
Abstract
Cells utilize calcium ions (Ca2+) to signal almost all aspects of cellular life, ranging from cell proliferation to cell death, in a spatially and temporally regulated manner. A key aspect of this regulation is the compartmentalization of Ca2+ in various cytoplasmic organelles that act as intracellular Ca2+ stores. Whereas Ca2+ release from the large-volume Ca2+ stores, such as the endoplasmic reticulum (ER) and Golgi apparatus, are preferred for signal transduction, Ca2+ release from the small-volume individual vesicular stores that are dispersed throughout the cell, such as lysosomes, may be more useful in local regulation, such as membrane fusion and individualized vesicular movements. Conceivably, these two types of Ca2+ stores may be established, maintained or refilled via distinct mechanisms. ER stores are refilled through sustained Ca2+ influx at ER-plasma membrane (PM) membrane contact sites (MCSs). In this review, we discuss the release and refilling mechanisms of intracellular small vesicular Ca2+ stores, with a special focus on lysosomes. Recent imaging studies of Ca2+ release and organelle MCSs suggest that Ca2+ exchange may occur between two types of stores, such that the small stores acquire Ca2+ from the large stores via ER-vesicle MCSs. Hence vesicular stores like lysosomes may be viewed as secondary Ca2+ stores in the cell.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ca2+ is a common second messenger in the cell that has been implicated in the regulation of virtually all aspects of cellular life, including cell growth, differentiation, motility and death (Clapham, 2007; Berridge, 2012). Upon binding to its effector proteins, such as calmodulin and synaptotagmins, Ca2+ regulates a variety of cellular processes, such as gene transcription, secretion and muscle contraction (Clapham, 2007; Berridge, 2012).
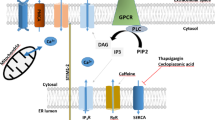
Ca2+ signaling is switched on and off via transient changes in cytosolic Ca2+ concentration ([Ca2+]cyt), either locally or globally. Under resting conditions, [Ca2+]cyt is low (~100 nmol/L), while [Ca2+]s are at least several thousand fold higher in the extracellular environment (~2 mmol/L) and in the lumen of organelles, such as the endoplasmic reticulum (ER, 0.3–0.7 mmol/L) and lysosomes (0.4–0.6 mmol/L), which serve as intracellular Ca2+ stores (Berridge et al., 2000; Morgan et al., 2011; Pizzo et al., 2011; Bengtson and Bading, 2012) (Fig. 1). Upon stimulation by extracellular transmitters and hormones, activation of Ca2+ influx channels in the plasma membrane (PM) or Ca2+ release channels in the ER, such as inositol 1,4,5-triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) (Clapham, 2007; Berridge, 2012), leads to rapid [Ca2+]cyt increases of 10–100 fold to μmol/L concentrations. Upon termination of Ca2+ signaling, [Ca2+]cyt returns quickly to a resting level, via primary, principally sarcoendoplasmic reticulum calcium transport ATPase (SERCA), pumps and secondary Ca2+ transporters in the PM and membranes of intracellular Ca2+-sequestering organelles (Clapham, 2007; Berridge, 2012).

Intracellular Ca2+ stores. Diagram of intracellular Ca2+ stores, illustrating Ca2+ release and uptake mechanisms. Large, continuous stores include the ER, the Golgi apparatus, mitochondria and the nucleus. Small, non-continuous vesicular stores, include endosomes, lysosomes, (auto)phagosomes and secretory vesicles, as well as vesicles in specialized cell types, such as tubulovesicles in parietal cells, melanosomes in melanocytes, synaptic vesicles in neurons and secretory granules in neurosecretory cells. TRPMLs, TPCs, P2X4, VGCCs, TRPA1 and TRPM2 are potential Ca2+ release channels in lysosomes. The H+ gradient in the lysosome is established and maintained by V-ATPases, and the Ca2+ gradient in lysosomes is established and maintained by a putative Ca2+ transporter/channel
The ER, which consists of interconnected and continuous tubules and cisternae and constitutes the largest membrane-bound organelle in the cell, is the most important intracellular Ca2+ storage site in the cell (Prakriya and Lewis, 2015; Phillips and Voeltz, 2016). Dys-regulation of intracellular Ca2+ homeostasis, either during signal initiation or termination, is associated with a number of genetic diseases (Berridge, 2012).
In addition to the ER, the Golgi apparatus, nucleus, and mitochondria also store Ca2+ (Rizzuto et al., 2012; Patel and Cai, 2015; Xu et al., 2015; Raffaello et al., 2016; Bagur and Hajnoczky, 2017) (Fig. 1). These membrane-bound organelles are interconnected and at least partially continuous in their lumens, providing a large storage capacity (Prakriya and Lewis, 2015; Phillips and Voeltz, 2016). The release and uptake mechanisms for these large stores have been studied and reviewed extensively (Prakriya and Lewis, 2015).
In contrast, intracellular vesicles, of which there are tens to hundreds in a cell, are a much less understood Ca2+ store organelles. Acidic stores (e.g., endosomes, lysosomes, secretory granules and lysosome-related organelles) can also undergo regulated Ca2+ release (Morgan et al., 2011) (Fig. 1). Compared with the large Ca2+ stores, vesicular Ca2+ storage and release has been technically challenging to study due to the relatively small signal amplitude that can be generated by the small-sized releasable Ca2+ pool in individual vesicles (Morgan et al., 2015; Xu et al., 2015). Fortunately, this roadblock has been partially removed with the recent development of organelle-targeted genetically-encoded Ca2+ indicators (GECIs) (Shen et al., 2012; Morgan et al., 2015; Xu et al., 2015; Garrity et al., 2016; Sahoo et al., 2017). Lysosomes are the cell’s recycling centers, playing essential roles in the basic cell biological processes of endocytosis, exocytosis and autophagy (Morgan et al., 2011; Patel and Cai, 2015; Xu and Ren, 2015).
In this review, we focus our discussion on the Ca2+ release and refilling mechanisms of the lysosome. We summarize the evidence supporting each of two distinct uptake hypotheses: the long known pH-dependent Ca2+ uptake hypothesis (Christensen et al., 2002; Morgan et al., 2011) and the recently introduced ER-dependent refilling hypothesis (Garrity et al., 2016; Wang et al., 2017). We will then extend the discussion to other vesicular Ca2+ stores in the cell, including cell-type-specific vesicles.
