500 μL digestion buffer (50 mmol/L Tris-HCl, pH 7.9; 5 mmol/L CaCl2; 100 μg/mL BSA) plus 1,400 U micrococcal nuclease (NEB) for 20 min at 37 °C, followed by adding 5 μL 0.5 mol/L EDTA and incubating on ice for 5 min. Sonicate samples in EP tubes on ice with power output 30%, 3 min, 0.5 s on, 0.5 s off. One percent of the sonicated lysate was used as the input. Sonicated lysates were diluted into 0.1% SDS using dilution buffer (50 mmol/L Tris-HCl, pH 7.6; 1 mmol/L CaCl2; 0.2% Triton X-100; 0.5% SDS) and incubated with 25 μL pre-washed protein G agarose beads plus 4 μg anti-hnRNPAB (Santa Cruz Biotechnology) or 2 μg anti-H3K4me3 (Active Motif) or anti-H3K27ac (Millipore) antibodies on rocker at 4 °C overnight. After wash with Wash Buffer I (20 mmol/L Tris-HCl, pH 8.0; 150 mmol/L NaCl; 2 mmol/L EDTA; 1% Triton X-100; 0.1% SDS), 4 times wash with Wash Buffer II (20 mmol/L Tris-HCl, pH 8.0; 500 mmol/L NaCl; 2 mmol/L EDTA; 1% Triton X-100; 0.1% SDS), 4 times wash with Wash Buffer III (10 mmol/L Tris-HCl, pH 8.0; 0.25 mol/L LiCl; 1 mmol/L EDTA; 1% deoxycholate; 1% NP-40), and 2 times wash with TE buffer, resuspend each pellet in 100 μL elution buffer (0.1 mol/L NaHCO3; 1% SDS) with 1 μL 20 mg/mL proteinase K. Cross-linked chromatin was reversed at 65 °C overnight. DNAs were purified using the PCR purification Kit (TianGen). Purified DNA was used for quantitative PCR analyses and was normalized to input chromatin. See Table S4 for qPCR primers.
Chromosome conformation capture (3C)
3C experiments were performed essentially as described previously (Hagege et al., 2007). Briefly, 1 × 107 Neuro-2a cells per experiment were crosslinked with 2% formaldehyde in the medium for 10 min at room temperature and quenched by 0.125 mol/L glycine for 5 min. Cells were then washed twice with ice-cold PBS. Cells were lysed in 3C lysis buffer (10 mmol/L Tris-HCl, pH 8.0; 10 mmol/L NaCl; 0.2% NP40; PMSF) for 1 h on rocker at 4 °C and nuclei were pelleted by centrifugation at 3,500 rpm for 10 min at 4 °C. Pellets were then resuspended in 500 μL 1.2× restriction enzyme buffer (NEB) with 0.3% SDS and incubated with rotation at 37 °C for 1 h. Triton X-100 was added to a final concentration of 2% followed by 1 h incubation at 37 °C with rotation. 400 U of highly concentrated EcoR I (NEB) was added and incubated overnight at 37 °C with rotation. The following day, SDS was added to a final concentration of 1.6%, and samples were incubated at 65 °C for 20 min. Samples were then brought up to a final volume of 7 mL in T4 ligase buffer (66 mmol/L Tris-HCl, pH 7.6; 6.6 mmol/L MgCl2; 10 mmol/L DTT; 100 μmol/L ATP) with 1% Triton X-100. Samples were rotated at 37 °C for 1 h. Samples were then chilled on ice for 5 min, and 700 U T4 DNA ligase (Takara Bio) was added, and samples were incubated at 16 °C for overnight followed by 30 min at room temperature. Next, 300 μg of proteinase K was added, and crosslinks were reversed at 65 °C overnight. The following day, an additional 300 μg of RNase A was added and incubated at 37 °C for 40 min. Finally, genomic DNA was purified by phenol-chloroform extraction followed by ethanol precipitation. Ligation events were detected using specific primers. qPCRs were performed on a CFX ConnectTM Real-Time PCR Detection System using iTaqTM Universal SYBR® Green Supermix (Bio-rad). Specificity and efficiency of all 3C primers were verified by performing digestion and ligation of the BAC DNA containing the regions of interest. Ligation products were then serially diluted in sheared genomic DNA, and the efficiency of each PCR reaction was verified. Amplicons from BAC qPCRs and actual 3C template were run on agarose gel to verify the production of a single band of the expected size. See Table S4 for qPCR primers.
