
Abstract
DRIFTS experiments such as CO adsorption, CO-TPSR and CO+H2 were designated to study the effect of Fe promoter on the key steps of C2 oxygenates formation from syngas. The CO adsorption results demonstrated that Fe weakened CO adsorption and especially the bridging adsorption. It was found in CO-TPSR experiments that the catalyst with lower Fe loading is more easily dissociated while the ones with higher Fe loading own stronger hydrogenation activity. Moreover, it was observed by CO+H2 experiments that Fe plays a role in stabilizing the lineally adsorbed CO species and decreasing the CO desorption rate. The catalytic performance results indicated that when Fe content is 4wt. %, the selectivity of total C2 oxygenates is the highest, which was in accordance with the DRIFTS results.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rhodium-based catalysts have been paid considerable attention due to its high selectivity to C2 oxygenates such as ethanol from syngas [1,2,3]. Meanwhile, it is well known that Fe is an efficient promoter of rhodium-based catalyst [4, 5]. For instance, a series of Rh-Fe/Al2O3 catalysts were prepared by Burch et al. [6] where they observed that Fe inhibited the formation of CH4 and increased the selectivity of C2 oxygenates as well. Chen et al. [7] found that the optimized Fe content was 4 wt % for obtaining the highest ethanol yield over Rh-Fe/Al2O3 catalysts. Yu et al. [8] reported that the optimized Fe loading over Rh-Mn–Li/SiO2 was 0.1 wt %. Furthermore, some possible mechanisms about the promotion of Fe.
Promoter had been discussed, although they were still controversial. Schunemann et al. [9] attributed the improved activity of rhodium-based catalyst to the highly dispersed Fe3+ (Fe2+) oxides in close contact with Rh particles, which favored the ethanol and ethyl acetate formation. Choi et al. [10] concluded that Fe increased the reaction energy barrier of methane formation, thereby increasing the ethanol selectivity.
Wang et al. [11] reported that the addition of the Fe promoter brought an increase in the interfacial region between Rh and Fe.
It is well accepted that mechanism of ethanol formation is determined by such steps such as CO adsorption, CO dissociation, CO hydrogenation and CO insertion [12]. Therefore, it is helpful to understand the effect of Fe promoter on the investigated catalysts if we know how it affects these mechanism steps. In situ diffuse reflectance infrared transform spectroscopy (DRIFTS) is widely used to study the interaction of probe molecular with catalyst surface, which was found to be very sensitive to adsorption sites, CO coverage and surface orientation [13,14,15]. In CO hydrogenation reaction, CO adsorption behavior on the Rh-based catalysts was investigated and some meaningful conclusions had been reached. For example, Yu et al. [16] concluded the addition of Mn to Rh-based catalysts enhanced CO adsorption and found CO conversion was related with CO adsorption type and intensity. Mo et al. [17] found Fe decreased CO adsorption but improved hydrogenation ability of Rh-based catalysts. Liu et al. [18] found that the doping of Fe changed CO dissociation behavior over Rh/CeO2 catalyst during the experiments of CO adsorption and CO temperature programmed surface reaction (TPSR). Therefore, DRIFTS technique would be a very useful tool to understand the promotion of promoter from the view of elementary steps of ethanol formation, and to explain the difference in catalytic activity from a different view. Till now, the effect of Fe addition to Rh/Al2O3 on these steps has not been studied in detail. In this paper, DRIFTS experiments such as CO adsorption, CO hydrogenation, CO-TPSR were designated to explore the promotion effect of Fe on the catalytic performance of Rh/Al2O3 catalysts for C2 oxygenates synthesis via syngas.
Experimental
Catalyst preparation
Rhodium-based catalysts supported on γ-Al2O3 with different Fe loading were prepared by a co-impregnation method with aqueous solutions of iron nitrate [Fe(NO3)3], and rhodium nitrate [Rh(NO3)3]. After impregnation, the samples were dried at 110 °C for 12 h and calcined at 500 °C for 4 h. The obtained catalysts are designated as 2Rh-xFe/Al2O3, in which x ranges from 2 to 10 and the number before the metal element is weight percent relative to the mass of the γ-Al2O3 support.
Catalyst characterization
