
Abstract
A computational approach for identifying functionally relevant SNPs in gene LIG1 has been proposed. LIG1 is a crucial gene which is involved in excision repair pathways and mutations in this gene may lead to increase sensitivity towards DNA damaging agents. A total of 792 SNPs were reported to be associated with gene LIG1 in dbSNP. Different web server namely SIFT, PolyPhen, CUPSAT, FASTSNP, MAPPER and dbSMR were used to identify potentially functional SNPs in gene LIG1. SIFT, PolyPhen and CUPSAT servers predicted eleven nsSNPs to be intolerant, thirteen nsSNP to be damaging and two nsSNPs have the potential to destabilize protein structure. The nsSNP rs11666150 was predicted to be damaging by all three servers and its mutant structure showed significant increase in overall energy. FASTSNP predicted twenty SNPs to be present in splicing modifier binding sites while rSNP module from MAPPER server predicted nine SNPs to influence the binding of transcription factors. The results from the study may provide vital clues in establishing affect of polymorphism on phenotype and in elucidating drug response.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Single nucleotide polymorphisms, often referred as SNP, are the most common DNA variations present throughout human genome with a frequency of one in thousand base pairs (Brookes 1999). SNPs present in coding region are either synonymous SNP (sSNP) in which any alteration in the codon does not result in coding of different amino acid or nonsynonymous SNP (nsSNP) where a change in codon results in coding of different amino acid. The missense mutations (a category of nsSNP) are of importance because of their ability to influence protein functions and many of them are linked to human inheritable diseases (krawczak et al. 2000; Tokuriki et al. 2008; Wang and Moult 2001). While SNPs present in other genomic regions, viz untranslated regions (UTR), intron and promoter regions have potential to influence gene regulation (Mooney 2005). SNPs in transcription factor binding site (TFBS) may disrupt the site (Boccia et al. 1996; Vasiliev et al. 1999) or may form a novel binding site (Knight et al. 1999; Piedrafita et al. 1996). Similarly, a SNP in micro RNA binding site may lead to repression of protein coding genes or activators of RNA degradation (Mishra et al. 2008). Furthermore, SNPs in splicing modifiers binding site (enhancers or silencers) may generate an unstable mRNA resulting in a defective or truncated protein (ElSharawy et al. 2006). Some SNPs are functional (Hardison 2003) and thus, their identification is crucial to understand molecular basis of complex traits and diseases in human (Shastry 2002).
The experimental techniques are most comprehensive and precise ones in distinguishing functional SNPs from neutral ones (Chen and Sullivan 2003). It is not feasible in terms of time and cost to perform laboratory experiments for all SNPs in human genome (or in single gene) and elucidate their functional importance while theoretical or computational methods aid in narrowing down the number of potentially functional SNPs present in a human gene (Ramensky et al. 2002). In this study, the authors have applied web-based computational tools to identify potentially functional SNPs influencing protein stability, binding of splicing modifiers, binding of transcription factors and binding of micro RNA in gene DNA Ligase I (LIG1, ATP-dependent). The two most important processes in which gene LIG1 participates are joining of Okazaki fragments during eukaryotic DNA replication and ligation of synthesized patch during base excision repair (BER) (Pascal et al. 2004; Vago et al. 2009; Goetz et al. 2005; Lee et al. 2008; Timson et al. 2000). DNA replication gene LIG1 also interacts with proliferation cell nuclear antigen (PCNA) (Levin et al. 1997; Montecucco et al. 1998; Liang et al. 2008) and loss in its ability to interact with PCNA jeopardises its normal functionality to join Okazaki fragments and to ligate synthesized patch during BER (Liang et al. 2008; Levin et al. 2000). SNPs in gene LIG1 may cause DNA Ligase I deficiency which results in immunodeficiency and increased sensitivity to DNA-damaging agents (Barnes et al. 1992). In this study, mutant protein structures were modelled and compared with native structure of gene product LIG1, for changes in energy and Root Mean Square Deviation (RMSD) values.