Biotin-labeled RNA pull-down
RNA pull-down was performed as described previously (Tsai et al., 2010). To make biotinylated RNA pull-down probes, the 5′-overhang of forward primer was modified with a T7 promoter (See Table S4 for the primers used in RNA pull-down probes). Biotinylated RNAs were transcribed using the Biotin-RNA Labeling Mix (Roche) and HiScribe™ T7 High Yield RNA Synthesis Kit (NEB) according to the manufacturer’s protocol. About 3 μg of biotinylated RNA was heated at 90 °C for 2 min, and then cooled down on ice for 2 min in RNA structure buffer (10 mmol/L Tris pH 7, 0.1 mol/L KCl, 10 mmol/L MgCl2), and then shifted to room temperature (RT) for 20 min. About 5 × 107 primary cells from E14.5 mouse cortices were used for each RNA pull-down experiment. Cells were resuspended in 2 mL PBS, 2 mL nuclear isolation buffer (1.28 mol/L sucrose; 40 mmol/L Tris-HCl pH 7.5; 20 mmol/L MgCl2; 4% Triton X-100), and 6 mL water for 20 min on ice. Nuclei were pelleted by centrifugation at 2,500 ×g for 15 min and resuspended in 1 mL RIP buffer [150 mmol/L KCl, 25 mmol/L Tris pH 7.4, 0.5 mmol/L DTT, 0.5% NP-40, 1 mmol/L PMSF and protease inhibitor cocktail (Biotool). Resuspended nuclei were sonicated on ice at 30% power output for 3 min (0.5 s on, 0.5 s off). Nuclear extracts were collected by centrifugation at 12,000 rpm for 10 min, and were pre-cleared by 40 μL streptavidin agarose (Thermo Fisher) for 20 min at 4 °C with rotation. Pre-cleared lysates were mixed with 3 μg folded biotinylated RNA and 20 μg yeast RNA at 4 °C overnight, followed by adding 60 μL washed streptavidin agarose beads to each binding reaction and incubating at RT for 1.5 h. After 4 × 10 min washes by RIP buffer (containing 0.5% sodium deoxycholate) at 4 °C, proteins bound to RNA were eluted in 1× sample buffer by heating at 100 °C for 10 min, and then subjected to SDS-PAGE, and further visualized by silver staining. Finally, proteins were identified by mass spectrometry.
Native RNA-protein complex immunoprecipitation
Native RNA-protein complex immunoprecipitation assays were carried out as described (Xing et al., 2017) with modifications. Dorsal forebrain (cortex) tissues were resected from E14.5 mouse embryos and homogenated in 1 mL lysis buffer [50 mmol/L Tris pH 7.4, 150 mmol/L NaCl, 0.5% NP-40, 1 mmol/L PMSF, 2 mmol/L RVC, protease inhibitor cocktail (Biotool)] followed by sonication with a 30% power output for 3 min (0.5 s on, 0.5 s off) on ice. After centrifuging at 12,000 rpm for 10 min at 4 °C, the supernatant was pre-cleared with 30 μL protein G agarose beads. The pre-cleared lysates were further incubated with 4 μg anti-hnRNPAB antibody (Santa Cruz) for 2 h at 4 °C. Then 50 μL protein G agarose beads (blocked with 1% BSA and 20 μg/mL yeast tRNA) were added to the mixture and incubated for another 1 h at 4 °C followed by washing with wash buffer [50 mmol/L Tris pH 7.4, 300 mmol/L NaCl, 0.05% sodium deoxycholate, 0.5% NP-40, 1 mmol/L PMSF, 2 mmol/L RVC, protease inhibitor cocktail (Biotool)]. RNAs were extracted with Trizol. For qRT-PCR, each RNA sample was treated with DNase I (Thermo Fisher) and reverse transcription was performed with PrimerScriptTM Reverse Transcriptase (Takara Bio) using random primers followed by qRT-PCR analysis. See Table S4 for qPCR primers.
