
Abstract
The oral bioavailability of poorly water soluble drug can be improved using nanosuspension. Nanosuspensions are fine dispersion of uniform-sized solid particles in an aqueous vehicle. The present work is aimed at the formulation and evaluation of nanosuspension of felodipine, a poorly water soluble antihypertensive drug. The nanosuspension of felodipine may increase the dissolution rate of drug to improve its oral bioavailability. The nanosuspensions were prepared by nanoprecipitation alone and in combination with ultrasonnication method using ethanol as solvent and water as antisolvent. The prepared nanosuspensions were characterised for particle size, zeta potential, polydispersity index, Scanning electron microscopy (SEM), Differential scanning calorimetry (DSC), X-ray diffraction (XRD) pattern and release behaviour. The effect of variable concentration of drug and stabiliser, ultrasonnication, and solvent to antisolvent ratio on the physical, morphological and dissolution properties of felodipine were studied. The average particle size of felodipine nanoparticles was found to be in the range of 60–330 nm. It was further confirmed by SEM photograph. The particle size varies with increase in concentration of drug and stabiliser. The preparations showed negative zeta potential and polydispersity index in the range of 0.3–0.8. DSC and XRD studies indicated that the crystallinity of precipitated felodipine nanoparticles was markedly lowered than the pure drug. The dissolution of prepared felodipine nanoparticles markedly increased as compared to the original drug. The dissolution profiles of nanosuspension formulation showed up to 79.67 % release in 4 h. It may be concluded that the nanoprecipitation with ultrasonnication have potential to formulate homogenous nanosuspensions with uniform-sized stable nanoparticles of felodipine. The prepared nanosuspension showed enhanced dissolution which may lead to enhanced oral bioavailability of felodipine.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The enhancement of the bioavailability of poorly water soluble drugs is one of the main targets of drug development during the last decades. Several techniques have been developed concerning the optimization of the dissolution rate of these drugs. Such methods include particle size reduction, solubilization, salt formation and preparation of solid dispersion systems. Nevertheless, there are several disadvantages and limitations in the use of these techniques. Specifically, the particle size reduction technique is practically limited regarding the minimum size, which could be achieved, while the production of very fine powders deteriorates their flow properties and wettability, while it advances the development of electrostatic forces, leading to problematic formulations. The solubilization techniques correspond to liquid preparations, which usually do not assure the patient’s acceptability and normally appear to have poor stability. Salt formation is a complicated process, while it is not feasible for neutral compounds. In addition, the dissolution enhancement is not always predictable due to the high correlation of the salt’s solubility with the pH value which appears to be a high variability in the gastrointestinal tract. (Kakrana et al. 2010).
In fact, for biopharmaceutic class II drugs, the bio-absorption process is rate-limited by dissolution in gastrointestinal fluids. According to the Noyes–Whitney equation, the dissolution rate of poorly water-soluble drugs could be increased by reducing the particle size to the micro or nano-scale thus increasing the interfacial surface area (Müller and Peters 1998; Douroumis and Fahr 2006). The conventional approaches to produce untrafine drug particles can be divided into top-down and bottom-up techniques (Keck and Müller 2006; Rabinow 2004). In the case of top-down techniques which include jet-milling, pear/ball milling and high pressure homogenising, the bulk drugs are comminuted into micro or nano-sized range using mechanical force (Rasenack et al. 2004). However, these techniques need high energy input and exhibit some disadvantages in practice such as contamination of drugs, variation of crystal structures, uncontrolled particle morphology, and broad particle size distribution (Keck and Müller 2006; Kharb et al. 2006). In the last decade, bottom-up techniques that rely on dissolving the drug in a solvent and precipitating it by the addition of a non-solvent, like supercritical fluid (SCF) technique and liquid precipitation, have been widely investigated to obtain ultrafine drug particles, such as cephradine (Zhong et al. 2005), cefuroxime axetil (Zhang et al. 2006), danazol (Rogers et al. 2002; Zhao et al. 2007), ibuprofen (Rasenack et al. 2004), etc.