The present in silico study focuses on identification of functional SNPs in most of genomic regions of human gene LIG1 as compared to the recent in silico studies which were more focussed on identification of deleterious nsSNPs (Doss et al. 2008a, b; Rajasekaran and Sethumadhavan 2010; Kanthappan and Sethumadhavan 2010).
Materials and methods
Dataset
The single nucleotide polymorphism database (dbSNP) (Sherry et al. 2001) cited at http://www.ncbi.nlm.nih.gov/SNP was used to retrieve SNPs and their related protein sequences for the gene LIG1.
Identification of deleterious nonsynonymous single nucleotide polymorphism by sequence homology based method
Sorting Intolerant from Tolerant (SIFT) tool accessible at http://sift.jcvi.org/ was applied to detect deleterious nonsynonymous SNPs (Ng and Henikoff 2001, 2002, 2003; Kumar et al. 2009). SIFT compiles a dataset of functionally linked protein sequences by searching protein database using PSI-BLAST algorithm. Then, it builds an alignment from the homologous sequences with the query sequence and scans all positions in the alignment and calculates the probabilities for amino acids at that position. The substitution at each position with normalized probabilities less than a tolerance index or SIFT score of 0.05 are predicted to be deleterious or intolerant while those equivalent or greater than 0.05 are predicted to be tolerant (Ng and Henikoff 2001). In this study RefSeq ID or GI number and substitution(s) was given as input to SIFT blink program (Kumar et al. 2009). The program was executed on default settings i.e., best BLAST hits for each organism were included and sequences greater than 90% identity to query were removed. A total of thirty-one nsSNPs in protein transcript (NP_000225.1) of gene LIG1 (NM_000234.1) were analysed for identification of deleterious variant(s).
Identification of damaging nonsynonymous single nucleotide polymorphism by structural-homology based method
Polymorphism Phenotyping tool (PolyPhen) available at http://coot.embl.de/PolyPhen/ uses structural and evolutionary characteristics to identify deleterious nsSNPs (Sunyaev et al. 2000; Ramensky et al. 2002). PolyPhen uses either amino acid sequence or SWall protein database ID (SPTR) or accession number with the two amino acid variants along with their position as inputs. The algorithm performs sequence-based characterization of the mutation site using a blend of various algorithms, followed by the identification and alignment of homologs to the query sequence and generating profile score. The amino acid residue substitution is then mapped to the known protein 3D structures and position-specific independent counts (PSIC) scores are calculated for each of the two amino acids. Finally, PSIC score difference is computed. A PSIC score difference more than or equal to 1.5 is considered to be damaging. Based on PSIC score difference, PolyPhen ranks nsSNP into one of the following three categories: (a) Benign (b) Possibly damaging and (c) Probably damaging. A total of thirty-one nsSNPs in protein transcripts (NP_000225.1) of gene LIG1 (NM_000234.1) were analysed for identification of deleterious variant(s).
Identification of nonsynonymous single nucleotide polymorphism influencing protein stability
Cologne University Protein Stability Analysis Tool (CUPSAT) (Parthiban et al. 2006, 2007a, b) available at http://cupsat.tu-bs.de/ was applied to analyse changes in protein stability upon point mutation. The computational method makes use of amino acid-atom potentials and torsion angle distribution to assess amino acid environment of the mutation site (Parthiban et al. 2007a, b). The overall stability is calculated from atom and torsion angle potentials. In case of unfavourable torsion angles, atom potentials may have higher impact on stability which results in stabilising mutation (Parthiban et al. 2007). The output comprises of information about mutational site, its structural features, and information regarding changes in protein stability for 19 possible substitutions at the give position. The structure of gene product LIG1 was acquired from Protein Data Bank (PDB) (Berman et al. 2000), having PDB id 1x9n (A chain). The protein structure, native amino acid residue and its position was given as an input to the tool. A total of sixteen nsSNPs were evaluated for their influence on protein stability.