Formaldehyde crosslinking RNA immunoprecipitation
Formaldehyde crosslinking RNA immunoprecipitation assays were carried out as described (Xing et al., 2017) with modifications. Dorsal forebrain (cortex) tissues were resected from E14.5 mouse embryos and fixed 10 mL PBS with 1% formaldehyde 10 min at room temperature followed by incubation with 0.25 mol/L glycine at room temperature for 5 min. After pelleting tissues at 1,000 rpm for 5 min, the pellet was homogenized in 1 mL RIPA buffer [50 mmol/L Tris pH 7.4, 150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mmol/L PMSF, 2 mmol/L RVC, protease inhibitor cocktail (Biotool)] followed by sonication with a 30% power output for 3 min (0.5 s on, 0.5 s off) on ice. After centrifuging at 12,000 rpm for 10 min at 4 °C, the supernatant was pre-cleared with 30 μL protein G agarose beads and 20 μg/mL yeast tRNA at 4 °C for 30 min. Then the pre-cleared lysate was incubated with 50 μL beads that were pre-coated with 4 μg anti-hnRNPAB antibody (Santa Cruz) for 4 h at 4 °C. The beads were washed 4 × 5 min with washing buffer I (50 mmol/L Tris pH 7.4, 1 mol/L NaCl, 1% NP-40, 1% sodium deoxycholate), and 4 × 5 min with washing buffer II (50 mmol/L Tris pH 7.4, 1 mol/L NaCl, 1% NP-40, 1% sodium deoxycholate, 1 mol/L urea). The RNA-protein complex was eluted from beads by adding 140 μL elution buffer (100 mmol/L Tris pH 8.0, 10 mmol/L EDTA, 1% SDS) at room temperature for 5 min. To reverse crosslinking, 4 μL 5 mol/L NaCl and 2 μL 10 mg/mL proteinase K were added into the RNA samples, and incubated at 42 °C for 1 h followed by incubation at 65 °C for one hour. The RNA was extracted with Trizol. For qRT-PCR, each RNA sample was treated with DNase I (Thermo Fisher) and then reverse transcription was performed with PrimerScriptTM Reverse Transcriptase (Takara Bio) using random primers followed by qRT-PCR analysis. See Table S4 for qPCR primers.
RNA-protein in vitro binding experiments
RNA-protein in vitro binding experiments (Dig-RNA pull-down assays) were carried out as described (Xing et al., 2017) with modifications. To make Dig-labeled LncKdm2b truncations and loop1 mutations, the 5′-overhang of forward PCR primer was modified with a T7 promoter (See Table S4 for the primers used in RNA pull-down probes). Digoxigenin labeled riboprobes were transcribed using the DIG-RNA Labeling Mix (Roche). 2 × 107 HEK293T cells (Two 10-cm dishes) expressing Flag-hnRNPAB were harvested and resuspended in 1 mL lysis buffer [50 mmol/L Tris pH 7.4, 150 mmol/L NaCl, 0.5% NP-40, 0.5 mmol/L PMSF, 2 mmol/L RVC, protease inhibitor cocktail (Roche)] followed by sonication on ice. After centrifuging at 12,000 rpm for 10 min at 4 °C, the supernatant was incubated with 50 μL anti-Flag agarose beads (Biotool). After immunoprecipitation and washing 4 × 5 min with TBS buffer, one fifth beads was saved for immunoblotting. The rest was equilibrated in binding buffer [50 mmol/L Tris-HCl at pH 7.4, 150 mmol/L NaCl, 0.5% NP-40, 1 mmol/L PMSF, 2 mmol/L RVC, protease inhibitor cocktail (Biotool)] and incubated with 300 ng Dig-labeled RNA (Dig-labeled RNAs were annealed by heating at 65 °C for 5 min followed with slowly cooling down to room temperature) for at 4 °C for 4 h. After washing 4 × 5 min with binding buffer, the bound RNA was extracted with Trizol and analyzed by Northern blot.