Large Ca2+ stores
The ER, which is the largest intracellular Ca2+ store in the cell (Phillips and Voeltz, 2016), has a luminal Ca2+ concentration ([Ca2+]ER) that is about 5,000 times higher than resting [Ca2+]cyt (Berridge et al., 2000; Prakriya and Lewis, 2015). SERCA pumps establish and maintain this very high [Ca2+]ER. Upon stimulation, ER releases Ca2+ into the cytoplasm via IP3Rs and RyRs (Prakriya and Lewis, 2015). A single stimulation event from an extracellular cue may only result in incomplete depletion of the store (Berridge et al., 2000; Prakriya and Lewis, 2015). Nevertheless, given the large volume of the ER’s interconnected tubules, substantial increases in global [Ca2+]cyt can be achieved with each stimulation event (Prakriya and Lewis, 2015), triggering various signal transduction cascades in the cell. In muscle cells, the opening of RyRs in the sarcoplasmic reticulum, a specialized type of ER in striated muscle cells, produces the massive increases in [Ca2+]cyt required for muscle contraction.
ER stores can be refilled by the well characterized process of store-operated Ca2+ entry (SOCE). SOCE relies on the collaborative actions of stromal interaction molecule (STIM) proteins, which serve as ER luminal Ca2+ sensors, and Orai proteins, which act as store-operated Ca2+ channels in the PM. Upon ER Ca2+ store depletion, STIM1 and STIM2 become activated and oligomerized (Prakriya and Lewis, 2015), favoring the formation of membrane contact sites (MCSs) between ER tubules and the PM (Stathopulos and Ikura, 2017). Orai proteins in the PM then accumulate through diffusion to the PM side of MCSs (Berridge et al., 2000; Prakriya and Lewis, 2015; Phillips and Voeltz, 2016). SOCE is triggered when STIM proteins bind directly to and thereby activate Orai channels, resulting in a sustained Ca2+ influx from the extracellular space that raises local [Ca2+]cyt in ER-PM MCS locations (Prakriya and Lewis, 2015). The imported Ca2+ is then taken up into the ER, resulting in ER Ca2+ store refilling, via high-affinity (low μmol/L range) SERCA pumps (Clapham, 2007). Detailed descriptions of the molecular mechanisms of ER Ca2+ channels and SOCE can be found in several recently-published excellent reviews (Prakriya and Lewis, 2015; Lopez et al., 2016; Putney et al., 2017; Stathopulos and Ikura, 2017).
Other intracellular organelles, including the nucleus, Golgi apparatus and mitochondria, serve as large Ca2+ stores. The nuclear envelope is continuous with ER membranes and contains IP3R and RyR Ca2+ release channels as well as SERCA Ca2+ uptake transporters (Bootman et al., 2009). SERCA, IP3Rs and RyRs are also expressed in Golgi apparatus membranes, which are partially interconnected but hold unevenly-distributed Ca2+ stores, ranging from ~130 μmol/L in the trans-Golgi cisterna to 250 μmol/L in the cis-Golgi cisterna (Pizzo et al., 2011). Besides SERCA pump-mediated Ca2+ uptake, Ca2+ can also be brought into the Golgi apparatus by way of secretory pathway Ca2+-ATPases (Pizzo et al., 2011). Finally, mitochondria are known to uptake cytosolic Ca2+ into their matrix under high [Ca2+]cyt conditions, making them, in essence, a cellular Ca2+ sink (De Stefani et al., 2016). Mitochondrial Ca2+ uptake is driven by a large negative membrane potential (∆ψ) in the inner membrane and mediated by voltage-dependent anion channels (VDACs) in the outer membrane and mitochondrial Ca2+ uniporters in the inner membrane (De Stefani et al., 2016).
Because of their collectively large luminal volumes, the ER, nucleus, Golgi apparatus and mitochondria function as the large Ca2+ stores of the cell. Mobilizing and emptying these large stores would result in substantial increases in [Ca2+]cyt, which in theory are preferentially suited for signal transduction.
Small Ca2+ stores
Lysosomes are acidic membrane-bound organelles responsible for degrading macromolecules from both intracellular and extracellular sources (Xu and Ren, 2015). Early studies detected Ca2+ release induced by nicotinic acid adenine dinucleotide phosphate (NAADP) in non-ER Ca2+ stores (Lee and Aarhus, 1995; Calcraft et al., 2009; Morgan et al., 2011). Glycyl-L-phenylalanine-naphthylamide (GPN), a di-peptide that is degraded in lysosomes by luminal cathepsins, induces Ca2+ release if applied alone and abolishes NAADP-induced Ca2+ release via osmotic swelling of lysosomes, giving rise to the notion that lysosomes can act as NAADP-targeted Ca2+ stores (Morgan et al., 2011). Calibration experiments employing lysosome-targeted, pH-corrected luminal Ca2+ indicators (e.g., Fura-Dextran dyes) have indicated that the Ca2+ concentration in the lysosome lumen ([Ca2+]Ly) is about 0.5 mmol/L, which is comparable to [Ca2+]ER (Christensen et al., 2002; Lloyd-Evans et al., 2008). Ca2+ release from individual lysosomes is limited by their small volume (typically <0.3 μm in diameter). Hence, lysosomes and other acidic stores, such as secretory granules, are referred to as small Ca2+ stores.
Ca2+ release channels of the lysosome
The recognition of lysosomes as intracellular Ca2+ stores and the potential roles of lysosomal Ca2+ release in regulating lysosomal membrane fusion and fission have prompted intense investigation of lysosomal Ca2+ release pathways (Patel and Cai, 2015). Using lysosome-targeted GECIs (Shen et al., 2012; Morgan et al., 2015), together with the recently-developed whole-lysosome patch-clamp technique, several candidate release channels have been identified, along with the cellular cues that activate them (Calcraft et al., 2009; Wang et al., 2012; Cang et al., 2013; Cao et al., 2015a, b; Xu and Ren, 2015). These cellular cues may serve as mobilizers of the lysosomal Ca2+ stores, similar to IP3R signaling in the ER.
Mucolipin subfamily of transient receptor potential (TRPML) channels
The TRPML channels, which consist of TRPML1, TRPML2 and TRPML3 (a.k.a. MCOLN1–3), are Ca2+-permeable cation channels expressed in endosome and lysosome membranes (Xu and Ren, 2015; Xiong and Zhu, 2016; Grimm et al., 2017) (Fig. 1). TRPML1, which is widely expressed in most cell types, is localized predominantly to late endosomes and lysosomes (Cheng et al., 2010). TRPML2 and TRPML3 are also localized to early and recycling endosomes in addition to late endosomes and lysosomes (Cheng et al., 2010). TRPML-mediated Ca2+ release may regulate Ca2+-dependent lysosomal membrane trafficking events involved in a variety of basic cell biological processes, including lysosomal exocytosis, autophagy and membrane repair (Xu and Ren, 2015; Xiong and Zhu, 2016; Grimm et al., 2017). In humans, loss-of-function mutations of TRPML1 cause type IV mucolipidosis (ML-IV), a lysosomal storage disease (LSD).
Phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2), an endolysosome-specific phosphoinositide, may serve as an endogenous TRPML agonist (Dong et al., 2010a, b). Reactive oxygen species have been shown to activate TRPML1 directly, triggering Ca2+ release and Ca2+-dependent lysosome biogenesis and autophagy (Zhang et al., 2016). Furthermore, mucolipin-specific synthetic agonists (ML-SAs) have been identified and shown to regulate various TRPML-dependent lysosomal functions by mimicking endogenous agonists (Shen et al., 2012; Xu and Ren, 2015; Grimm et al., 2017). Recent cryo-electron microscope structural images of TRPML1 and TRPML3 revealed that ML-SA1 binds to residues in the S5 and S6 helices of these TRPMLs (Schmiege et al., 2017; Zhou et al., 2017), which form an activation gate. Hence, cellular cues, or synthetic agonists, can induce lysosomal Ca2+ release via direct binding to TRPML channels. Furthermore, because TRPML currents are strongly rectifying at the inward direction, cellular cues can also regulate TRPML-mediated Ca2+ release by modulating lysosomal ∆ψ a driving force for Ca2+ release (Cheng et al., 2010; Dong et al., 2010a, b). Indeed, recently identified lysosome Na+ and K+ channels that regulate lysosome ∆ψ were shown to modulate the Ca2+ release from TRPML1 (Cao et al., 2015a, b; Xu and Ren, 2015; Xiong and Zhu, 2016; Wang et al., 2017).
Two-pore channels (TPC) channels
TPC1 and TPC2 channels, encoded by TPCN1 and TPCN2, respectively, are localized on endosomal and lysosomal membranes (Calcraft et al., 2009) (Fig. 1). Both TPC1 and TPC2 are ubiquitously expressed in mammalian cells. Whole-lysosome patch-clamping studies suggested that mammalian TPCs are Na+-selective with limited Ca2+ permeability (Wang et al., 2012; Cang et al., 2013). However, studies from multiple laboratories reported that TPC overexpression promoted lysosomal Ca2+ release (Brailoiu et al., 2009; Calcraft et al., 2009; Pitt et al., 2010; Ruas et al., 2010; Grimm et al., 2017), suggesting that the relatively small Ca2+ permeability of TPCs is physiologically significant.
NAADP (in nmol/L ranges) is the most potent Ca2+-mobilizing second messenger regulating intracellular Ca2+ stores (Lee and Aarhus, 1995) and TPCs are, thus far, the most promising candidate receptors for NAADP (Brailoiu et al., 2009; Calcraft et al., 2009; Pitt et al., 2010; Ruas et al., 2010; Grimm et al., 2017). However, radiolabeled NAADP has also been reported to bind other unidentified proteins in TPC knockout cells (Lin-Moshier et al., 2012; Walseth et al., 2012). Hence, resolving how Na+-selective TPCs are involved in NAADP-induced lysosomal Ca2+ release will require further investigation. On the other hand, PI(3,5)P2 can also activate whole-lysosome TPC currents (Wang et al., 2012; Xu and Ren, 2015). The endogenous protein kinase C inhibitor sphingosine has been reported to induce TPC1-dependent lysosomal Ca2+ release (Hoglinger et al., 2015). However, whether sphingosine activates TPCs directly has yet to be confirmed with direct whole-lysosome recording. The relative contributions of TRPMLs and TPCs in PI(3,5)P2-induced lysosomal Ca2+ release remain to be established (Wang et al., 2012; Xu and Ren, 2015).
Other lysosomal Ca2+ channels
P2X4 (purigenic receptor X4), an ATP-gated cation channel first discovered in the PM of various cell types, also resides on the lysosomal membranes of Cos1 cells where it can be activated by luminal ATP and alkalization (Qureshi et al., 2007; Huang et al., 2014) (Fig. 1). Ca2+ release through lysosomal P2X4 has been implicated in lysosomal membrane fusion in a calmodulin-dependent manner (Cao et al., 2015a, b). It is not clear whether P2X4 is expressed ubiquitously in mammalian lysosomes, or restrictively in certain cell types, as has been reported in tissue distribution studies (Qureshi et al., 2007).
TRPA1 (transient receptor potential ankyrin 1) is a Ca2+-permeable non-selective cation channel in somatosensory neurons that is activated by plant-derived chemicals, such as allyl isothiocyanate (a major ingredient of mustard oil) (Jordt et al., 2004). Recently, it was reported that TRPA1 is also expressed on peripheral lysosomes in somatosensory neurons, where it mediates allyl isothiocyanate-induced dense-core vesicle exocytosis and neuropeptide release (Shang et al., 2016).
TRPM2, a Ca2+ permeable non-selective cation channel gated by ADP ribose and Ca2+, is expressed on the PMs of neurons, pancreatic cells, and immune cells (Lange et al., 2009). TRPM2 is also localized on lysosomes in pancreatic β cells, leading to the proposition that TRPM2-mediated lysosomal Ca2+ release may regulate insulin secretion (Lange et al., 2009).
In central nervous system neurons, P/Q-type voltage-gated Ca2+ channels (VGCCs), encoded by CACNA1, mediate the Ca2+ entry that triggers neurotransmitter release. In a recent study, Tian et al., found that the α1A subunit of VGCCs is also present on lysosomal membranes in both fruit flies and mice, and is required for autophagosome-lysosome fusion (Tian et al., 2015).
In summary, both ubiquitous and cell-type-specific lysosomal expression of Ca2+ release channels have been described. They are activated by diverse cellular cues. Some are lysosome-committed channels, while others are dually expressed on lysosomal membranes and PMs.
Ca2+-dependent membrane trafficking of individual lysosomes
Generally, mammalian cells each have several hundred lysosomes, which are heterogeneous in size and morphology, as well as in their ionic and lipid compositions (Xu and Ren, 2015). Under physiological conditions, lysosomal Ca2+ channel-activating cellular signals are likely only present in a subset of lysosomes. Hence, lysosomal Ca2+ release from individual lysosomes may not be synchronized in a manner that gives rise to global increases in [Ca2+]cyt. However, such localized Ca2+ release may be sufficient to regulate local membrane trafficking events, such as fusion and fission (Xu and Ren, 2015). Theoretically, the decision to fuse vesicles should be determined based on the luminal cargo contents of individual vesicles (Xu and Ren, 2015). Hence, under physiological conditions in intact cells, lysosomal Ca2+ release is likely conducted by individual lysosomes depending on need. Notwithstanding, in some experimental settings, synchronized lysosomal Ca2+ release may be amplified by ER Ca2+ release triggering further cell signaling transduction (Kilpatrick et al., 2016a, b); it is not known whether such Ca2+-induced Ca2+ release occurs under physiological conditions.