H2-TPR as well as H2-TPD profile of the catalysts was recorded using a Micromeritics AutoChem II 2920 instrument. For H2-TPR experiments, the samples (0.2 g, 40–60 μm) were purged in argon stream at 500 °C for 30 min to remove traces of water and then cooled to room temperature. After that, the catalyst sample was exposed to 50 mL/min of 10% H2/Ar flow. Then, the sample was reduced from room temperature to 800 °C with an increase of 10 °C/min, and the TPR profile was recorded according to H2 consumption. As to H2-TPD measurements, the sample (0.2 g, 40–60 μm) was reduced in 50 mL/min of 10% H2/Ar flow at 350 °C for 2 h and was purged in He flow for another 30 min. After cooling to room temperature, H2 was introduced into the catalyst bed until saturation in a pulse mode. Subsequently, the catalyst bed was purged by He flow for 30 min and heated from room temperature to 800 °C under He flow at a ramp rate of 10 °C/min, while the desorbed products were detected with the TCD detector.
High-resolution scanning transmission electron microscopy (STEM) measurements were performed on a Tecnai G2 F30 S-TWIN electron microscope with 300 kV accelerating voltage via high-angle annular dark-field (HAADF). Point energy-dispersive X-ray spectroscopy (EDS) was taken in an area within 5 nm diameter.
DRIFTS experiments were carried out with a Nicolet 6700 spectrometer equipped with an MCT-A detector (Thermo, USA) in the range of 4000–650 cm−1. The DRIFTS experiments contain CO adsorption, CO temperature programmed surface reduction (CO-TPSR) and CO+H2. For the above experiments, sample pre-treatment was done as follows: the sample was put in the in situ cell and purged by pure nitrogen at 180 °C for 30 min. After that, the catalyst bed was ramped to 350 °C with a rise of 2 °C/min and reduced in flowing hydrogen for 2 h, and then, background spectra were collected at designated temperature under 10−4 mbar vacuum.
For CO adsorption, a flow of 5% CO/He (v/v) was introduced to the reduced catalyst for 30 min. The IR spectra were recorded after flushed by N2 for 30 min. For CO-TPSR, the IR spectra were recorded under 10% H2/Ar (v/v) flow with the temperate linearly increased from 30 to 260 °C after CO adsorption. For CO hydrogenation, the IR spectra were recorded under a mix gas flow of 5% CO/10%H2/85%He with the temperature linearly increased from 30 to 260 °C. All the spectra were recorded with 64 scans and a resolution of 4 cm−1.
Catalyst activity test
CO hydrogenation experiments were carried out in a fixed bed reactor at temperature of 260 °C, pressure of 2.0 MPa, space velocity of 3600 mL/(g. h) and H2/CO of 2:1. Prior to reaction, the catalysts (1 g, 40–60 mesh) were reduced in a H2 flow for 10 h at temperature of 350 °C and pressure of 0.1 MPa. Then, syngas was switched into the system. After steady state, the reaction was kept for 24 h to collect liquid samples. Outlet gases were online analyzed by two chromatographs (Agilent GC7890A). One was equipped with two thermal conductivity detector (TCD) to analyze CO, CO2, N2 and H2 using a 5-A molecular sieve column. The other one was one hydrogen flame ionization detector (FID) to analysis C1–C6 hydrocarbons using Plot Q column. The liquid products were offline analyzed with the chromatograph (Agilent GC7890A) fitted with one FID and one TCD using Plot Q column to separate C1–C6 liquid products and water.
Results and discussion
Physicochemical properties
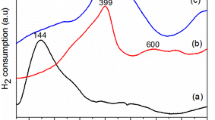
Figure 1 shows the TPR profile of 2Rh/Al2O3, 2Fe/Al2O3 and 2Rh-2Fe/Al2O3 catalyst, respectively. It can be observed that the board hydrogen consumption peak of 2Rh/Al2O3 (black curve) was located at 50 ~ 300 °C, corresponding to the reduction from surface rhodium to bulk rhodium [6]. The maximum reduction peak centered at 398 °C and 606 °C for 2Fe/Al2O3 (blue curve) can be assigned to the reduction of Fe3+→Fe2+ and subsequent Fe2+→Fe0. In the TPR profile of 2Rh-2Fe/Al2O3 (red curve), the Rh reduction peak moves to the lower temperature compared to that of 2Rh/Al2O3, which indicated that the addition of Fe boosts the transfer of Rh species from the bulk to the surface. In addition, according to the computation of H2 consumption of Rh/Al2O3, Rh3+ was not fully reduced [19]. Due to the inhibition effect of Fe promoter, Rh3+ and Rh0 must co-exist on the Fe promoted Rh-based catalysts.