The nanoprecipitation technique (or solvent displacement method) for nanoparticle manufacture was first developed and patented by Fessi and co-workers (Fessi et al. 1988, 1992). This technique presents numerous advantages, in that it is a straight forward technique, rapid and easy to perform. The nanoparticle formation is instantaneous and the entire procedure is carried out in only one step. Briefly, it requires two solvents that are miscible. Ideally, both the polymer and the drug must dissolve in the first one (the solvent), but not in the second system (the non-solvent). Nanoprecipitation occurs by a rapid desolvation of the polymer when the polymer solution is added to the non-solvent. Indeed, as soon as the polymer-containing solvent has diffused into the dispersing medium, the polymer precipitates, involving immediate drug entrapment. The rapid nanoparticle formation is governed by the so-called Marangoni effect, which is due to interfacial turbulences that take place at the interface of the solvent and the non-solvent and result from complex and cumulated phenomena such as flow, diffusion and surface tension variations (Quintanar-Guerrero et al. 1998). Nanoprecipitation often enables the production of small nanoparticles (100–300 nm) with narrow unimodal distribution. This technique is mostly suitable for compounds having a hydrophobic nature such as indomethacin, which is soluble in ethanol or acetone, but has very limited solubility in water. Consequently, reduced or even zero drug leakage toward the outer medium led to nanoparticles with entrapment efficiency values reaching 100 % (Fessi et al. 1988; Barichello et al. 1999).
Felodipine is a dihydropyridine calcium antagonist widely used as a potent antihypertensive drug (Saltiel et al. 1988). However, the oral bioavailability of felodipine extensively metabolised in the gut and the liver, and is excreted almost entirely as metabolites. About 70 % of each dose is excreted in the urine; the remainder appears in the faeces (Dunselman and Edgar 1991; Edgar et al. 1987). Felodipine is a compound with a strong lipophilic character whilst it appears as a crystalline powder practically insoluble in aqueous solutions (solubility <0.5 mg/l) (Karavas et al. 2005; Abrahamsson et al. 1994). On the contrary, Felodipine is rapidly absorbed by the gastrointestinal tract after dissolution. However, even though it is highly permeable through biological membranes (Diez et al. 1991), the physicochemical characteristics of felodipine indicate that its dissolution profile is the limiting factor of its bioavailability (Wingstrand et al. 1990). In order to increase its dissolution rate, several attempts were carried out in the past (Kerc et al. 1991; Kim et al. 2005; Lee et al. 2003) Other methods of solubility enhancement like solid dispersions have also been explored for enhancement of solubility of felodipine (Dong-Han et al. 2005).
The aim of this study is to investigate the possibility of producing a nanosuspension of felodipine, by controlled precipitation using ultrasonnication technique to enhance the dissolution and thereby oral bioavailability of the drug. The impact of experimental parameters for particle formation including solvent–antisolvent flow rate, drug concentrations, type and concentration of stabiliser and stirring time was studied. Characterization and physical stability of the obtained nanosuspension were also carried out.
Materials and methods
Preparation of nanoparticles
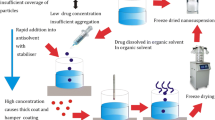
Felodipine nanoparticles were produced by precipitation–ultrasonnication technique. The required amount of drug was completely dissolved in water-miscible solvent. Different concentrations of drug in solvent (20, 40, 60, 80, 100 mg/ml) were used. The obtained drug solution was then injected into the water containing the stabiliser under stirring at 1,000 rpm. Precipitation of solid drug particles occurred immediately upon mixing. The suspension was then ultrasonicated for 15 min under cold condition.
Size measurement and zeta potential analysis
The particle size and the polydispersity Index (PI) of the formed drug particles was measured immediately after precipitation by dynamic laser light scattering (Zetasizer Ver. 6.11 Malvern). The measurement was done in triplicate and size Z-Average (d.nm) and the polydispersity Index PI was reported (Yuancai et al. 2010). The zeta potential of the preparations was also measured using (Zetasizer Malvern).
Scanning electron microscopy (SEM)
Particle morphology was observed using scanning electron microscopy (SEM) JSM-6360 (JEOL Inc., Japan). The samples’ small drop of the suspension was air dried followed by oven drying and was fixed on an SEM stub using double-sided adhesive tape and coated with Au at 20 mA for 6 min through a sputter-coater (Ion sputter JFC 1100). A scanning electron microscope with a secondary electron detector was used to obtain digital images of the samples at an accelerating voltage of 15 kV. (Yuancai et al. 2010).
Drug entrapment efficiency (DEE)
The freshly prepared nanosuspension was centrifuged at 20,000 rpm for 20 min at 5 °C temperature using cool ultracentrifuge. The amount of unincorporated drug was measured by taking the absorbance of the appropriately diluted 25 ml of supernatant solution at 363 nm using UV spectrophotometer against blank/control nanosuspension. DEE was calculated by subtracting the amount of free drug in the supernatant from the initial amount of drug taken. The experiment was performed in triplicate for each batch and the average was calculated (Mandal et al. 2010).