Electrophoretic mobility shift assay (EMSA)
For RNA EMSAs, digoxigenin labeled riboprobes (LncKdm2b loop1, 463–625 nt) were transcribed using the DIG-RNA Labeling Mix (Roche). EMSA experiments were conducted according to the manufacturer’s protocol with a Light Shift Chemiluminescent RNA EMSA kit (Thermo Scientific). The Dig-labeled RNAs were annealed by heating at 65 °C for 5 min followed with slowly cooling down to room temperature. One fmol labeled RNAs were used for each EMSA reaction. For detection of dose-dependent binding of protein to RNA, different doses of Flag-hnRNPAB (1 μg, 3 μg or 10 μg) were used. Unlabeled probe was used for competitive reaction. Binding reactions were incubated in binding buffer [10 mmol/L HEPES pH 7.5, 20 mmol/L KCl, 1 mmol/L MgCl2, 1 mmol/L DTT] at room temperature for 25 min, then immediately loaded onto a 2% nondenaturing agarose 0.5× TBE (Tris-borate-EDTA) gel. After transfer to a nylon membrane, labeled probes were cross-linked by UV, probed with Anti-Digoxigenin-AP antibody, and incubated with detection substrates.
Real-time quantitative reverse transcription PCR (qRT-PCR)
Total RNAs (0.5–1 μg) were reverse-transcribed at 42 °C using PrimerScriptTM Reverse Transcriptase (Takara Bio). Then iTaqTM Universal SYBR® Green Supermix (Bio-rad) was employed to perform quantitative PCR on a CFX ConnectTM Real-Time PCR Detection System (Bio-rad). Gene expressions were determined using the 2-ΔΔCt method, normalizing to housekeeping genes Gapdh. See Table S4 for qPCR primers.
In utero electroporation of developing cerebral cortices
In utero microinjection and electroporation were performed essentially at E13.5 as described (Li et al., 2017). In brief, pregnant CD-1 mice were anesthetized by intraperitoneal injection of pentobarbital (70 mg/kg). The uteri were exposed through a 2 cm midline abdominal incision. Embryos were carefully pulled out using ring forceps through the incision and placed on sterile and irrigated drape. Intermittently wet uterine walls with saline to prevent drying. Supercoiled plasmid DNA (prepared using Endo Free plasmid purification kit, Tiangen) mixed with 0.05% Fast Green (Sigma) was injected through the uterine wall into the telencephalic vesicle of 3–4 embryos at intervals using pulled borosilicate needles (WPI). Electric pulses (36 V, 50 ms duration at 1 s intervals for 5 times) were generated using CUY21VIVO-SQ (BEX) and delivered across the uterine wall using 5 mm forceps-like electrodes (BEX). The uteri were then carefully put back into the abdominal cavity and incisions were sutured. The whole procedure was completed within 30 min. Mice were warmed on a heating pad until they woke up and given analgesia treatment (Ibuprofen) in drinking water until sacrifice.