H2-TPR profile of catalysts a 2Rh/Al2O3; b 2Fe/Al2O3; c 2Rh-2Fe/Al2O3
To further understand the effect of Fe on the reduction behavior of Rh-based catalysts, TPR profiles of Rh-Fe/Al2O3 catalysts with 2 ~ 10 wt. % Fe loading were performed and shown in Fig. 2. As can be seen from Fig. 2, a significant hydrogen consumption peak can be observed at 50 ~ 200 °C (low temperature) for all the Fe-promoted catalysts, and the peak intensity increases with the increase of Fe content, which confirms that the low temperature peak is attributed to the co-reduction of rhodium oxide and iron oxide. In addition, the low temperature reduction peak of Rh–Fe catalysts with 2 ~ 6 wt % Fe content is lower than un-promoted Rh catalysts, and get sharpen at higher Fe loading. In the range of 400 ~ 600 °C, no obvious peaks were observed for the catalysts with 2 ~ 6 wt. % Fe loading but distinct peaks centered at 495 °C for the catalysts with 8 ~ 10 wt. % Fe loading. The Fe loading-dependent TPR curves were sourced from the strong interaction between Fe and Rh. As we know, the strong interaction lead to a significant reduction temperature decrease for Fe3+ to Fe2+ and a reduction temperature raising up for Rhδ+ to Rh0. With low Fe loading, taking 2Rh-2Fe/Al2O3– (Fig. 2a) as an example, the interaction lead to a wide reduction peak relative to reduction of both Fe3+ to Fe2+ and Rhδ+ to Rh0. By raising up the Fe loading, in particular for 2Rh-8Fe/Al2O3– (Fig. 2d) and 2Rh-10Fe/Al2O3– (Fig. 2e), the more accumulate interaction caused the reduction temperatures of Fe3+ to Fe2+ and Rhδ+ to Rh0 further closer, leading to a sharp reduction peak.