The entrapment efficiency (EE %) could be achieved by the following equation:
X-ray diffraction studies (XRD)
X-ray diffraction analysis was employed to detect the effect on crystallinity of precipitated felodipine nanopartilces which was conducted using a XRD-6000 diffractometer (Shimadzu, Japan). The powder was placed in a glass sample holder. CuK radiation was generated at 30 mA and 40 kV. Samples were scanned from 10° to 90° with a step size of 0.02° (Martine et al. 1998).
Fourier transforms infrared spectroscopy (FT-IR)
Drug excipients interactions were studied by FTIR spectroscopy (Hai-Xia et al. 2009). The spectra were recorded for Felodipine, HPMC (Hydroxy propyl methyl cellulose), Felodipine–HPMC Physical Mixture (1:5) and the dried nanoparticles. Samples were prepared in KBr discs (2 mg drug in 8 mg KBr) with a hydrostatic press at a force of 8 t cm−2 for 2 min. The scanning range was 450–4,000 cm−1 and resolution was 2 cm−1.
Differential scanning calorimetry (DSC)
The DSC analysis of pure drug, HPMC, Felodipine–HPMC Physical Mixture (1:5) and the dried nanoparticles was carried out using Mettler Toledo (Model SW 810) to evaluate any possible drug–excipients interaction. Samples (5.5–8 mg) were weighed accurately using a single pan electronic balance and heated in sealed aluminium pan at rate of 5 °C/min from 25 to 450 °C temperature range under a nitrogen flow of 35 ml/min. (Hany et al. 2009).
In vitro release kinetic experiments
The dialysis membrane diffusion technique was used. One millilitre of the nanosuspensions was placed in the dialysis membrane (Mw cutoff 12,000–14,000 Hi-media), fixed in a Kisery chien apparatus of surface area and receptor volume of 20 ml. A solvent system of Ethanol–water (50:50) was used as receptor medium. The entire system was kept at 37 °C with continuous magnetic stirring. Samples (1 ml) were withdrawn from the receptor compartment at predetermined time intervals and replaced by fresh medium. The amount of drug dissolved was determined with UV spectrophotometry at 363 nm.
Physical stability study
Three temperature conditions were applied in the stability study of the nanosuspensions: 4 °C (refrigerator), room temperature and 40 °C (Stability Chamber). Physical stability of the nanosuspensions was evaluated after 6 months of storage. The measurement of particle size was performed and settling behaviour by visual examination (Eerdenbrugh et al. 2008).
Results and discussion
Preparation of nanoparticles
Felodipine nanoparticles were produced by precipitation–ultrasonnication technique. The aqueous phase containing a suitable stabiliser has been used as the antisolvent and the use of different water miscible solvents (ethanol, acetone) having good solubility of felodipine has been explored as solvent. The effect of various variables like diffusing drug concentration, solvent:antisolvent ratio (S:NS), type of stabiliser, concentration of stabiliser, stirring time and ultrasonnication has been observed. The results have been summarised in Table 1.
Acetone and ethanol have been tried as solvent for the preparations. But acetone-based formulations show aggregation of particles and settling on storage. The viscosity of the preparation increases indication flocculation behaviour. Hence acetone has not been used for further studies. The preparations may require use of suitable stabilisers. Ethanol as solvent produces very uniform-sized nanoparticles on precipitation.
From the preliminary studies no specific effect on particle sizes has been observed at different solvent:antisolvent ratios (1:10, 1:15, 1:20). Preparations with S: NS 1:20 were a bit more homogenous, hence were taken for the preparation as suitable S:NS ratio. The effect of stirring time (5, 10, 30, 60 min) on the particle size was studied. No sign of aggregation due to stirring has been observed, and the particle size does not depend on stirring time.
HPMC and PVA, at various concentrations (0.1, 0.25, 0.5, 1.0 %), have been used as stabiliser for the preparations. From the preliminary studies, 0.5 % concentration was found to be optimum for both PVA (poly vinyl alcohol) and HPMC. The PVA-based formulations at concentration 0.5 % gave smaller and more uniform nanoparticles as observed by particle size analysis and SEM whereas preparations based on 0.5 % HPMC as stabiliser were a bit bigger and cuboidal in size.
