
Abstract
In this study, a series of fused-heterocyclic derivatives were systematically designed and synthesized using an efficient route, and evaluated in terms of GLP-1R agonist activity. We employed short synthetic steps and reactions that are tolerant of the presence of various functional groups and suitable for parallel operations to enable the rapid generation of libraries of diverse and structurally complex small molecules. Of the compounds synthesized, 3-(8-chloro-6-(trifluoromethyl)imidazo[1,2-a] pyridin-2-yl)phenyl methanesulfonate (8e) was the most potent agonist with an EC50 of 7.89 μM, and thus is the compound with the greatest potential for application. These findings represent a valuable starting point for the design and discovery of small-molecule GLP-1R agonists that can be administered orally.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus (DM2), a state of hormonal disruption and incretin deficiency, is increasingly becoming a worldwide epidemic (Kwak and Ha 2013). Current drugs utilized in the treatment of DM2 have well-established shortcomings: (1) increasing body weight and (2) increasing loss of β-cell function (Whitehouse 1997; Giugliano et al. 2009). However, the recent emergence of incretin-based therapies, which focus on glucagon-like peptide-1 (GLP-1), has attracted much interest.
GLP-1 is a peptide hormone of 30 amino acid residues. As a peptide, it has a very short half-life (<2 min) (Deacon et al. 1995). Such a short half-life has limited the utility of native GLP-1 in the treatment of DM2. The effort to identify GLP-1 analogues has resulted in the development of the drugs exenatide (Sennik et al. 2011; Buse et al. 2004) and liraglutide (Sjöholm 2010; Hribal and Sesti 2010). However, the requirement for injection limits the clinical utility of these peptide drugs. Therefore, orally active, small-molecule agonists of the GLP-1 receptor (GLP-1R) are highly sought after (Murphy and Bloom 2007).
Figure 1 shows synthetic small molecule agonists reported by several groups (Teng et al. 2000; Wang et al. 2009; Teng et al. 2007; Kopin 2004; Gong et al. 2010). Compound 6b, characterized by a novel imidazopyridine hit core, was identified from a library of 10,000 heterocyclic small molecules (Gong et al. 2010). As a small and drug-like active molecule, it represents an interesting starting point for the development of novel drugs. Therefore, we selected this compound as a model. In an effort to move away from the labile ester group of the phenol, we planned a synthetic pathway of new derivatives of imidazo[1,2-α]pyridine-based molecules (Fig. 2). To evaluate the structure–activity relationship, we designed and synthesized a series of heterocyclic derivatives containing a ring-junction nitrogen using a three-dimensional (3D) pharmacophore model reported previously (Gong et al. 2010) (Fig. 2). For the first stage, only combinations of five- and six-membered rings are considered, including imidazo[1,5-α]pyridine, imidazo[1,2-α]pyrimidine and imidazo[1,2-α]pyrazine. We employed short synthetic steps and reactions that are tolerant of the presence of various functional groups and suitable for parallel operations to enable the rapid generation of libraries of diverse, structurally complex, small molecules.

Known ago-allosteric modulators of GLP-1R

Structures of synthesized compounds. a Synthesized imidazo[1,2-a]pyridine-based molecules. b Other synthesized heterocycle-series compounds
Materials and methods
Chemistry
All the chemicals used in synthesis were supplied by Aldrichand TCI, and were used without further purification. All solvents were purified and stored in a dry condition. Reaction progress was determined by thin-layer chromatography (TLC) on Merck TLC Silica gel 60 F245 plates. Column chromatography was carried out using a silica gel 60 (63–200 mesh, Merck). NMR spectra were recorded on Agilent 400 instruments operating at 400 MHz for 1H and 100 MHz for 13C, and Agilent 500 instruments operating at 500 MHz for 1H and 125 MHz for 13C. Chemical shifts are expressed as parts per million (ppm) with tetramethylsilane as the internal standard. MS spectra were recorded on an Agilent G6530A Q-TOF.