H2-TPR profile of catalysts a 2Rh-2Fe/Al2O3; b 2Rh-4Fe/Al2O3; c 2Rh-6Fe/Al2O3; d 2Rh-8Fe/Al2O3; e 2Rh-10Fe/Al2O3
To verify the idea that the addition of Fe boosts the transfer of Rh species from the bulk to the surface, the STEM images of the catalysts of un-promoted and 4wt. % Fe promoted Rh based Are shown in Fig. 3. It is vividly seen in Fig. 3a for 2Rh/Al2O3 catalyst, no clear metal particles were observed. The possible reason is that only a small portion of Rh species is on the surface of the support. However, it was observed from Fig. 3b for 2Rh-4Fe/Al2O3 catalyst that the particles can be clearly seen, which verified our guess. The EDS spectrum of random point in the particle region and the particle-free region of the 2Rh4Fe catalyst (corresponding to points P1 and P2, respectively) was presented in Fig. 4. It can be seen that Rh and Fe are both contained in the particle with about 2 nm diameter, indicating that Rh is in close contact with Fe. In addition, only the weaker Fe signal was observed in the particle-free region, demonstrated the presence of isolated Fe species.

HRTEM images of a 2Rh/Al2O3 and b 2Rh-4Fe/Al2O3 catalyst

EDS spectra on 2Rh-4Fe/Al2O3 catalyst
Figure 5 shows hydrogen desorption behavior of Rh-based catalysts with different Fe loading. It can be observed desorption peaks in the temperature range of 50 ~ 300 °C and 400 ~ 500 °C for all the catalysts, corresponding to weak adsorption and strong adsorption of hydrogen, respectively. There are different opinions on the attribution of H2-TPD peaks for rhodium-based catalysts. Hilmen et al. [20] attributed the lower temperature peak to the desorption of chemisorbed hydrogen, and the higher temperature peak to the desorption of hydrogen spillover from support surface. However, Luo et al. [21] ascribed the higher temperature peak to the H2 desorption on larger Rh particles and the lower one to the H2 desorption on highly dispersed Rh particles. Figure 5 also displays that the desorption peaks intensity of 2Rh-2Fe/Al2O3 is nearly three times of that of 2Rh/Al2O3, and the total desorption peak area is increased when Fe content is changed from 2 to 10 wt %. Therefore, in our case, we only can conclude that the increase of desorption peak area is due to the increase of the hydrogen adsorption sites on the catalyst surface [22, 23].

H2-TPD profiles of Rh catalysts with different Fe loadings. a 2Rh/Al2O3; b 2Rh-2Fe/Al2O3; c 2Rh-4Fe/Al2O3; d 2Rh-6Fe/Al2O3; e 2Rh-8Fe/Al2O3; f 2Rh-10Fe/Al2O3
Figure 6 shows the infrared spectra of CO adsorption on Rh–Fe catalysts at different Fe contents (0 ~ 10 wt %). The bands at 2050 cm−1 in Fig. 6 were assigned to linearly adsorbed CO (CO (l)) on Rh0 sites. The double peaks centered at 2086 and 2018 cm−1 were attributed to the symmetric and asymmetric stretching vibration peak of Rh+(CO)2 (CO (gem)) on Rh+ sites. The band at 1860 cm−1 was assigned to bridged CO (CO (b)) on Rh0 sites [24]. Based on the DRIFTS spectra, it was found that the increase of Fe content inhibits CO adsorption intensity, especially the bridged one. Early studies have demonstrated that the Lewis acid sites of promoters are associated with CO adsorption in a bridged form [5]. Therefore, this can be explained that the strong Lewis acid nature of surface Fe3+ (Fe2+) can withdraw electron clouds from the Rh0 sites, consequently, the bridged CO band was weakened.

DRIFTS spectra of chemisorbed CO on catalysts at 25 °C. a 2Rh/Al2O3; b 2Rh-2Fe/Al2O3; c 2Rh-4Fe/Al2O3; d 2Rh-6Fe/Al2O3; e 2Rh-8Fe/Al2O3; f 2Rh-10Fe/Al2O3
To further understand the effect of Fe promoter on CO adsorption/desorption behaviors of Rh-based catalysts, IR spectra of adsorbed CO on 2Rh/Al2O3 and 2Rh-4Fe/Al2O3 catalyst at 260 °C during N2 purging were recorded in Figs. 7 and 8, respectively. The absorption peaks of gas phase CO at 2180 and 2125 cm−1 disappear after purging in N2 stream for 5 min. With the purging time increasing, the adsorption band of chemisorbed CO is shifted to lower frequency. In contrast to 2Rh/Al2O3 catalyst, the desorption rate of CO (l) and CO (gem) on the 2Rh-4Fe/Al2O3 catalyst is more slow. It means that the introduction of Fe inhabited the desorption rate of adsorbed CO species, which is in accordance with the conclusion from the work of Mo et al. [25]. It is noted that the band of bridged CO gets weaker for 2Rh/Al2O3 catalyst while nearly disappearing at reaction temperature (260 °C) for 2Rh-4Fe/Al2O3, indicating that the CO adsorbed on bridged site is very active and may be more easily involved in hydrogenation reaction.

DRIFTS spectra of adsorbed CO on 2Rh/Al2O3 catalyst at 260 °C with N2 purging

DRIFTS spectra of adsorbed CO on 2Rh-4Fe/Al2O3 catalyst at 260 °C with N2 purging
On the other hand, with the purpose to investigate the effect of Fe promoter on hydrogenation behaviors of adsorbed CO, IR spectra of adsorbed CO on 2Rh/Al2O3 and 2Rh-4Fe/Al2O3 catalyst at 260 °C during H2 purging were recorded in Figs. 9 and 10, respectively. We found that CO(b) and CO(l) decreased very rapidly in the presence of H2, indicating that the adsorbed CO species were partly involved in the hydrogenation reaction. In contrast to Fig. 9, it can be observed from Fig. 10 that the rate of CO (l) decreases more slowly on the 2Rh-4Fe/Al2O3, revealing that the addition of Fe stabilized CO(l) species under H2 flow conditions and subsequently increases non-dissociative adsorbed CO, and thus provides more active sites for CO insertion.