The preparations have been made with different diffusing drug concentration (20, 40, 60, 80, 100 mg/ml). At lower concentrations of 5 and 10 mg/ml the precipitated particles were quite bigger and mostly in millimetre range. The particle size varies with the change in drug concentration. The study indicates that sufficient supersaturation is required for diffusing drugs to get precipitated in nanoparticulate range. Preparations with 80 mg/ml diffusing drug concentration were found to be optimum for PVA-based formulations whereas in case of HPMC it was found at 40 mg/ml.
The effect of ultrasonnication has been studied since the preparation obtained by precipitation alone showed less homogeneity and particles were obtained in bigger size range with average particles size at 600–800 nm as shown in Fig. 1a. The particle size was considerably decreased and more uniform in case of precipitation followed by ultrasonnication for 15 min under cold condition as shown in Fig. 1b. Hence, precipitations with ultrasonnication have been used for further preparations of nanoparticles of felodipine.

a Particle size distribution of nanoparticles formed by precipitation without sonication. b Particle size distribution of PVA based nanoparticles formed by precipitation with sonication. c Particle size distribution of HPMC based nanoparticles formed by precipitation with sonication. d Zeta potential measurement of HPMC based nanoparticles formed by precipitation with sonication
Size measurement and zeta potential analysis
The particle size and the polydispersity Index (PI) of the formed drug particles was measured immediately after precipitation by dynamic laser light scattering (Zetasizer Ver. 6.11Malvern). The average particle size of felodipine nanoparticles was found to be in the range of 60–330 nm. The formulations containing 0.5 % HPMC as stabiliser showed particle size in the range of 320–410 nm at various drug concentrations. The formulations were homogeneous as indicated by polydispersity index of 0.5 ± 0.1. The average particle diameter was found to decrease further in formulations containing PVA as stabiliser at concentrations of 0.5 %. The average diameter ranged in between 108 and 191 nm at various drug concentrations. The particle size distributions were found to be more uniform as the polydispersity index narrowed down to 0.3–0.5. The zeta potential of the nanoparticles was found to be negative which may be due to the presence of terminal carboxylic groups. High potential values should be achieved to ensure a high-energy barrier and favour a good stability. The results have been shown in Table 1.
Drug entrapment efficiency
The formulations showed drug entrapment in the range of 84–92 %. Formulation (FF6) containing drug concentration 80 mg/ml stabilised with PVA showed highest entrapment up to 92 % w/v. Formulations containing PVA showed better entrapment in comparison to HPMC. The results have been shown in Table 1.
Scanning electron microscopy (SEM)
Morphology of precipitated drug particles in the suspension after air drying followed by oven-drying is shown in Fig. 2a and b. The drug particles precipitated with the PVA as stabiliser are spherical in shape and the size ranges from 60 to 200 nm. The particles are discrete and uniform in size and there is no sign of agglomerations. The drug particles precipitated with the HPMC as stabiliser are slightly bigger and spherical and somewhat cuboidal and the size ranges from 100 to 300 nm.

a SEM photomicrograph of PVA based felodipine Nanoparticles (X 43,000). Scale bar = 0.5 μm. b SEM photomicrograph of HPMC based felodipine Nanoparticles (X 27,000). Scale bar = 0.5 μm
Differential scanning calorimetry (DSC)
The DSC analysis of pure drug, HPMC, Felodipine–HPMC Physical Mixture (1:5) and the dried nanoparticles was carried. The pure drug felodipine showed a melting point peak at 145 °C which was not changed in its physical mixture (1:5) with HPMC. The DSC thermogram however showed that after being precipitated as nanoparticles, its melting point was decreased to 133 °C, indicating reduced crystallinity. The results are shown in Fig. 3.

DSC thermograms of (red line) Pure drug felodipine (blue line) HPMC (green line) Felodipine and HPMC (physical mixture) (violet line) precipitated felodipine nanoparticles
X-ray diffraction studies (XRD)
The representative X-ray diffraction patterns of the pure Felodipine powder and oven dried nanosuspensions are shown in Fig. 4. The figures indicated the changes in the drug crystal structure. The X-ray patterns of the pure Felodipine displayed the presence of numerous distinct peaks at 10.19°, 16.51°, 20.47°, 23.27°, 24.51°, 25.37°, 26.45°, and 32.65°, which suggested that the drug was of crystalline form. The precipitated nanoparticles samples showed the similar diffraction pattern but with lower peak intensity, suggesting the crystallinity of precipitated nanoparticles decreased during the precipitation process.