Primary culture of embryonic cortical neural progenitor cells (NPCs)
Primary culture of embryonic cortical NPCs was performed as described (Li et al., 2017). In brief, E11.5 or E12.5 mouse cortices (dorsal forebrain) tissues were washed with and minced in filter-sterilized hibernation buffer (30 mmol/L KCl; 5 mmol/L NaOH; 5 mmol/L NaH2PO4; 5.5 mmol/L glucose; 0.5 mmol/L MgCl2; 20 mmol/L Na-pyruvate; 200 mmol/L Sorbitol, pH 7.4) followed by dissociating into single cells using pre-warmed Papain (Worthington Biochemical) enzyme solution (1× DMEM; 1 mmol/L Na-pyruvate; 1 mmol/L L-glutamine; 1 mmol/L N-acetyl-L-cysteine; 20 U/mL Papain; 12 μg/mL DNase I). Dissociated cells were cultured using serum-free media consisting of DMEM/F12 media (Life Technologies), N2 and B27 supplements (1×, Life Technologies), 1 mmol/L Na-pyruvate, 1 mmol/L N-acetyl-L-cysteine (NAC), human recombinant FGF2 and EGF (20 ng/mL each; Life Technologies). For adherent cortical cultures in Figs. 2C, S3A and S3B, cells were maintained on poly-L-lysine coated plates with the presence of 20 ng/mL FGF2 for 24 h followed by differentiation (FGF2 withdrawal) for 48 h. For sphere culture in Fig. 3F, cells were cultured with the presence of EGF and FGF2 for 1 week. For clonal culture in Fig. 6A–G, cells were maintained 72 h with the presence of 20 ng/mL FGF2 for 72 h (4 × 104 cells per well in 24-well plates).
Quantification and statistical analysis
Data were presented as the mean ± SEM unless otherwise indicated. Statistical analyses were conducted using GraphPad Prism (version 6.01). Statistical significance was determined using unpaired 2-tailed Student’s t test, 1-way ANOVA followed by Tukey post hoc test, 2-way ANOVA followed by the Bonferroni post hoc test. P ≤ 0.05 was considered statistically significant. ‘ * ’; P values less than 0.01, or 0.001 was marked as ‘ ** ’, and ‘ *** ’ respectively.
REFERENCES
Aprea J, Prenninger S, Dori M, Ghosh T, Monasor LS, Wessendorf E, Zocher S, Massalini S, Alexopoulou D, Lesche M et al (2013) Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. EMBO J 32:3145–3160
Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK et al (2006) NFAT dysregulation by increased dosage of DSCR2 and DYRK1A on chromosome 21. Nature 441:595–600
Ayala R, Shu T, Tsai LH (2007) Trekking across the brain: the journey of neuronal migration. Cell 128:29–43
Ayoub AE, Oh S, Xie Y, Leng J, Cotney J, Dominguez MH, Noonan JP, Rakic P (2011) Transcriptional programs in transient embryonic zones of the cerebral cortex defined by high-resolution mRNA sequencing. Proc Natl Acad Sci USA 108:14950–14955
Baek ST, Kerjan G, Bielas SL, Lee JE, Fenstermaker AG, Novarino G, Gleeson JG (2014) Off-target effect of doublecortin family shRNA on neuronal migration associated with endogenous microRNA dysregulation. Neuron 82:1255–1262
Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W et al (2014) Considerations when investigating lncRNA function in vivo. Elife 3:e03058
Bauer M, Kinkl N, Meixner A, Kremmer E, Riemenschneider M, Forstl H, Gasser T, Ueffing M (2009) Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Ther 16:142–147
Belgard TG, Marques AC, Oliver PL, Abaan HO, Sirey TM, Hoerder-Suabedissen A, Garcia-Moreno F, Molnar Z, Margulies EH, Ponting CP (2011) A transcriptomic atlas of mouse neocortical layers. Neuron 71:605–616
Benavides-Piccione R, Dierssen M, Ballesteros-Yanez I, Martinez de Lagran M, Arbones ML, Fotaki V, DeFelipe J, Elston GN (2005) Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/- mouse. Neurobiol Dis 20:115–122
Berghoff EG, Clark MF, Chen S, Cajigas I, Leib DE, Kohtz JD (2013) Evf2 (Dlx6as) lncRNA regulates ultraconserved enhancer methylation and the differential transcriptional control of adjacent genes. Development 140:4407–4416
Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D (2012) A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149:819–831
Cheng M, Jin X, Mu L, Wang F, Li W, Zhong X, Liu X, Shen W, Liu Y, Zhou Y (2016) Combination of the clustered regularly interspaced short palindromic repeats (CRISPR)-associated 9 technique with the piggybac transposon system for mouse in utero electroporation to study cortical development. J Neurosci Res 94:814–824
Corish P, Tyler-Smith C (1999) Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng 12:1035–1040
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I et al (2011) A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9:e1000582
Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, Galter D, Cookson MR (2013) mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci 16:499–506
Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539:452–455
Fietz SA, Huttner WB (2011) Cortical progenitor expansion, self-renewal and neurogenesis-a polarized perspective. Curr Opin Neurobiol 21:23–35
Fotaki V, Dierssen M, Alcantara S, Martinez S, Marti E, Casas C, Visa J, Soriano E, Estivill X, Arbones ML (2002) Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol 22:6636–6647
Fu XD (2014) Non-coding RNA: a new frontier in regulatory biology. Natl Sci Rev 1:190–204
Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN et al (2008) An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455:351–357
Ghosh A, Greenberg ME (1995) Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15:89–103
Goecks J, Nekrutenko A, Taylor J, Galaxy T (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86
Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD (2013) Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci 14:755–769
Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, Macura K, Blass G, Kellis M, Werber M et al (2013) The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 24:206–214
Guo C, Eckler MJ, McKenna WL, McKinsey GL, Rubenstein JL, Chen B (2013) Fezf2 expression identifies a multipotent progenitor for neocortical projection neurons, astrocytes, and oligodendrocytes. Neuron 80:1167–1174
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C et al (2010) Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28:503–510
Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forne T (2007) Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc 2:1722–1733
Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW et al (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112
Homem CC, Repic M, Knoblich JA (2015) Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 16:647–659
Imayoshi I, Kageyama R (2014) bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 82:9–23
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S et al (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152:570–583
Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN (2011) FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell 43:285–298
Kwan KY, Sestan N, Anton ES (2012) Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139:1535–1546
Labonne JD, Lee KH, Iwase S, Kong IK, Diamond MP, Layman LC, Kim CH, Kim HG (2016) An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum Genet 135:757–771
Lepoivre C, Belhocine M, Bergon A, Griffon A, Yammine M, Vanhille L, Zacarias-Cabeza J, Garibal MA, Koch F, Maqbool MA et al (2013) Divergent transcription is associated with promoters of transcriptional regulators. BMC Genomics 14:914
Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X et al (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498:516–520
Li Y, Wang W, Wang F, Wu Q, Li W, Zhong X, Tian K, Zeng T, Gao L, Liu Y et al (2017) Paired related homeobox 1 transactivates dopamine D2 receptor to maintain propagation and tumorigenicity of glioma-initiating cells. J Mol Cell Biol 9:302–314
Lin N, Chang KY, Li Z, Gates K, Rana ZA, Dang J, Zhang D, Han T, Yang CS, Cunningham TJ et al (2014) An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell 53:1005–1019
Liu B, Ye B, Yang L, Zhu X, Huang G, Zhu P, Du Y, Wu J, Qin X, Chen R et al (2017) Long noncoding RNA lncKdm2b is required for ILC3 maintenance by initiation of Zfp292 expression. Nat Immunol 18:499–508
Lu J, Sheen V (2005) Periventricular heterotopia. Epilepsy Behav 7:143–149
Luo S, Lu JY, Liu L, Yin Y, Chen C, Han X, Wu B, Xu R, Liu W, Yan P et al (2016) Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell 18:637–652
Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, Mehler MF (2010) Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci 11:14
Molyneaux BJ, Goff LA, Brettler AC, Chen HH, Hrvatin S, Rinn JL, Arlotta P (2015) DeCoN: genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron 85:275–288
Ng SY, Bogu GK, Soh BS, Stanton LW (2013) The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol Cell 51:349–359
Noonan FC, Goodfellow PJ, Staloch LJ, Mutch DG, Simon TC (2003) Antisense transcripts at the EMX2 locus in human and mouse. Genomics 81:58–66
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q et al (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58
Perino M, Veenstra GJ (2016) Chromatin control of developmental dynamics and plasticity. Dev Cell 38:610–620
Ponjavic J, Oliver PL, Lunter G, Ponting CP (2009) Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet 5:e1000617