DRIFTS spectra of CO + H2 on 2Rh/Al2O3 catalyst 260 °C and 0.1 MPa

DRIFTS spectra of CO + H2 on 2Rh-4Fe/Al2O3 catalyst at 260 °C and 0.1 MPa
Temperature-programmed surface reduction (TPSR) is one of the most effective methods to study hydrogenation activity of chemically adsorbed CO [26, 27]. Methane is easily desorbed and its formation includes the steps as CO dissociation and hydrogenation, so its formation temperature and peak intensity is usually used as a tool to measure the CO dissociation ability and hydrogenation capacity of catalyst [7]. Here, three Rh–Fe catalysts were selected to investigate the effect of Fe loading on hydrogenation behavior of adsorbed CO.
Figures 11, 12, 13 show the TPSR spectra of 2Rh-2Fe/Al2O3, 2Rh-4Fe/Al2O3 and 2Rh-10Fe/Al2O3 catalysts, respectively. The peak at 3015 cm−1 is assigned to plane vibrations of gas phase CH4. It can be seen that the CH4 formation temperature of 2Rh-2Fe/Al2O3 catalyst is lower than that of 2Rh-4Fe/Al2O3 and 2Rh-10Fe/Al2O3, indicating that CO adsorbed on 2Rh-2Fe/Al2O3 is more easily dissociated. To compare the intensity of CH4 peak at reaction temperature, CH4 peaks at 260 °C of the three catalysts were plotted together and presented in Fig. 14. It can be observed from Fig. 14 that the CH4 peak intensity of 2Rh-10Fe/Al2O3 is the highest, which means it has the strongest hydrogenation activity. Furthermore, it can be obtained that the CO desorption rate on the 2Rh-10Fe/Al2O3 catalyst is the fastest by comparison of the spectra in the range of 1750 ~ 2200 cm−1 in Figs. 11, 12, 13.

CO-TPSR DRIFTS spectra of 2Rh-2Fe/Al2O3

CO-TPSR DRIFTS spectra of 2Rh-4Fe/Al2O3

CO-TPSR DRIFTS spectra of 2Rh-10Fe/Al2O3

CO-TPSR DRIFTS spectra of catalysts at 260 °C in the range of 2600–3200 cm−1. a 2Rh-10Fe/Al2O3; b 2Rh-4Fe/Al2O3; c 2Rh-2Fe/Al2O3
Catalytic performance of catalysts
Table 1 shows the results of CO hydrogenation over Fe promoted Rh-based catalysts. It can be seen from Table 1 that with the increase of Fe loading, the selectivity of methanol, hydrocarbon (HC) and CO2 increased. According to widely accepted mechanism of ethanol formation as shown in Fig. 15 [24], methanol is formed by the hydrogenation of non-dissociative adsorbed CO, hydrocarbon is derived from hydrogenation or chain growth of CHx species which begins with CO dissociation and hydrogenation. We suppose that the bridge CO was mainly responsible for the formation of CH4. The introduction of Fe inhibits CO adsorption especially bridge adsorption, which decreased the selectivity of CH4 greatly and correspondingly the total hydrocarbons selectivity. According to our CO-TPSR results, the increase of Fe content suppressed CO dissociation and increased hydrogenation activity, which provided evidences for more methanol formation. On the other hand, it was found that existed stronger Fe0 peak in the Rh–Fe catalysts at higher Fe loading from TPR profiles. Combined with activity data, it can be concluded that Fe0 content was related to methanol and CO2 activity, which is in accordance with the conclusion of Burch [6].