X-ray diffraction patterns of (blue line) Pure felodipine drug (red line) Precipitated felodipine nanoparticles
Fourier transforms infrared spectroscopy (FT-IR)
Infra red spectra of Felodipine and physical mixture (1:5) of Felodipine and HPMC and the precipitated nanoparticles were comparable and the peaks of Felodipine in the physical mixture are of lower intensity than pure drug. The characteristic peak of 993, 1,431, 1,672, 2,935 and 3,134 cm−1 which corresponds to C–Cl (aromatic), aromatic ring stretch, C=O (ester form), C–H stretch and heterocyclic secondary amine stretch respectively were found to be intact in the physical mixture as well as the formulation which indicates the absence of HPMC and drug interactions. Very less stretching or broadening of peaks of drug was observed in mixture form. The results have been shown in Fig 5.

FTIR spectras of a Pure drug felodipine b HPMC c Precipitated felodipine nanoparticles
In vitro release kinetic experiments
It needs to be verified in vitro that nanoparticles were able to release incorporated drugs to achieve a biological effect. Membrane diffusion techniques are the most widely used experimental methods for the study of the in vitro release profiles of drugs incorporated in nanoparticles. The release profile of felodipine nanosuspensions in ethanol–water (50:50) solvent system which shows up to 79.67 % release in 4 h. The dissolution of prepared felodipine nanoparticles markedly increased as compared to the original drug. The comparative release results of selected formulations have been shown in Fig 6. Formulations based on PVA showed better release than that of HPMC as stabiliser.

Cumulative percentage of felodipine released from various formulations
Physical stability study
The particle size of the formulations after 6 months was observed. The formulations at 4 °C remained stable, whereas that at room temperature showed slight increase in particle size. In the case of HPMC formulations, only those stored at higher temperatures (40 °C) exhibited an increase in mean particle size. The increase in size in the HPMC formulation stored at 40 °C was attributed to dehydration of the HPMC chains and subsequent loss of protection of the nanoparticles. The particle size data are given in Table 2. Sedimentation was observed in all the conditions but the preparations were easily redispersed on shaking.
Conclusion
It may be concluded that the nanoprecipitation with ultrasonnication have potential to formulate homogenous nanosuspensions with uniform-sized stable nanoparticles of felodipine. The reduced particle size and decline in crystallinity of the precipitated particles may enhance the solubility of felodipine. The study also showed that the nanosuspension showed enhanced dissolution of felodipine. Since the limited oral bioavailability of felodipine is due to its poor dissolution hence the increase solubility, and thereby the dissolution of felodipine in the form of nanosuspension may enhance the oral bioavailability of felodipine. The preparations were found to physically stable with PVA and HPMC as stabiliser.
References
Abrahamsson B, Johansson D, Torstensson A, Wingstrand K (1994) Evaluation of solubilizers in the drug release of hydrophilic matrix extended-release tablets of felodipine. Pharm Res 11:1093–1097
Barichello JM, Morishita M, Takayama K, Nagai T (1999) Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev Ind Pharm 25:471–476
Diez I, Colom H, Moreno J, Obach R, Peraire C, Domenech JA (1991) Comparative in vitro study of transdermal absorption of a series of calcium channel antagonists. J Pharm Sci 80:931–934
Dong-Han W, Min-Soo K, Sibeum L, Jeong-Sook P, Sung-Joo H (2005) Improved physicochemical characteristics of felodipine solid dispersion particles by supercritical anti-solvent precipitation process Int. J. Pharm 301:199–208
Douroumis D, Fahr A (2006) Nano- and micro-particulate formulations of poorly water-soluble drugs by using a novel optimized technique. Eur J Pharm Biopharm 63:173–175
Dunselman PH, Edgar B (1991) Felodipine clinical pharmacokinetics. Clin Pharmacokinet 21:418–430
Edgar B, Collste P, Haglund K, Regardh CG (1987) Pharmacokinetics and haemodynamic effects of felodipine as monotherapy in hypertensive patients. Clin Invest Med 10:388–394