Possible H+-dependent Ca2+ uptake mechanisms in the lysosome
The mechanisms that establish and maintain the massive 5,000-fold Ca2+ concentration gradient across the lysosomal membrane are of great interest. The prevailing view in the literature is that the lysosomal H+ gradient is essential for lysosomal Ca2+ store maintenance and refilling. Using both cytosolic and luminal Ca2+ dyes, researchers have shown that manipulations that cause lysosomal pH dissipation, such as V-ATPase inhibition, lead to lysosomal Ca2+ release, while restoration of the acidic luminal pH is accompanied Ca2+ store replenishment (Christensen et al., 2002; Lloyd-Evans et al., 2008; Calcraft et al., 2009; Dickson et al., 2012; Shen et al., 2012). Hence, it was proposed that a Ca2+/H+ exchanger (CAX) may drive pH-dependent Ca2+ uptake into lysosomes (Christensen et al., 2002; Morgan et al., 2011). CAXs are well known for their expression on vacuoles (lysosome-like organelles in yeast and plants) (Pittman, 2011). CAXs were long thought to be absent from metazoans. Although CAX genes have been identified more recently in some echinoderm, mollusk, fish, amphibian and non-placental mammal species, they have not been found in placental mammals thus far (Melchionda et al., 2016), suggesting that they might not be responsible for lysosomal Ca2+ uptake, at least not in placental mammals (Patel and Docampo, 2010). It is also possible that pH gradients may drive Ca2+ uptake indirectly in the absences of CAXs, such as through Na+/H+ exchangers and Na+/Ca2+ exchangers in series. That being said, Na+/H+ exchangers have thus far been demonstrated to be expressed in endosomes, but not lysosomes (Morgan et al., 2011).
The fact that CAXs have been remained putative in placental mammals for more than a decade encourages a revisiting of the evidence that led to the pH hypothesis. Notably, all presumed lysosomal Ca2+-mobilizing agents in earlier studies (i.e., GPN, Baf-A1, and NAADP) also cause lysosomal H+ release (Yoshimori et al., 1991; Morgan and Galione, 2007; Scott and Gruenberg, 2011; Appelqvist et al., 2012). Due to the pH sensitivities of most cytosolic Ca2+ dyes and probes (Rudolf et al., 2003), the presumed Ca2+ signals in the earlier studies may have contained, in some portion, pH signals or other unidentified pH-mediated non-Ca2+-dependent signals. Indeed, GPN-induced or Baf-A1-induced Ca2+ signals were found to be remain largely intact in the presence of a potent intracellular Ca2+ chelator, namely BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)], in a recent study (Garrity et al., 2016). In other words, the observed sensitivity to V-ATPase blockers in earlier lysosomal Ca2+ content studies might have contained a contaminating pH component. Hence, it is possible that secondary changes in Ca2+ buffering and ionic composition in the lysosome lumen consequent to pH changes may have led to a misinterpretation of previous data (Dickson et al., 2012; Garrity et al., 2016).
In situ assay of lysosomal Ca2+ refilling
The low pH environment in the lysosome lumen makes it challenging to measure and monitor [Ca2+]LY accurately. Thus, there has been a need for an assay that can detect Ca2+ release and refilling without interfering with lysosomal pH. To monitor lysosomal Ca2+ release, Shen et al. (Shen et al., 2012) fused a GECI (i.e., GCaMP) to the cytosolic N-terminus of TRPML1. Using this lysosome-targeted GECI, Garrity et al. (Garrity et al., 2016) developed a robust lysosomal Ca2+ refilling assay, in which consecutive applications of ML-SAs trigger consecutive bouts of Ca2+ release at 5-min intervals. The initial application of ML-SA depletes lysosomal Ca2+ stores, such that the response to the second application depends on lysosomal Ca2+ refilling during the 5-min interval (Garrity et al., 2016). To rule out the possibility that the GECI also detects ML-SA1-induced Ca2+ responses from organelles other than endolysosomes, control experiments were performed in which lysosomal Ca2+ stores were depleted with GPN. Importantly, a TRPML1 agonist, whose specificity was confirmed with mouse knockouts, was shown to be cell permeable with reversible effects (Shen et al., 2012; Garrity et al., 2016). In addition, the specificity of ML-SAs ensures that one is observing real changes in lysosomal Ca2+, as opposed to pH changes associated with other Ca2+-mobilizing reagents acting on lysosomes (Garrity et al., 2016). Indeed, ML-SA1-induced responses were completely abolished by BAPTA-AM treatment, consistent with Ca2+-specificity of the signal (Garrity et al., 2016). In contrast, GPN- or Baf-A1-induced presumed-to-be-Ca2+-specific responses persisted in the presence of BAPTA-AM (Garrity et al., 2016). Therefore, these results suggest that although GPN mobilizes more than just Ca2+, it is a very specific lysosome-disrupting agent. Hence, it can be used as a powerful tool to confirm the lysosome-specificity of other Ca2+-mobilizing reagents, such as ML-SA1.
The aforementioned lysosome-targeted GCaMP and ML-SA assay is the first robust and sensitive method developed with the ability to measure lysosomal Ca2+ release directly, independent of intracellular pH. It allows for time-lapse examination of lysosomal Ca2+ store depletion and refilling with acute (<5 min) application of various pharmacological reagents, which has many advantages, including amenability to prolonged treatment protocols, as have been used predominantly in previous studies.
Using this powerful refilling assay, Garrity et al., found that dissipation of the proton gradient in the lysosome (e.g., by V-ATPase inhibitors) has little to no impact on naïve Ca2+ stores or their refilling (2016). This result is inconsistent with predictions based on the prevailing pH-dependent hypothesis and, instead, suggests that lysosomal Ca2+ refilling is likely to be pH independent. It is possible that V-ATPase blockers in earlier studies (e.g., (Christensen et al., 2002; Calcraft et al., 2009; Morgan et al., 2011)) abolished the indirect effects of lysosomal H+ release on pH-sensitive Ca2+ dyes and probes (2016).
ER Ca2+ and IP3Rs are required for lysosomal Ca2+ refilling
In sharp contrast to the lack of V-ATPase effects, depletion of ER Ca2+ stores by SERCA inhibitors was shown to abolish lysosomal Ca2+ refilling (2016). Furthermore, inhibition of IP3Rs, but not RyRs, on the ER membrane blocked refilling (2016). Notably, IP3R inhibition induces lysosome dysfunction and LSD-like phenotypes in cells (2016). The inhibition of refilling by IP3R inhibitors argues against the possibility of GCaMP signaling being mediated by ER Ca2+ release and then amplified by lysosomal Ca2+ (Kilpatrick et al., 2016a, b). The inference that Ca2+ store refilling is mediated by IP3Rs, but not RyRs, suggests that ER Ca2+ release induced by lysosomal Ca2+ release may not operate through Ca2+-induced Ca2+ release because RyRs are better suited for this role than IP3Rs. Consistently, when Ca2+ levels in the lysosome lumen were measured with lysosome-targeted Fura-Dextran dye, it was shown that depleting ER Ca2+ or inhibiting IP3Rs also blocked refilling (2016). In another recently published independent study in which lysosomal Ca2+ was monitored with a pH-insensitive aequorin-based probe fused with a cathepsin protein, lysosomal Ca2+ was not refilled if SERCA activity was inhibited (Ronco et al., 2015). Taken together, these studies employing both juxta- and intra-lysosomal Ca2+ sensors/dyes suggest that lysosome stores are refilled with Ca2+ from the ER, independent of lysosomal pH.