Identification of single nucleotide polymorphism in splicing modifier binding site
FASTSNP (Yuan et al. 2006) a web-based tool, available at http://FASTSNP.ibms.sinica.edu.tw was used to determine polymorphism(s) in coding (nsSNP and sSNP) and in UTR regions of gene LIG1 influencing splicing regulation. FASTSNP is based on a decision tree principle and uses three web services: (i) ESEfinder (Cartegni et al. 2003; Smith et al. 2006) (ii) ESE-RESCUE (Fairbrother et al. 2002), and (iii) FAS-ESS (Wang et al. 2004) to predict impact of SNPs present in splicing modifier binding sites. SNPs present in Exonic Splicing Enhancer (ESE) sites are identified by ESEfinder and ESE-RESCUE tools. ESEfinder aids in identification of sSNPs located in ESE sites that will potentially weaken the binding site and ESE-RESCUE provides cross reference to the results from ESEfinder. While SNPs present in Exonic Splicing Silencer (ESS) site are identified by FAS-ESS tool. It also aids in identification of coding SNPs that will potentially abolish ESS sites. FASTSNP also computes a score based on the level of risk i.e., 0, 1, 2, 3, 4 and 5 indicating No, Very Low, Low, Medium, High and Very High risk.
Identification of single nucleotide polymorphism in transcription factor binding site and in micro-RNA binding site
The authors used rSNP module from MAPPER web server available at http://genome.ufl.edu/mapper/mapper-main to identify SNPs present in binding site of one or more transcription factors in gene LIG1. The tool identifies TFBS in multiple genomes, by combining TRANSFAC (Matys et al. 2003, 2006) and JASPAR (Sandelin et al. 2004; Bryne et al. 2008; Portales-Casamar et al. 2010) data with profile hidden Markov model (HMMs) (Marinescu et al. 2005a, b) The gene LIG1 was given as an input to rSNP module and models from all available three libraries i.e., TRANSFAC matrices, TRANSFAC factors and JASPAR matrices were selected. The result comprises of a list of SNPs in TFBSs along with computed scores, these scores indicate changes in binding affinity of transcription factors. Furthermore, the tool does not limit its prediction to 5′UTR and promoter region but also extends it to intron region (Jun and Jing 2010).
Database of all miRNA binding sites within 200 nucleotides of a SNP (dbSMR) which may influence binding of miRNA, available at http://miracle.igib.res.in/polyreg/ was used to detect these SNPs (Hariharan et al. 2009). Both options present in database i.e., polymorphisms around predicted miRNA binding sites and polymorphisms around validated miRNA binding sites, were executed to identify SNPs influencing binding of miRNA to its target sites in gene LIG1.
Modelling nsSNPs on protein structure and determining alterations in energy and RMSD
The structure of the gene product LIG1 was acquired from PDB, having PDB id 1x9n (A chain). The Swiss-PDB Viewer (Kaplan and Littlejohn 2001) was used for mapping mutations on structure. Selenomethionine residues present in the protein structures (native and mutant) were modified as Methionine using protein preparation wizard, Schrodinger, maestro (Schrodinger Inc. USA). The native and mutated structures were parameterized with amber03 force field and energy minimization was performed using GROMACS (Hess et al. 2008) (version 4.5.1) employing steepest descent algorithm. The RMSD values were computed using structural superimposition program from the Schrodinger suite. A total of seven nsSNPs were mapped onto the protein structure and analysed for change in energy and RMSD values from native structure.
Results
Dataset
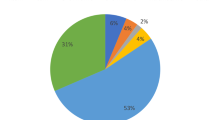
The SNPs related to LIG1 gene were acquired from dbSNP database, build 132 (Sherry et al. 2001). Out of 792 SNPs, 52 (6.56%) SNPs were present in coding region of the gene (31 nsSNP and 21 sSNP), 3 SNPs (0.37%) in 3′UTR, 736 SNPs (92.92%) in intron region, 1 SNP (0.12%) in 5′UTR.