Mechanism of ethanol formation
From Table 1, it also can be seen that CO conversion increases with increasing Fe loading. The higher CO conversion of 2Rh-10Fe/Al2O3 should be assigned to the stronger hydrogenation capacity and the faster CO desorption rate according to CO-TPSR results. On the other hand, the selectivity of C2 oxygenates increases first and then decreases with the increase of Fe loading. The selectivity of C2 oxygenates present the highest with Fe proportion of 4 wt. %. Previous work suggests that Rh–Fe3+–O species are active sites for the generation of C2 oxygenates such as ethanol [5, 9]. Considering the contribution of Rh0 and Rh+ to CO insertion ability, the active site for ethanol formation over Rh-Fe/Al2O3 catalyst was assumed to the (Rh0−xRh+y) -O-Fe3+ (Fe2+) sites with reference to that of Rh-Mn catalysts [18]. These active sites could be formed by the close contact between Rh and Fe. From the results of TPR and STEM-EDS, we know that a part of Fe species was in close contact with Rh and a part of Fe existed as isolated Fe species. At the lower Fe loading, the catalyst has good synergistic effect between Rh and Fe and more Rh+ center which is mainly responsible for CO insertion. These increases Rh–Fe interface and provides more active sites for the formation of C2 oxygenates. However, the formation of C2 oxygenates should be a balance of CO dissociation, CO insertion and hydrogenation. 2Rh-2Fe/Al2O3 catalyst is more easily dissociated than 2Rh-4Fe/Al2O3 may be responsible for its slightly lower C2 oxygenate selectivity.
Conclusion
Promotion of Rh catalyst with Fe in the range of 2–10 wt. % was investigated. The addition of Fe facilitated the transfer of rhodium from bulk to surface and enhanced the hydrogen adsorption sites. The introduction of Fe inhibits CO adsorption especially bridge adsorption, correspondingly, the formation of total hydrocarbons decreases. CO adsorbed on the catalyst with lower Fe loading is more easily dissociated and CO adsorbed on the catalyst with higher Fe loading exhibited the strongest hydrogenation activity. The introduction of Fe also inhibits the desorption rate of CO (gem) and stabilizes CO (l) species.
Fe is an effective promoter to suppress the formation of hydrocarbons especially methane, and to shift selectivity to methanol and C2 oxygenates. However, it promotes water gas shift reaction, which leads to the increase in CO2 selectivity. The selectivity to C2 oxygenates in the products shows a rapid increase and then a slow decrease with the increase of Fe loading and passes through a maximum at 4wt. % of Fe loading.
References
Xu DD, Ma HF, Qian WX, Ying WY (2017) Effect of Ce promoter on Rh-Fe/TiO2 catalysts for ethanol synthesis from syngas. Catal Commun 98:93–98
Spivery J, Harrison D, Earle J, Goodwin J, Bruce D, Mo X, Torres W, Viswanathan JAV, Sadok R, Overbury S (2011) Catalytic process for the conversion of coal-derived syngas to ethanol. Board Of Supervisors of Louisiana State University, US
Yan C, Zhang HT, Ma HF, Qian WX, Jin FY, Ying WY (2018) Direct conversion of syngas to ethanol over Rh–Fe/γ-Al2O3 catalyst: promotion effect of Li. Catal Lett 148:1–8
Chen WM, Ding YJ, Song XG, Wang T, Luo HY (2011) Promotion effect of support calcination on ethanol production from CO hydrogenation over Rh/Fe/Al2O3 catalysts. Appl Catal A 407:231–237
Fukushima T, Arakawa H, Ichikawa M (1985) In situ high-pressure FT-IR studies on the surface species formed in carbon monoxide hydrogenation on silicon dioxide-supported rhodium-iron catalysts. J Phys Chem 89:4440–4443
Burch R, Hayes MJ (1997) The preparation and characterisation of Fe-promoted Al2O3-supported Rh catalysts for the selective production of ethanol from syngas. J Catal 165:249–261