Ramos AD, Diaz A, Nellore A, Delgado RN, Park KY, Gonzales-Roybal G, Oldham MC, Song JS, Lim DA (2013) Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell 12:616–628
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166
Roberts TC, Hart JR, Kaikkonen MU, Weinberg MS, Vogt PK, Morris KV (2015) Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat Protoc 10:1198–1211
Saba LM, Flink SC, Vanderlinden LA, Israel Y, Tampier L, Colombo G, Kiianmaa K, Bell RL, Printz MP, Flodman P et al (2015) The sequenced rat brain transcriptome—its use in identifying networks predisposing alcohol consumption. FEBS J 282:3556–3578
Sarkisian MR, Bartley CM, Rakic P (2008) Trouble making the first move: interpreting arrested neuronal migration in the cerebral cortex. Trends Neurosci 31:54–61
Scruggs BS, Adelman K (2015) The importance of controlling transcription elongation at coding and noncoding RNA loci. Cold Spring Harb Symp Quant Biol 80:33–44
Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, Adelman K (2015) Bidirectional transcription arises from two distinct hubs of transcription factor binding and active chromatin. Mol Cell 58:1101–1112
Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, Temple S (2006) The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci 9:743–751
Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis CC et al (2013) Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci USA 110:2876–2881
Sinnamon JR, Waddell CB, Nik S, Chen EI, Czaplinski K (2012) Hnrpab regulates neural development and neuron cell survival after glutamate stimulation. RNA 18:704–719
Spigoni G, Gedressi C, Mallamaci A (2010) Regulation of Emx2 expression by antisense transcripts in murine cortico-cerebral precursors. PLoS ONE 5:e8658
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, Aifantis I (2014) Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 158:593–606
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693
Venkov CD, Link AJ, Jennings JL, Plieth D, Inoue T, Nagai K, Xu C, Dimitrova YN, Rauscher FJ, Neilson EG (2007) A proximal activator of transcription in epithelial-mesenchymal transition. J Clin Invest 117:482–491
Vescovi AL, Reynolds BA, Fraser DD, Weiss S (1993) bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron 11:951–966
Vickers TA, Koo S, Bennett CF, Crooke ST, Dean NM, Baker BF (2003) Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J Biol Chem 278:7108–7118
Vierstra J, Rynes E, Sandstrom R, Zhang M, Canfield T, Hansen RS, Stehling-Sun S, Sabo PJ, Byron R, Humbert R et al (2014) Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 346:1007–1012
Walder RY, Walder JA (1988) Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc Natl Acad Sci USA 85:5011–5015
Wang A, Wang J, Liu Y, Zhou Y (2017) Mechanisms of long non-coding RNAs in the assembly and plasticity of neural circuitry. Front Neural Circuits 11:76
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA et al (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472:120–124
Wang Y, He L, Du Y, Zhu P, Huang G, Luo J, Yan X, Ye B, Li C, Xia P et al (2015) The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 16:413–425
Wu M, Wang PF, Lee JS, Martin-Brown S, Florens L, Washburn M, Shilatifard A (2008) Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol 28:7337–7344
Xing YH, Yao RW, Zhang Y, Guo CJ, Jiang S, Xu G, Dong R, Yang L, Chen LL (2017) SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 169:664–678.e616
Ye B, Liu B, Yang L, Zhu X, Zhang D, Wu W, Zhu P, Wang Y, Wang S, Xia P et al (2018) LncKdm2b controls self-renewal of embryonic stem cells via activating expression of transcription factor Zbtb3. EMBO J 37:e97174
Yin Y, Yan P, Lu J, Song G, Zhu Y, Li Z, Zhao Y, Shen B, Huang X, Zhu H et al (2015) Opposing roles for the lncRNA haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell 16:504–516
Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD et al (2014) A comparative encyclopedia of DNA elements in the mouse genome. Nature 515:355–364
Zhou ZJ, Dai Z, Zhou SL, Hu ZQ, Chen Q, Zhao YM, Shi YH, Gao Q, Wu WZ, Qiu SJ et al (2014) HNRNPAB induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by transcriptionally activating SNAIL. Cancer Res 74:2750–2762
AUTHOR CONTRIBUTIONS
Conceptualization, W.L., W.S. and Y.Z.; methodology, W.L. and W.S.; investigation, W.L., W.S., B.Z., K.T., Y.L., L.M., Z.L, X.Z., and Y.L.; writing-original draft, W.L. and Y.Z.; writing-review & editing, W.L. and Y.Z.; funding acquisition, Y.L. and Y.Z.; resources, X.W.; supervision, Y.Z.