General synthetic procedure for (6a–b)
To a stirred solution of bromomethylketone 3 (1.21 g, 4.7 mmol) and 2-amino-5-trifuoromethylpyridine 4 (0.61 g, 4.7 mmol) or 2-amino-3-chloro-5-trifluoromethylpyridine 5 (0.92 g, 4.7 mmol) in EtOH (50 mL) was added NaHCO3 (0.31 g, 4.7 mmol) at room temperature. The reaction mixture was heated to reflux and monitored by TLC (hexane/ethyl acetate: 2/1) until completion. After removing EtOH, the residue was extracted with ethyl acetate and water. The combined organic phases were washed with water, 1 N HCl, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/hexane = 10–20 %, Rf = 0.23).
3-(6-(Trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl acetate (6a)
Pale yellow solid; Yield: 64 %;1H NMR (500 MHz, CDCl3): δ 2.33 (s, 3H), 7.10 (d, J = 10.1 Hz, 1H), 7.32 (d, J = 11.9 Hz, 1H), 7.45 (t, J = 10.0 Hz, 1H), 7.70–7.73 (m, 2H), 7.80 (d, J = 9.7 Hz, 1H), 7.94(s, 1H), 8.49 (s, 1H);13C NMR (125 MHz, CDCl3); δ 21.1, 109.8, 117.0, 117.3, 118.0, 119.5, 121.1, 121.8, 123.6, 124.8, 129.9, 134.2, 145.2, 146.2, 151.2, 169.6; EI-HRMS calculated for (C16H11F3N2O2+H)+ 321.0851, found 321.0860.
3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl acetate (6b)
Pale yellow solid; Yield: 29 %;1H NMR (500 MHz, CDCl3): δ 2.34 (s, 3H), 7.10 (dd, J = 10.1, 2.1 Hz, 1H), 7.41–7.47 (m, 2H), 7.76 (t, J = 2.2 Hz, 1H), 7.81 (d, J = 10.2 Hz, 1H), 7.98 (s, 1H), 8.43 (s, 1H);13C NMR (125 MHz, CDCl3): δ 21.2,111.2, 119.62, 119.65, 119.68, 122.0, 123.3, 123.4, 123.7, 124.3, 129.8, 134.0, 142.7, 147.0, 151.1, 169.6; EI-HRMS calculated for (C16H10ClF3N2O2+H)+ 355.0461, found 355.0470.
General synthetic procedure for (8a and 8d)
To a mixture of 7a (99 mg, 0.36 mmol) or 7b (113 mg, 0.36 mmol) and K2CO3 (250 mg, 1.81 mmol) in acetone (10 mL) was added 1-chloroacetone (1 mL, 34.83 mmol) at room temperature. The reaction mixture was heated to reflux for 6 h. After removing acetone and 1-chloroacetone, the residue was extracted with ethyl acetate and water. The combined organic phases were washed with water and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/hexane = 10–20 %, Rf = 0.25).
General synthetic procedure for (8b and 8e)
To a solution of 7a (98 mg, 0.35 mmol) or 7b (121 mg, 0.45 mmol) in pyridine (5 mL) was added methanesulfonyl chloride (66 mg, 0.60 mmol) dropwise with stirring overnight in an ice bath. The reaction mixture was quenched with water in an ice bath and extracted with ethyl acetate (3 × 30 mL). The combined organic phases were washed with water, 1 N HCl, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 4/1, Rf = 0.25).
General synthetic procedure for (8c and 8f)
To a solution of 7a (90 mg, 0.32 mmol) or 7b (100 mg, 0.32 mmol) in pyridine (10 mL) was added toluenesulfonyl chloride (80 mg, 0.42 mmol) dropwise in an ice bath. After stirring for 2 h at room temperature, the reaction mixture was quenched with water in an ice bath and then extracted with ethyl acetate (3 × 30 mL). The combined organic phases were washed with water, 1 N HCl, and brine, dried, and filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 8/1, Rf = 0.23).
1-(3-(6-(Trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenoxy)propan-2-one (8a)
Pale yellow solid; Yield: 63 %;1H NMR (500 MHz, CDCl3): δ 2.32 (s, 3H), 4.64 (s, 2H), 6.91 (d, J = 10.1 Hz, 1H), 7.32–7.40 (m, 2H), 7.55–7.57 (m, 2H), 7.73 (d, J = 11.8 Hz, 1H), 7.94 (s, 1H), 8.50 (s, 1H);13C NMR (125 MHz, CDCl3): δ 26.7, 73.0, 109.6, 112.2, 114.8, 118.1, 119.6, 120.7, 122.4, 124.6, 126.7, 130.1, 134.6, 145.2, 147.1, 158.2, 205.5; EI-HRMS calculated for (C17H13F3N2O2+H)+ 335.1007, found 335.1019.