Deleterious nonsynonymous single nucleotide polymorphisms predicted by SIFT server
Eleven nsSNPs were predicted to be deleterious with a tolerance index below 0.05. Lower the tolerance index or SIFT score, greater functional consequence an amino acid residue substitution is expected to have (Ng and Henikoff 2001). Four nsSNPs (rs111507847, rs3730947, rs34087182, rs11666150) had a tolerance index of 0.00, four nsSNPs (rs113944619, rs55686525, rs117019444, rs55950593) had a tolerance index of 0.01, two nsSNPs (rs3730863, rs3731003) had a tolerance index of 0.02, and the remaining one nsSNP (rs4987181) in the deleterious category had a tolerance index of 0.03. Seven nsSNPs (rs113944619, rs4987181, rs3730863, rs3730947, rs117019444, rs3731003, rs11666150) out of eleven nsSNPs predicted to be deleterious had a validated status (Table 1).
Damaging nonsynonymous single nucleotide polymorphism predicted by PolyPhen
Thirteen nsSNPs out of thirty-one nsSNPs were predicted to be either possibly damaging or probably damaging and had PSIC score difference in the range of 1.548 and 2.840 (Table 1). Out of these thirteen nsSNPs, eight nsSNPs (rs113944619, rs4987181, rs12981963, rs11879148, rs55686525, rs111507847, rs34087182, and rs11666150) were put into the category of probably damaging and the remaining five nsSNPs (rs41555118, rs3730863, rs4987070, rs79897727, rs55950593) were put into the category of possibly damaging by the program. Eight nsSNPs (rs41555118, rs3730863, rs4987070, rs79897727, rs113944619, rs4987181, rs12981963, rs11879148) out of thirteen nsSNPs predicted to be in the category of either possibly damaging or probably damaging had validated status. It was observed that six nsSNPs (rs113944619, rs4987181, rs55686525, rs111507847, rs34087182, rs11666150) predicted to be probably damaging by PolyPhen server were also predicted deleterious by SIFT server. While two nsSNPs (rs3730863, rs55950593) predicted to be possibly damaging by PolyPhen server were also predicted to be deleterious by SIFT server. This shows a significant level of correlation between the results from evolutionary-based approach (SIFT) and structural-based approach (PolyPhen). The highly damaging nsSNP (rs34087182) had a PSIC score difference of 2.840 and SIFT score 0.00.
Nonsynonymous single nucleotide polymorphism responsible for destabilising protein structure
CUPSAT identified two nsSNPs (rs3731003 and rs11666150) out of sixteen nsSNPs to be influencing over all stability of the protein structure. Ten nsSNPs (rs3730933, rs111846131, rs111507847, rs3730947, rs3730966, rs4987068, rs112555243, rs74929288, rs55950593, rs11668325) only exhibited unfavourable changes in torsion angles with no influence on overall stability of protein (Table 2). The nsSNP rs11666150 predicted to be destabilising protein structure was also predicted damaging by SIFT server (SIFT score 0.00) and PolyPhen server (PSIC score difference 2.307).
Functional single nucleotide polymorphism in splicing modifiers binding site
FASTSNP predicted twenty SNPs to be influencing splicing regulation by their presence in splicing modifiers (enhancers and silencers) binding site (Table 3) (krawczak et al. 2000). Sixteen SNPs predicted to be influencing splicing regulation had a risk in range of 2–3 (low to medium) and remaining four SNPs with a risk in range of 3–4 (medium to high). Interestingly, two SNPs rs20581 and rs20580 were also highlighted in recent studies for their functional importance (Chang et al. 2008; Lee et al. 2008; Liu et al. 2009). None of the SNPs in UTR were reported to be present in splicing modifier binding sites.