Chen G, Guo CY, Huang Z, Yuan G (2011) Synthesis of ethanol from syngas over iron-promoted Rh immobilized on modified SBA-15 molecular sieve: effect of iron loading. Chem Eng Res Des 89:249–253
Yu J, Mao D, Han L, Guo Q, Lu G (2013) CO hydrogenation over Fe-promoted Rh–Mn–Li/SiO2 catalyst: the effect of sequences for introducing the Fe promoter. Fuel Process Technol 112:100–105
Schunemann V, Trevino H, Lei G, Tomczak D, Sachtler W, Fogash K, Dumesic J (1995) Fe promoted Rh-clusters in zeolite NaY: characterization and catalytic performance in CO hydrogenation. J Catal 153:144–157
Choi YM, Liu P (2009) Mechanism of ethanol synthesis from syngas on Rh (111). J Am Chem Soc 131:13054–13061
Wang JJ, Zhang QH, Wang Y (2011) Rh-catalyzed syngas conversion to ethanol: studies on the promoting effect of FeOx. Catal Today 171:257–265
Chuang SSC, Stevens RW, Khatri R (2005) Mechanism of C2+ oxygenate synthesis on Rh catalysts. Top Catal 32:225–232
Fang L, Qian WX (2017) Effect of Fe impregnation sequence on ethanol synthesis from syngas over Mn and Fe promoted Rh/γ-Al2O3. Appl Petrochem Res 7:161–167
Subramanian ND, Kumar CSSR, Watanabe K, Fischer P, Tanaka R, Spivey JJ (2012) A DRIFTS study of CO adsorption and hydrogenation on Cu-based core–shell nanoparticles. Catal Sci Technol 2:621–631
Zhang HB, Dong X, Lin GD, Liang XL, Li HY (2005) Carbon nanotube-promoted Co–Cu catalyst for highly efficient synthesis of higher alcohols from syngas. Chem Commun 40:5094–5096
Yu J, Mao D, Dan D, Guo X, Lu G (2016) New insights into the effects of Mn and Li on the mechanistic pathway for CO hydrogenation on Rh-Mn-Li/SiO2 catalysts. J Mol Catal A 423:151–159
Mo XH, Gao J, Umnajkaseam N, Goodwin JG (2009) La, V, and Fe promotion of Rh/SiO2 for CO hydrogenation: effect on adsorption and reaction. J Catal 267:167–176
Liu W, Wang S, Sun T, Wang S (2015) The promoting effect of Fe doping on Rh/CeO2 for the ethanol synthesis. Catal Lett 145:1741–1749
Li F, Zhang HT, Ying WY, Fang DY (2014) Ethanol synthesis from syngas on Mn-and Fe-promoted Rh/γ-Al2O3. C R Chimie 17:1109–1115
Hilmen AM, Xu MT, Gines MJL, Iglesia E (1998) Synthesis of higher alcohols on copper catalysts supported on alkali-promoted basic oxides. Appl Catal A 169:355–372
Luo HY, Zhang W, Zhou HW, Huang SY, Lin PZ, Ding YJ, Lin LW (2001) A study of Rh-Sm-V/SiO2 catalysts for the preparation of C2-oxygenates from syngas. Appl Catal A 214:161–166
Mahdavi V, Peyrovi M, Islami M, Mehr JY (2005) Synthesis of higher alcohols from syngas over Cu-Co2O3/ZnO, Al2O3 catalyst. Appl Catal A 281:259–265
Underwood RP, Bell AT (1988) Lanthana-promoted RhSiO2: I. Studies of CO and H2 adsorption and desorption. J Catal 109:61–75
Ichikawa M, Fukushima T (1985) Mechanism of syngas conversion into C2-oxygenates such as ethanol catalysed on a SiO2-supported Rh–Ti catalyst. J Chem Soc 6:321–323
Mo XH, Gao J, Goodwin JG Jr (2009) Role of promoters on Rh/SiO2 in CO hydrogenation: a comparison using DRIFTS. Catal Today 147:139–149
Chen G, Guo CY, Zhang X, Huang Z, Yuan G (2011) Direct conversion of syngas to ethanol over Rh/Mn-supported on modified SBA-15 molecular sieves: effect of supports. Fuel Process Technol 92:456–461
Chen GG, Zhang XH, Guo CY, Yuan GQ (2010) Manganese-promoted Rh supported on a modified SBA-15 molecular sieve for ethanol synthesis from syngas. Effect of manganese loading. C R Chimie 13:1384–1390
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31671797).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, F., Qian, W. DRIFTS study of Fe promoter effect on Rh/Al2O3 catalyst for C2 oxygenates synthesis from syngas. Appl Petrochem Res 9, 211–219 (2019). https://doi.org/10.1007/s13203-019-00238-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-019-00238-3