ACKNOWLEDGEMENTS
We thank Drs. Haohong Li, Jiekai Chen, Baoming Qin, Xiaoqun Wang, Xiang Lv, Wei Jiang, Min Wu and Haining Du for sharing reagents and technical supports. We thank Drs. Weimin Zhong, Qin Shen and Xiaohua Shen, and all members in Zhou lab for critical reading of the manuscript. This work was supported by grants from National Key R&D Program of China (2018YFA0800700), National Natural Science Foundation of China (Grant Nos. 31671418 and 31471361), National Natural Science Foundation of Hubei Province (2018CFA016), Fundamental Research Funds for the Central Universities (2042017kf0242) and Wuhan University Experiment Technology Project Funding (WHU-2018-SYJS-01).
ABBREVIATIONS
3C, chromosome conformation capture; ASO, antisense oligonucleotide; ChIP, chromatin immunoprecipitation; CP, cortical plate; Ctx, cortex; DIG, digoxigenin; EMSA, electrophoretic mobility shift assay; FISH, fluorescent RNA in situ hybridization; hnRNPAB, heterogeneous nuclear ribonucleoprotein A/B; IF, immunofluorescence; ILC3, group 3 innate lymphoid cell; IPC, intermediate progenitor cell; ISH, in situ hybridization; IZ, intermediate zone; Kdm2b, lysine (K)-specific demethylase 2B; KUS, Kdm2b upstream sequence; KUSR, Kdm2b upstream sequence, reversed; lncRNA, long non-coding RNA; Luc, luciferase; LV, lateral ventricle; MB, midbrain; mESC, mouse embryonic stem cell; MS, mass spectrometry; NPC, neural progenitor cell; NRO, nuclear run-on; NSPCs, neural stem/progenitor cells; NURF, nuclear remodeling factor; pAS, polyadenylation sequence; PN, projection neuron; Pol II, RNA polymerase II; PRC1, polycomb repressor complex 1; qRT-PCR, quantitative reverse transcription PCR; RGPC, radial glial progenitor cell; RIP, RNA immunoprecipitation experiments; SPF, specific-pathogen-free; SVZ/IZ, subventricular and intermediate zone; SVZ, subventricular zone; TF, transcription factor; TSS, transcription start sites; VZ, ventricular zone; WT, wild-type
COMPLIANCE WITH ETHICS GUIDELINES
Wei Li, Wenchen Shen, Bo Zhang, Kuan Tian, Yamu Li, Lili Mu, Zhiyuan Luo, Xiaoling Zhong, Xudong Wu, Ying Liu and Yan Zhou declare that they have no conflict of interest. This article does not contain any studies with human subjects performed by any of the authors. All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, W., Shen, W., Zhang, B. et al. Long non-coding RNA LncKdm2b regulates cortical neuronal differentiation by cis-activating Kdm2b. Protein Cell 11, 161–186 (2020). https://doi.org/10.1007/s13238-019-0650-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-019-0650-z