Functional single nucleotide polymorphism in transcription factor binding site, micro RNA binding site, and in promoter region
Gene LIG1 contains binding sites for a number of transcription factors which may mediate increased expression in dormant cells in response to growth factors (Noguiez et al. 1992). The presence of transcription factor binding site is not limited to 5′UTR or to promoter region but it also extends to intronic region (Jun and Jing 2010). Nine SNPs were predicted to be present in transcription factor binding site. Five SNPs (rs3730842, rs75696040, rs74747924, rs7246696 and rs3730840) in intron and four SNPs (rs3730838, rs752084, rs3730836 and rs79501686) in promoter region were predicted to be present in TFBS. Two SNPs (rs75696040 and rs74747924) were predicted to be present in the binding site of MZF1 transcription factor in chromosomal region between 48,673,165 to 48,673,177 on chromosome 19. Other than SNP rs79501686, all SNPs gave a score difference of more than 2, indicating the presence of SNP substantially influences binding affinity of transcription factors (Table 4). None of the SNPs by dbSMR were reported to be influencing binding of micro RNA in gene LIG1.
Mapping and analysis of mutants on protein structure
Seven nsSNPs (rs11666150, rs55950593, rs34087182, rs3731003, rs117019444, rs3730947 and rs111507847) predicted to be deleterious by SIFT or PolyPhen server and present between the residue number 262 and 901 were mapped on the protein structure (PDB id: 1x9n, A chain) of gene product LIG1. The amino acid residue substitution was performed using Swiss-PDB Viewer to get seven mutant modelled protein structures for SNPs rs117019444, rs111507847, rs3730947, rs3731003, rs34087182, rs55950593, and rs11666150. The total energy of the native structure (1x9n, A chain, Fig. 1) and the seven mutant modelled protein structures for SNPs rs117019444, rs111507847, rs3730947, rs3731003, rs34087182, rs55950593, and rs11666150 was −52745.32, −62163.56, −40160.3672, −59187.7773, −57279.74, −59290.78, −53570.56 and −41863.19 kJ/mol, respectively (Table 5). It can be observed from Table 5 that the RMSD values fall in range of 0.00522673–0.0361993 and do not suggest much deviation while significant changes in energy of mutant structures can be observed. The mutant protein models for SNPs rs11666150 (Fig. 2) and rs111507847 showed an increase in energy compared to the energy of native structure. The result for nsSNP rs11666150 correlates with results given by SIFT, PolyPhen and CUPSAT servers. The native and mutant protein molecule structures were visualised using Visual Molecular Dynamics (VMD) program (Humphrey et al. 1996).

Native structure of protein DNA Ligase I

Mutant model of mapped nsSNP (rs11666150) on the structure of protein DNA Ligase I
Discussion
Laboratory-based techniques are most accurate and conclusive in distinguishing functional SNPs from non-functional SNPs (Chen and Sullivan 2003). But large number of SNPs present in human genome makes execution of laboratory techniques very demanding in terms of time, cost, and labour. On contrary, in silico methods can help in distinguishing potentially functional SNPs from neutral SNPs present in a gene.
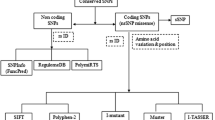
The computational pipeline (Fig. 3) was applied to all SNPs linked to gene LIG1as cited in dbSNP. Eleven and thirteen nsSNPs were predicted to be deleterious by SIFT and PolyPhen server, respectively. Eight nsSNPs were predicted to be deleterious by both SIFT and PolyPhen server. Evaluation of protein stability upon point mutation by CUPSAT server showed two nsSNPs (rs11666150 and rs3731003) to be able to destabilize protein structure. Out of seven mutant models of nsSNPs only two nsSNPs (rs11666150 and rs111507847) mutant models demonstrated significant change in energy compared to native structure of protein. Interestingly, one nsSNP (rs11666150) was predicted to be intolerant, probably damaging and destabilizing by SIFT, PolyPhen and CUPSAT servers, respectively, and also its mutant structure showed a significant change in energy level. FASTSNP web server predicted twenty SNPs to be influencing splicing regulation and four were predicted with a risk in range of 3–4 (medium to high). Nine SNPs from intron and promoter region were predicted by rSNP module from MAPPER to be influencing binding of transcription factor. The in silico study was well-focussed on SNPs present in all regions of gene LIG1 as regulatory region SNPs may also be disease causatives (Hudson 2003; Yan et al. 2002). Furthermore, results of the study were in concordance with the results from recent studies (Chang et al. 2008; Lee et al. 2008; Liu et al. 2009; Ryu et al. 2009).