ER-dependent three-step model of lysosomal Ca2+ refilling
A possible critical role of the ER in lysosomal Ca2+ refilling is reinforced by the structurally intimate localization of the ER and lysosomes at ER-lysosome MCSs (Phillips and Voeltz, 2016). An interesting testable hypothesis is that lysosomal Ca2+ refilling may be triggered directly by lysosomal Ca2+ release and may be dependent on ER-lysosome interactions that are dynamically regulated by lysosomal Ca2+. A similar model was proposed to explain ER store refilling wherein ER Ca2+ release triggers ER-PM functional coupling via STIM and Orai proteins (Saheki and De Camilli, 2017). Hence, lysosomal refilling may be a regulated, three-step process (Fig. 2): 1) triggering, by increased peri-lysosomal Ca2+ and/or decreased [Ca2+ ]LY; 2) docking, involving the formation of ER-lysosome MCSs; and 3) fueling, wherein Ca2+ is transported from the ER to lysosomes through functional ER-lysosome MCSs.

A three-step working model of lysosomal refilling. Lysosome Ca2+ stores are depleted upon cellular stimulation triggering lysosomal Ca2+ release. An increase in juxta-lysosomal [Ca2+]Cyt or a decrease in [Ca2+]Ly triggers refilling. In the docking step, MCSs are formed by both constitutive tethers, including endolysosome-localized ORP1L, STARD3, Protrudin, NPC1, and ER-localized ORP5, and VAP ([vesicle-associated membrane protein]-associated ER protein), as well as by putative Ca2+-sensitive tethers (e.g., E-syt1). In the fueling step, ER and lysosomal membranes are brought closer (within 5 nm). Meanwhile, both IP3Rs and putative uptake channel/transporters are enriched in ER-lysosome MCSs. Ca2+ released from lysosomes induces a conformational change of E-syt1-like protein on ER membranes, which in turn triggers the binding of E-syt1 with PI(4,5)P2, or other phosphoinositide, on lysosomal membranes, creating a functional ER-lysosome contact site for refilling. Ca2+ is then released from the ER via IP3Rs, causing a steep Ca2+ gradient that drives the influx of Ca2+ via an unidentified lysosomal uptake channel/transporter
Docking: formation of ER-lysosome MCSs
MCSs are close (typically <30 nm) appositions with tethering, but not fusion of membranes between organelles (Phillips and Voeltz, 2016). That is, they provide physical platforms for material exchange between organelles via a direct, non-fusion mechanism (English and Voeltz, 2013; Phillips and Voeltz, 2016; Saheki and De Camilli, 2017). Although ER-lysosome MCSs are well documented (Phillips and Voeltz, 2016), their functional significance is not clear. In comparison, ample evidence supports the involvement of ER-PM and ER-mitochondrial MCSs in Ca2+ exchange. In ER-PM MCSs, STIM and Orai proteins are concentrated (Saheki and De Camilli, 2017), and the oligomerization of ER-localized STIM1 activates Ca2+ influx via PM-localized Orai1 channels, thereby enabling refilling of ER Ca2+ stores (Saheki and De Camilli, 2017). Likewise, in ER-mitochondria MCSs, a protein complex is formed by mitochondrion-outer-membrane-localized VDAC channels, ER-localized IP3Rs, and the tethering protein Grp75, facilitating Ca2+ uptake from the ER to mitochondria (De Stefani et al., 2016; Krols et al., 2016; Phillips and Voeltz, 2016). Generally speaking, the short (<30 nm) distance between the ER and lysosomal membranes in MCSs should enable quiescent ER-to-lysosome Ca2+ transport without causing global [Ca2+]Cyt increases.
ER-lysosome MCS formation requires several tethering proteins to keep the two opposing membranes in apposition, including oxysterol-binding protein-related protein 1L (ORP1L) (Rocha et al., 2009), protrudin (Raiborg et al., 2016), stAR-related lipid transfer protein 3 (STARD3) and oxysterol-binding protein-related protein 5 (ORP5)/Niemann-Pick C1 protein (NPC1) (Du et al., 2011; van der Kant and Neefjes, 2014; Phillips and Voeltz, 2016) (Fig. 2). Given that lysosome Ca2+ refilling requires peri-lysosomal increases in [Ca2+]Cyt, it is likely that at least some tethering events may be regulated by peri-lysosomal Ca2+ (Wang et al., 2017). MCS gaps could be reduced, from 20–30 nm to within 5–15 nm, to provide a functional conformation highly amenable to Ca2+ exchange (Phillips and Voeltz, 2016). Several E-Syts (extended synaptotagmin-like proteins) have been confirmed to act as tethers at ER-PM MCSs (Min et al., 2007; Giordano et al., 2013). E-Syts have an N-terminal β-hairpin embedded in the ER membrane and multiple C2 domains in the C-terminal, which contains binding sites for both Ca2+ and phospholipids (Min et al., 2007; Giordano et al., 2013). Similar Ca2+ sensor proteins may play equivalent roles in ER-lysosome MCSs. Both PI(4,5)P2 and PI(3,5)P2 have been observed in lysosomes (Xu and Ren, 2015). Thus, upon Ca2+ release from lysosomes, a Ca2+- and phospholipid-dependent interaction between the two membranes may, in addition to the pre-existing tethers, help bring ER and lysosomes even closer, further supporting the functionality of ER-lysosome MCSs (Fig. 2).
Fueling: Ca2+ transport in ER-lysosome ECS
After docking, a steep gradient between Ca2+-loaded ER and Ca2+-depleted lysosomes can drive the transfer of Ca2+ from the ER to lysosomes. This process appears to involve the coordinated actions of IP3R-mediated Ca2+ release from the ER and lysosomal Ca2+ uptake via a putative uptake channel or transporter (Fig. 2). Although accumulation of IP3Rs through lateral diffusion in ER-lysosome MCSs has not yet been demonstrated directly, constitutive Ca2+ release mediated by local enriched IP3Rs has been reported on ER-mitochondrial MCSs (Szabadkai et al., 2006; Cardenas et al., 2010). Because IP3Rs are constitutively active, refilling is plausible given very high local Ca2+ concentrations in MCSs, without a widespread Ca2+ release (Rizzuto et al., 1998).