Eerdenbrugh BV, Froyen L, Humbeck JV, Martens JA, Augustins P, Mooter GV (2008) Drying of crystalline drug nanosuspensions-the importance of surface hydrophobicity on dissolution behaviour upon redispersion. Eur Journal Pharm sci 35:127–135
Fessi H, Puissieux F, Devissaguet JP, Thies C (1988) Process for the preparation of dispersible colloidal systems of a substance in the form of nanoparticles. US Patent US5118528, 2 June
Fessi H, Devissaguet JP, Puisieux F, Thies C (1992) Process for the preparation of dispersible colloidal systems of a substance in the form of nanoparticles. US Patent 593 522
Hai-Xia Z, Jie-X W, Zhi-Bing Z, Yuan L, Zhi-Gang S, Jian-Feng C (2009) Micronization of atorvastatin calcium by antisolvent precipitation process. Int J Pharm 374:106–113
Hany SMA, Peter Y, Nicholas B (2009) Preparation of hydrocortisone nanosuspension through a bottom-up nanoprecipitation technique using microfluidic reactors. Int J Pharm 375:107–113
Kakrana M, Sahoo NG, Lia L, Judeh Z, Wang Y, Chong K, Loh L (2010) Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int J Pharm 383:285–292
Karavas E, Ktistis G, Xenakis A, Georgarakis E (2005) Miscibility behaviour and formation mechanism of stabilized felodipine polivinylpyrrolidone amorphous nanodispersions. Drug Dev Ind Pharm 31:473–489
Keck CM, Müller RH (2006) Drug nanocrystals of poorly soluble drugs produced by high pressure homogenization. Eur J Pharm Biopharm 62:3–16
Kerc J, Srcic S, Mohar M, Smid-Korbar J (1991) Some physicochemical properties of glassy felodipine. Int J Pharm 68:25–33
Kharb V, Bhatia M, Dureja H, Kaushik D (2006) Nanoparticle technology for the delivery of poorly water-soluble drugs. Pharm Technol 2:82–92
Kim BK, Hwang SJ, Park JB, Park HJ (2005) Characteristics of felodipine-located poly(epsilon-caprolactone) microparticles. J. Microenc 22:193–203
Lee DW, Hwang SJ, Park JB, Park HJ (2003) Preparation and release characteristics of polymer-coated and blended alginate microspheres. J Microenc 20:179–192
Mandal B, Alexander KS, Riga AT (2010) Sulfacetamide loaded eudragit RL100 nanosuspension with potential for ocular delivery. J Pharm Pharmaceut Sci 13(4):510–523
Martine LV, Laurence F, Young-Il K, Maurice H, Philippe M (1998) Preparation and characterization of nanoparticles containing an antihypertensive agent. Eur J Pharm Biopharm 46:137–143
Müller RH, Peters K (1998) Nanosuspensions for the formulation of poorly soluble drugs. I. Preparation by a size-reduction technique. Int J Pharm 160:229–237
Quintanar-Guerrero D, All’emann E, Fessi H, Doelker E (1998) Preparation techniques and mecanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm 24:1113–1128
Rabinow BE (2004) Nanosuspensions in drug delivery. Drug Discov 3:785–796
Rasenack N, Steckel H, Müller BW (2004) Preparation of microcrystals by in situ micronization. Powder Technol 143–144:291–296
Rogers TL, Hu J, Yu ZS, Johnston KP, Williams RO III (2002) A novel particle engineering technology: spray-freezing into liquid. Int J Pharm 242:93–100
Saltiel E, Ellrodt AG, Monk JP, Langley MS (1988) Felodipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension. Drugs 36:387–428
Wingstrand K, Abrahamson B, Edgar B (1990) Bioavailability from felodipine extended-release tablets with different dissolution properties Int. J Pharm 60:151–156
Yuancai D, Wai KN, Jun H, Shoucang S, Reginald BHT (2010) A continuous and highly effective static mixing process for antisolvent precipitation of nanoparticles of poorly water-soluble drugs. Int J Pharm 386:256–261
Zhang X, Xia Q, Gu N (2006) Preparation of all-trans retinoic acid nanosuspensions using a modified precipitation method. Drug Dev Ind Pharm 32:857–863
Zhao H, Wang JX, Wang QA, Chen JF, Yun J (2007) Controlled liquid antisolvent precipitation of hydrophobic pharmaceutical nanoparticles in a microchannel reactor. Ind Eng Chem Res 46:8229–8235
Zhong J, Shen ZG, Yang Y, Chen JF (2005) Preparation and characterization of uniform nanosized cephradine by combination of reactive precipitation and liquid anti-solvent precipitation under high gravity environment. Int J Pharm 301:286–293
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Sahu, B.P., Das, M.K. Nanosuspension for enhancement of oral bioavailability of felodipine. Appl Nanosci 4, 189–197 (2014). https://doi.org/10.1007/s13204-012-0188-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-012-0188-3