Workflow along with the tools and databases used to identify potential functional SNPs in human gene LIG1
A large variety of tools are freely available for identification of potentially functional SNPs in a gene and each tool has different perspective for same biological problem (Thusberg and Vihinen 2009). The choice of computational tools to be used in an analysis is made on the nature of functional SNP to be identified and the amount of data and information being available for a given gene.
Conclusion
In this study nsSNP rs11666150 was found damaging by all the functional nsSNP prediction servers used. Further, its mutant structure demonstrated significant overall energy change as compared to the native structure. In this analysis, SNPs influencing binding of transcription factor and splicing modifier binding site are also predicted. However, studies will be required for in vitro validation of potentially functional SNPs in LIG1 and eventually will lead to development of better drugs against DNA ligase I deficiency (MIM: 126391). The authors suppose that the computational pipeline used in this study may also apply to any other human gene to identify potentially functional SNPs in it.
References
Barnes DE, Tomkinson AE, Lehmann AR, Webster AD, Lindahl T (1992) Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell 69(3):495–503
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Boccia LM, Lillicrap D, Newcombe K, Mueller CR (1996) Binding of the Ets factor GA-binding protein to an upstream site in the factor IX promoter is a critical event in transactivation. Mol Cell Biol 16(5):1929–1935
Brookes AJ (1999) The essence of SNPs. Gene 234(2):177–186
Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A (2008) JASPAR, the open access database of transcription factor binding profiles: new content and tools in 2008 update. Nucleic Acids Res 36:D102–D106
Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR (2003) ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 31(13):3568–3571
Chang JS, Wrensch MR, Hansen HM, Sison JD, Aldrich MC, Quesenberry CP Jr, Seldin MF, Kelsey KT, Kittles RA, Silva G, Wiencke JK (2008) Nucleotide excision repair genes and risk of lung cancer among San Francisco Bay Area Latinos and African Americans. Int J Cancer 129(9):2095–2104
Chen X, Sullivan PF (2003) Single nucleotide polymorphism genotyping: biochemistry, protocol, cost and throughput. Pharmacogenomics J 3(2):77–96
Doss CGP, Rajasekaran R, Sudandiradoss C, Ramanathan K, Purohit R, Sethumadhavan R (2008a) A novel computational and structural analysis of nsSNPs in CFTR gene. Genomic Med 2(1–2):23–32
Doss CGP, Sudandiradoss C, Rajasekaran R, Purohit R, Ramanathan K, Sethumadhavan R (2008b) Identification and structural comparison of deleterious mutations in nsSNPs of ABL1 gene in chronic myeloid leukemia: a bio-informatics study. J Biomed Inform 41(4):607–612
ElSharawy A, Manaster C, Teuber M, Rosenstiel P, Kwiatkowski R, Huse K, Platzer M, Becker A, Nürnberg P, Schreiber S, Hampe J (2006) SNPSplicer: systematic analysis of SNP-dependent splicing in genotyped cDNAs. Hum Mutat 27(11):1129–1134
Fairbrother WG, Yeh RF, Sharp PA, Burge CB (2002) Predictive identification of exonic splicing enhancers in human genes. Science 297(5583):1007–1013
Goetz JD, Motycka TA, Han M, Jasin M, Tomkinson AE (2005) Reduced repair of DNA double-strand breaks by homologous recombination in a DNA ligase I-deficient human cell line. DNA Repair (Amst) 4(6):649–654
Hardison RC (2003) Comparative genomics. PLoS Biol 1(2):156–160
Hariharan M, Scaria V, Brahmachari SK (2009) dbSMR: a novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinform 10(1):108
Hess B, Kutzner C, Van Der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447