Theoretically, any Ca2+-permeable channel, exchanger or pump could mediate lysosomal Ca2+ uptake. The slow nature of the refilling process suggests that it involves either a low affinity Ca2+ transporter or a rectifying Ca2+ channel (2016). Interestingly, low-affinity (mmol/L range) Ca2+ transporters have been observed in isolated lysosomes (Lemons and Thoene, 1991). It is also possible that a putative VDAC-like channel in lysosomes might mediate the Ca2+ uptake (van der Kant and Neefjes, 2014).
Regulation of lysosomal Ca2+ refilling
Lysosomal ∆ψ appears to play a role in refilling (Wang et al., 2017). So-called big potassium (BK) channels, which regulate ∆ψ in excitable cells, exhibit functional expression in lysosomes (Cao et al., 2015a, b; Wang et al., 2017). Hence, Ca2+ activation of voltage-dependent, K+-selective conductance via BK channels may facilitate lysosomal Ca2+ release and refilling (Cao et al., 2015a, b; Wang et al., 2017). Hence, although there is no direct evidence, it is conceivable that lysosomal ∆ψ could affect refilling directly or indirectly. For example, it was reported recently that membrane potential can affect phosphoinositide dynamics (Zhou et al., 2015); and phosphoinositides are known to influence the interaction of lysosomes with other organelles, including peroxisomes and the ER (Chu et al., 2015). Hence, lysosomal Ca2+ refilling may require the action of multiple Ca2+ effectors in the triggering step, such as lysosome-localized BK channels, ER-localized IP3Rs and ER-localized E-Syt-like proteins (Fig. 2).
Diseases associated with lysosomal Ca2+ store defects
Dys-regulation of lysosome Ca2+ homeostasis causes LSDs and lysosome-related diseases. Notably, ML-IV is associated with impaired lysosomal Ca2+ release (Kiselyov et al., 2010). Additionally, lysosomal Ca2+ stores have been reported to be reduced in Niemann-Pick, type C cells (Lloyd-Evans et al., 2008). Moreover, in Niemann-Pick, type C cells (containing NPC1 mutation), TRPML1 activity was found to be inhibited by cholesterol accumulation in lysosomes, and increasing TRPML1 activity alleviated lysosomal storage in these cells (Shen et al., 2012). Indeed, compromised TRPML1 activity has been implicated in a number of LSDs (De Leo et al., 2016; Zhong et al., 2017). Additionally, lysosomal Ca2+ store defects are implicated in common neurodegenerative diseases, such as familial Alzheimer’s disease (Coen et al., 2012; Lee et al., 2015). A recent report showed that Parkinson disease patients’ cells with GBA1 or LRRK2 mutations (common risk factors of the disease) exhibit dysregulated lysosomal Ca2+ stores (Hockey et al., 2015; Kilpatrick et al., 2016a, b). It would not be surprising if more lysosome-related diseases associated with defects in lysosomal Ca2+ signaling are discovered (for detailed reviews, please see (Kiselyov et al., 2010; Morgan et al., 2011; Feng and Yang, 2016).
Other vesicular Ca2+ stores
There are various types of common cellular vesicles with Ca2+-regulated membrane trafficking, including early endosomes, recycling endosomes, phagosomes, autophagosomes, secretory vesicles and peroxisomes (Fig. 1), as well as additional Ca2+-regulated vesicles in specialized cell types, such as synaptic vesicles in neurons, melanosomes in melanocytes (Bellono and Oancea, 2014), tubulovesicles in parietal cells (Sahoo et al., 2017) and lytic granules in cytotoxic T-cells (Clark and Griffiths, 2003; Patel and Cai, 2015). These organelles are capable of storing and releasing Ca2+ and, thus, are also considered to be small Ca2+ stores (Fig. 1).
Studying Ca2+ channels in these atypical Ca2+ stores remains a challenge. Tubulovesicles have been long proposed as vesicular Ca2+ stores that undergo exocytosis, bringing H+/K+-ATPase proton pumps to the apical membranes of parietal cells upon histamine stimulation. Recently, Sahoo et al. (2017) demonstrated that TRPML1 is localized on the tubulovesicular membranes of parietal cells, and that upon histamine-protein kinase A pathway activation, Ca2+ is released, inducing tubulovesicle exocytosis. Whereas TRPML1 knockout mice and ML-IV patients are achlorhydric (lacking acid secretion), TRPML1 overexpressing transgenic exhibits constitutive acid secretion (Sahoo et al., 2017). This work established TRPML1-mediated Ca2+ release as a missing link between histamine-protein kinase A signaling and tubulovesicle exocytosis (Sahoo et al., 2017). However, the refilling mechanisms for these vesicles are completely unknown.
Conclusions and future directions
Although it is supposed that the ER may refill lysosomal Ca2+ stores, the underlying mechanisms of such refilling have not been delineated. Experiments showing that inhibition of ER Ca2+ and, more specifically, IP3Rs can abolish lysosomal store refilling support the notion that the ER may be the primary source of lysosomal Ca2+ under physiological conditions. Hence, small secondary stores, including lysosomes, may acquire Ca2+ from large primary stores, which in turn are refilled by extracellular Ca2+. In the near future, we expect to see advancements in the following areas:
-
Identification of more regulators of ER-lysosome MCS formation.
-
Revelation of lysosomal ∆ψ and Ca2+ roles in the regulation of ER-lysosome MCS formation.
-
Development of organelle-targeted voltage dyes and luminal Ca2+ sensors enabling the study of ER-lysosome MCS dynamics with super-resolution live imaging.
-
Identification of low-affinity uptake channels or transporters in the lysosome.
-
Discovery of additional small, vesicular and mobile Ca2+ stores, together with their Ca2+ release channels and Ca2+ uptake transporters in these vesicles.