Hudson TJ (2003) Wanted: regulatory SNPs. Nat Genet 33:439–440
Humphrey W, Dalke A, Schulten K (1996) VMD-visual molecular dynamics. J Molec Graphics 14:33–38
Jun H, Jing Z (2010) Co-occurrence of core of binding sites for transcription factors in intronic region of Saccharomyces cerevisiae ribosomal protein genes. International Conference on Bioinformatics and on Biomedical Technology, Chengdu, pp 88–91. doi:10.1109/ICBBT.2010.5479005
Kanthappan R, Sethumadhavan R (2010) Identification of structurally and functionally significant deleterious nsSNPs of GSS gene: in silico analysis. Adv Biosci Biotechnol 1:361–366
Kaplan W, Littlejohn TG (2001) Swiss-PDB viewer (Deep View). Brief Bioinform 2:195–197
Knight JC, Udalova I, Hill AV, Greenwood BM, Peshu N, Marsh K, Kwiatkowski D (1999) A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat Genet 22(2):145–150
Krawczak M, Ball EV, Fenton I, Stenson PD, Abeysinghe S, Thomas N, Cooper DN (2000) Human gene mutation database-a biomedical information and research resource. Hum Mutat 15(1):45–51
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(8):1073–1082
Lee YC, Morgenstern H, Greenland S, Tashkin DP, Papp J, Sinsheimer J, Cao W, Hashibe M, You NC, Mao JT, Cozen W, Mack TM, Zhang ZF (2008) A case-control study of the association of the polymorphisms and haplotypes of DNA ligase I with lung and upper-aerodigestive-tract cancers. Int J Cancer 122(7):1630–1638
Levin DS, Bai W, Yao N, O’Donnell M, Tomkinson AE (1997) An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc Natl Acad Sci USA 94(24):12863–12868
Levin DS, McKenna AE, Motycka TA, Matsumoto Y, Tomkinson AE (2000) Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr Biol 10(15):919–922
Liang L, Deng L, Nguyen SC, Zhao X, Maulion CD, Shao C, Tischfield JA (2008) Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res 36(10):3297–3310
Liu Y, Scheurer ME, El-Zein R, Cao Y, Do KA, Gilbert M, Aldape KD, Wei Q, Etzel C, Bondy ML (2009) Association and interactions between DNA repair gene polymorphisms and adult glioma. Cancer Epidemiol Biomarkers Prev 18(1):204–214
Marinescu VD, Kohane IS, Riva A (2005) The MAPPER database: a multi-genome catalog of putative transcription factor binding sites. Nucleic Acids Res 33(Database Issue): D91–D97
Marinescu VD, Kohane IS, Riva A (2005b) MAPPER: a search engine for the computational identification of putative transcription factor binding sites in multiple genomes. BMC Bioinform 6(1):79
Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E (2003) TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31(1):374–378
Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34:D108–D110
Mishra PJ, Banerjee D, Bertino JR (2008) MiRSNPs or MiR-polymorphisms, new players in microRNA mediated regulation of the cell: introducing microRNA pharmacogenomics. Cell Cycle 7(7):853–858
Montecucco A, Rossi R, Levin DS, Gary R, Park MS, Motycka TA, Ciarrocchi G, Villa A, Biamonti G, Tomkinson AE (1998) DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories. EMBO J 17:3786–3795
Mooney S (2005) Bioinformatics approaches and resources for single nucleotide polymorphism functional analysis. Brief Bioinform 6(1):44–56
Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874
Ng PC, Henikoff S (2002) Accounting for human polymorphisms predicted to affect protein function. Genome Res 12(3):436–446
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Noguiez P, Barnes DE, Mohrenweiser HW, Lindahl T (1992) Structure of the human DNA ligase I gene. Nucleic Acids Res 20(15):3845–3850
Prathiban V, Gromiha MM, Schomburg D (2006) CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res 34(2):W239–W242