References
Appelqvist H, Johansson AC, Linderoth E, Johansson U, Antonsson B, Steinfeld R, Kagedal K, Ollinger K (2012) Lysosome-mediated apoptosis is associated with cathepsin D-specific processing of bid at Phe24, Trp48, and Phe183. Ann Clin Lab Sci 42(3):231–242
Bagur R, Hajnoczky G (2017) Intracellular Ca2+ sensing: its role in calcium homeostasis and signaling. Mol Cell 66(6):780–788
Bellono NW, Oancea EV (2014) Ion transport in pigmentation. Arch Biochem Biophys 563:35–41
Bengtson CP, Bading H (2012) Nuclear calcium signaling. Adv Exp Med Biol 970:377–405
Berridge MJ (2012) Calcium signalling remodelling and disease. Biochem Soc Trans 40(2):297–309
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1(1):11–21
Bootman MD, Fearnley C, Smyrnias I, MacDonald F, Roderick HL (2009) An update on nuclear calcium signalling. J Cell Sci 122(Pt 14):2337–2350
Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S (2009) Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol 186(2):201–209
Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459(7246):596–600
Cang C, Zhou Y, Navarro B, Seo YJ, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham DE, Ren D (2013) mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell 152(4):778–790
Cao Q, Zhong XZ, Zou Y, Murrell-Lagnado R, Zhu MX, Dong XP (2015a) Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Biol 209(6):879–894
Cao Q, Zhong XZ, Zou Y, Zhang Z, Toro L, Dong XP (2015b) BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev Cell 33(4):427–441
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142(2):270–283
Cheng X, Shen D, Samie M, Xu H (2010) Mucolipins: intracellular TRPML1-3 channels. FEBS Lett 584(10):2013–2021
Christensen KA, Myers JT, Swanson JA (2002) pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 115(Pt 3):599–607
Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL, Song BL (2015) Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161(2):291–306
Clapham DE (2007) Calcium signaling. Cell 131(6):1047–1058
Clark R, Griffiths GM (2003) Lytic granules, secretory lysosomes and disease. Curr Opin Immunol 15(5):516–521
Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W (2012) Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol 198(1):23–35
De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Di Campli A, Polishchuk E, Di Tullio G, Morra V, Levtchenko E, Oltrabella F, Starborg T, Santoro M, Di Bernardo D, Devuyst O, Lowe M, Medina DL, Ballabio A, De Matteis MA (2016) Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol 18(8):839–850
De Stefani D, Rizzuto R, Pozzan T (2016) Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85:161–192
Dickson EJ, Duman JG, Moody MW, Chen L, Hille B (2012) Orai-STIM-mediated Ca2+ release from secretory granules revealed by a targeted Ca2+ and pH probe. Proc Natl Acad Sci USA 109(51):E3539–E3548
Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H (2010a) PI(3,5)P(2) controls membrane traffic by direct activation of mucolipin ca release channels in the endolysosome. Nat Commun 1(4):38
Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H (2010b) PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun 1:38
Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H (2011) A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol 192(1):121–135
English AR, Voeltz GK (2013) Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol 5(4):a013227
Feng X, Yang J (2016) Lysosomal calcium in neurodegeneration. Messenger 5:56–65
Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H (2016) The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife. https://doi.org/10.7554/eLife.15887
Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P (2013) PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 153(7):1494–1509
Grimm C, Butz E, Chen CC, Wahl-Schott C, Biel M (2017) From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium 67:148–155
Hockey LN, Kilpatrick BS, Eden ER, Lin-Moshier Y, Brailoiu GC, Brailoiu E, Futter CE, Schapira AH, Marchant JS, Patel S (2015) Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci 128(2):232–238
Hoglinger D, Haberkant P, Aguilera-Romero A, Riezman H, Porter FD, Platt FM, Galione A, Schultz C (2015) Intracellular sphingosine releases calcium from lysosomes. Elife 4:e10616
Huang P, Zou Y, Zhong XZ, Cao Q, Zhao K, Zhu MX, Murrell-Lagnado R, Dong XP (2014) P2X4 forms functional ATP-activated cation channels on lysosomal membranes regulated by luminal pH. J Biol Chem 289(25):17658–17667
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427(6971):260–265
Kilpatrick BS, Magalhaes J, Beavan MS, McNeill A, Gegg ME, Cleeter MW, Bloor-Young D, Churchill GC, Duchen MR, Schapira AH, Patel S (2016a) Endoplasmic reticulum and lysosomal Ca(2)(+) stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium 59(1):12–20
Kilpatrick BS, Yates E, Grimm C, Schapira AH, Patel S (2016b) Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J Cell Sci 129(20):3859–3867
Kiselyov K, Yamaguchi S, Lyons CW, Muallem S (2010) Aberrant Ca2+ handling in lysosomal storage disorders. Cell Calcium 47(2):103–111
Krols M, Bultynck G, Janssens S (2016) ER-Mitochondria contact sites: a new regulator of cellular calcium flux comes into play. J Cell Biol 214(4):367–370
Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R (2009) TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci Signal 2(71):ra23
Lee HC, Aarhus R (1995) A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem 270(5):2152–2157
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA (2015) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12(9):1430–1444
Lemons RM, Thoene JG (1991) Mediated calcium transport by isolated human fibroblast lysosomes. J Biol Chem 266(22):14378–14382
Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, Marchant JS (2012) Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem 287(4):2296–2307
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14(11):1247–1255
Lopez JJ, Albarran L, Gomez LJ, Smani T, Salido GM, Rosado JA (2016) Molecular modulators of store-operated calcium entry. Biochim Biophys Acta 1863(8):2037–2043
Melchionda M, Pittman JK, Mayor R, Patel S (2016) Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J Cell Biol 212(7):803–813
Min SW, Chang WP, Sudhof TC (2007) E-Syts, a family of membranous Ca2+-sensor proteins with multiple C2 domains. Proc Natl Acad Sci USA 104(10):3823–3828
Morgan AJ, Galione A (2007) NAADP induces pH changes in the lumen of acidic Ca2+ stores. Biochem J 402(2):301–310
Morgan AJ, Platt FM, Lloyd-Evans E, Galione A (2011) Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J 439(3):349–374
Morgan AJ, Davis LC, Galione A (2015) Imaging approaches to measuring lysosomal calcium. Methods Cell Biol 126:159–195
Patel S, Cai X (2015) Evolution of acidic Ca(2)(+) stores and their resident Ca(2)(+)-permeable channels. Cell Calcium 57(3):222–230
Patel S, Docampo R (2010) Acidic calcium stores open for business: expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol 20(5):277–286
Phillips MJ, Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17(2):69–82
Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, Ganesan A, Gosain R, Churchill GC, Zhu MX, Parrington J, Galione A, Sitsapesan R (2010) TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. J Biol Chem 285(45):35039–35046
Pittman JK (2011) Vacuolar Ca(2+) uptake. Cell Calcium 50(2):139–146
Pizzo P, Lissandron V, Capitanio P, Pozzan T (2011) Ca(2+) signalling in the Golgi apparatus. Cell Calcium 50(2):184–192
Prakriya M, Lewis RS (2015) Store-operated calcium channels. Physiol Rev 95(4):1383–1436
Putney JW, Steinckwich-Besancon N, Numaga-Tomita T, Davis FM, Desai PN, D’Agostin DM, Wu S, Bird GS (2017) The functions of store-operated calcium channels. Biochim Biophys Acta 1864(6):900–906
Qureshi OS, Paramasivam A, Yu JC, Murrell-Lagnado RD (2007) Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J Cell Sci 120(Pt 21):3838–3849
Raffaello A, Mammucari C, Gherardi G, Rizzuto R (2016) Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 41(12):1035–1049
Raiborg C, Wenzel EM, Pedersen NM, Stenmark H (2016) ER-endosome contact sites in endosome positioning and protrusion outgrowth. Biochem Soc Trans 44(2):441–446
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370):1763–1766
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13(9):566–578
Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 glued and late endosome positioning. J Cell Biol 185(7):1209–1225
Ronco V, Potenza DM, Denti F, Vullo S, Gagliano G, Tognolina M, Guerra G, Pinton P, Genazzani AA, Mapelli L, Lim D, Moccia F (2015) A novel Ca(2)(+)-mediated cross-talk between endoplasmic reticulum and acidic organelles: implications for NAADP-dependent Ca(2)(+) signalling. Cell Calcium 57(2):89–100
Ruas M, Rietdorf K, Arredouani A, Davis LC, Lloyd-Evans E, Koegel H, Funnell TM, Morgan AJ, Ward JA, Watanabe K, Cheng X, Churchill GC, Zhu MX, Platt FM, Wessel GM, Parrington J, Galione A (2010) Purified TPC isoforms form NAADP receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr Biol 20(8):703–709
Rudolf R, Mongillo M, Rizzuto R, Pozzan T (2003) Looking forward to seeing calcium. Nat Rev Mol Cell Biol 4(7):579–586
Saheki Y, De Camilli P (2017) Endoplasmic reticulum-plasma membrane contact sites. Annu Rev Biochem 86:659–684
Sahoo N, Gu M, Zhang X, Raval N, Yang J, Bekier M, Calvo R, Patnaik S, Wang W, King G, Samie M, Gao Q, Sahoo S, Sundaresan S, Keeley TM, Wang Y, Marugan J, Ferrer M, Samuelson LC, Merchant JL, Xu H (2017) Gastric acid secretion from parietal cells is mediated by a Ca2+ efflux channel in the tubulovesicle. Dev Cell 41(3):262–273 e266
Schmiege P, Fine M, Blobel G, Li X (2017) Human TRPML1 channel structures in open and closed conformations. Nature 550(7676):366–370
Scott CC, Gruenberg J (2011) Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. BioEssays 33(2):103–110
Shang S, Zhu F, Liu B, Chai Z, Wu Q, Hu M, Wang Y, Huang R, Zhang X, Wu X, Sun L, Wang Y, Wang L, Xu H, Teng S, Liu B, Zheng L, Zhang C, Zhang F, Feng X, Zhu D, Wang C (2016) Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J Cell Biol 215(3):369–381
Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3:731
Stathopulos PB, Ikura M (2017) Store operated calcium entry: from concept to structural mechanisms. Cell Calcium 63:3–7
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175(6):901–911
Tian X, Gala U, Zhang Y, Shang W, Nagarkar Jaiswal S, di Ronza A, Jaiswal M, Yamamoto S, Sandoval H, Duraine L, Sardiello M, Sillitoe RV, Venkatachalam K, Fan H, Bellen HJ, Tong C (2015) A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol 13(3):e1002103
van der Kant R, Neefjes J (2014) Small regulators, major consequences—Ca(2)(+) and cholesterol at the endosome-ER interface. J Cell Sci 127(Pt 5):929–938
Walseth TF, Lin-Moshier Y, Jain P, Ruas M, Parrington J, Galione A, Marchant JS, Slama JT (2012) Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J Biol Chem 287(4):2308–2315
Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H (2012) TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell 151(2):372–383
Wang W, Zhang X, Gao Q (2017) A voltage-dependent K+ channel in the lysosome is required for refilling lysosomal Ca2+ stores. J Cell Biol 216(6):1715–1730
Xiong J, Zhu MX (2016) Regulation of lysosomal ion homeostasis by channels and transporters. Sci China Life Sci 59(8):777–791
Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77:57–80
Xu H, Martinoia E, Szabo I (2015) Organellar channels and transporters. Cell Calcium 58(1):1–10
Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266(26):17707–17712
Zhang X, Yu L, Xu H (2016) Lysosome calcium in ROS regulation of autophagy. Autophagy 12(10):1954–1955
Zhong XZ, Zou Y, Sun X, Dong G, Cao Q, Pandey A, Rainey JK, Zhu X, Dong XP (2017) Inhibition of transient receptor potential channel mucolipin-1 (TRPML1) by lysosomal adenosine involved in severe combined immunodeficiency diseases. J Biol Chem 292(8):3445–3455
Zhou Y, Wong CO, Cho KJ, van der Hoeven D, Liang H, Thakur DP, Luo J, Babic M, Zinsmaier KE, Zhu MX, Hu H, Venkatachalam K, Hancock JF (2015) Signal transduction. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 349(6250):873–876
Zhou X, Li M, Su D, Jia Q, Li H, Li X, Yang J (2017) Cryo-EM structures of the human endolysosomal TRPML3 channel in three distinct states. Nat Struct Mol Biol 24(12):1146–1154
Acknowledgements
We apologize to colleagues whose works are not cited due to space limitations. The authors are supported by NIH Grants (NS062792, AR060837 and DK115471) and funds from the Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals.
Abbreviations
BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester); BK, big potassium; CAX, Ca2+/H+ exchanger; ER, endoplasmic reticulum; GECIs, genetically-encoded Ca2+ indicators; GPN, glycyl-Lphenylalanine-naphthylamide; IP3Rs, inositol 1,4,5-triphosphate receptors; MCSs, membrane contact sites; ML-IV, type IV mucolipidosis; ML-SAs, mucolipin-specific synthetic agonists; LSD, lysosomal storage disease; NAADP, nicotinic acid adenine dinucleotide phosphate; NPC1, Niemann-Pick C1 protein; ORP1L, oxysterol-binding protein-related protein 1L; ORP5, oxysterol-binding protein-related protein 5; P2X4, purigenic receptor X4; PI(3,5)P2, phosphatidylinositol 3,5-bisphosphate; PI(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PM, plasma membrane; RyRs, ryanodine receptors; SERCA, sarcoendoplasmic reticulum calcium transport ATPase; SOCE, store-operated Ca2+ entry; STARD3, stAR-related lipid transfer protein 3; STIM, stromal interaction molecule; TPC, two-pore channels; TRPA1, transient receptor potential ankyrin 1; TRPML, transient receptor potential; VAP, vesicle-associated membrane protein; VDACs, voltage-dependent anion channels; VGCCs, voltage-gated Ca2+ channels.
Compliance with Ethics Guidelines
Junsheng Yang, Zhuangzhuang Zhao, Mingxue Gu, Xinghua Feng, and Haoxing Xu declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Junsheng Yang and Zhuangzhuang Zhao contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yang, J., Zhao, Z., Gu, M. et al. Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 10, 8–19 (2019). https://doi.org/10.1007/s13238-018-0523-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-018-0523-x