Parthiban V, Gromiha MM, Abhinandan M, Schomburg D (2007a) Computational modeling of protein mutant stability: analysis and optimization of statistical potentials and structural features reveal insights into prediction model development. BMC Struct Biol 7:54
Parthiban V, Gromiha MM, Hoppe C, Schomburg D (2007b) Structural analysis and prediction of protein mutant stability using distance and torsion potentials: role of secondary structure and solvent accessibility. Proteins 66(1):41–52
Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T (2004) Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature 432(7016):473–478
Piedrafita FJ, Molander RB, Vansant G, Orlova EA, Pfahl M, Reynolds WF (1996) An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J Biol Chem 271(24):14412–14420
Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A (2010) JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38:D105–D110
Rajasekaran R, Sethumadhavan R (2010) In Silico identification of significant detrimental missense mutations of EGFR and their effect with 4-anilinoquinazoline-based drugs. Appl Biochem Biotechnol 160(6):1723–1733
Ramensky V, Pork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30(17):3894–3900
Ryu GM, Song P, Kim KW, Oh KS, Park KJ, Kim JH (2009) Genome-wide analysis to predict protein sequence variations that change phosphorylation sites or their corresponding kinases. Nucleic Acids Res 37(4):1297–1307
Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B (2004) JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32:D91–D94
Shastry BS (2002) SNP alleles in human disease and evolution. J Hum Genet 47(11):561–566
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) The NCBI database of genetic variation. Nucleic Acids Res 29(1):308–311
Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR (2006) An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 15(16):2490–2508
Sunyaev S, Ramensky V, Bork P (2000) Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet 16(4):198–200
Thusberg J, Vihinen M (2009) Pathogenic or not? And if so, then how? Studying the effect of missense mutations using Bioinformatics methods. Hum Mutat 130(5):703–714
Timson DJ, Singleton MR, Wigley DB (2000) DNA ligases in the repair and replication of DNA. Mutat Res 460(3–4):301–318
Tokuriki N, Stricher F, Serrano L, Tawfik DS (2008) How protein stability and new functions trade off. PLos Comput Biol 4(2):e1000002
Vago R, Leva V, Biamonti G, Montecucco A (2009) DNA ligase I and Nbs1 proteins associate in a complex and colocalize at replication factories. Cell Cycle 8(16):2600–2607
Vasiliev GV, Merkulov VM, Kobzev VF, Merkulova TI, Ponomarenko MP, Kolchanov NA (1999) Point mutations within 663–666 bp of intron 6 of the human TDO2 gene, associated with a number of psychiatric disorders, damage the YY-1 transcription factor binding site. Fed Eur Biochem Soc Lett 462(1–2):85–88
Wang Z, Moult J (2001) SNPs, protein structure, and disease. Hum Mutat 17(4):263–270
Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB (2004) Systematic identification and analysis of exonic splicing silencers. Cell 119:831–845
Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW (2002) Allelic variation in human gene expression. Science 297:1143
Yuan HY, Chiou JJ, Tseng WH, Liu CH, Liu CK, Lin YJ, Wang HH, Yao A, Chen YT, Hsu CN (2006) FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res 34(2):W635–W641
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Singh, A.A., Sivakumar, D. & Somvanshi, P. Cataloguing functionally relevant polymorphisms in gene DNA ligase I: a computational approach. 3 Biotech 1, 47–56 (2011). https://doi.org/10.1007/s13205-011-0006-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13205-011-0006-8