3-(6-(Trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl methanesulfonate (8b)
Pale yellow solid; Yield: 44 %;1H NMR (500 MHz, CDCl3): δ 3.20 (s, 3H), 7.29–7.36 (m, 2H), 7.50 (t, J = 9.9 Hz, 1H), 7.72 (d, J = 11.7 Hz, 1H), 7.89–7.92 (m, 2H), 7.98 (s, 1H), 8.51 (s, 1H);13C NMR (125 MHz, CDCl3): δ 37.6, 109.9, 118.3, 119.7, 121.09, 121.11, 122.0, 124.77, 124.81, 125.0, 130.5, 135.3, 145.3, 145.9, 149.8; EI-HRMS calculated for (C15H11F3N2O3S + Na)+ 379.0340, found 379.0360.
3-(6-(Trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl 4-methylbenzenesulfonate (8c)
Pale yellow solid; Yield: 64 %;1H NMR (500 MHz, CDCl3): δ 2.43 (s, 3H), 6.91 (d, J = 8.2 Hz, 1H), 7.30–7.35 (m, 4H), 7.62 (s, 1H), 7.67 (m, 1H), 7.75 (d, J = 7.4 Hz, 2H), 7.84–7.89 (m, 2H), 8.48(s, 1H);13C NMR (125 MHz, CDCl3): δ 21.7, 109.8, 118.2, 120.2, 120.9, 122.0, 124.8, 128.5, 129.8, 130.0, 132.2, 134.9, 145.3, 145.5, 146.1, 150.1; EI-HRMS calculated for (C21H15F3N2O3S + Na)+ 455.0653, found 455.0656.
1-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenoxy)propan-2-one (8d)
Pale yellow solid; Yield: 50 %;1H NMR (500 MHz, CDCl3): δ 2.32 (s, 3H), 4.64 (s, 2H), 6.89 (dd, J = 8.2, 2.7 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.40 (s, 1H), 7.54–7.57 (m, 2H), 7.97 (s, 1H), 8.43 (s, 1H);13C NMR (125 MHz, CDCl3): δ 26.8, 73.0, 111.3, 112.6, 115.0, 116.6, 116.9, 119.6, 119.7, 123.3, 124.3, 130.1, 134.1, 142.7, 147.6, 158.1, 205.5; EI-HRMS calculated for (C17H12ClF3N2O2+H)+ 369.0618, found 369.0669.
3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl methanesulfonate (8e)
Pale yellow solid;Yield: 45 %;1H NMR (500 MHz, CDCl3): δ 3.20 (s, 3H), 7.31 (d, J = 10.2 Hz, 1H), 7.43 (s, 1H), 7.50 (t, J = 10.0 Hz, 1H), 7.91 (m, 1H), 7.94 (d, J = 9.8 Hz, 1H), 8.03 (s, 1H), 8.45(s, 1H);13C NMR (125 MHz, CDCl3): δ 37.7, 111.5, 116.9, 117.2, 120.0, 122.2, 123.38, 123.43, 124.5, 125.3, 130.4, 134.8, 142.8, 146.4, 149.7; EI-HRMS calculated for (C15H10ClF3N2O3S+H)+ 391.0131, found 391.0135.
3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl 4-methylbenzene sulfonate (8f)
Pale yellow solid; Yield: 38 %;1H NMR (500 MHz, CDCl3): δ 2.43 (s, 3H), 6.91 (d, J = 8.2 Hz, 1H), 7.27–7.33 (m, 3H), 7.39 (m, 1H), 7.61 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.87 (d, J = 7.8 Hz, 1H), 7.94 (s, 1H), 8.48(s, 1H);13C NMR (125 MHz, CDCl3): δ 21.7, 111.5, 119.8, 120.3, 122.2, 123.5, 124.3, 125.1, 126.9, 128.5, 130.0, 132.3, 134.4, 138.6, 145.6, 145.8, 146.5, 149.8, 150.0. EI-HRMS calculated for (C21H14ClF3N2O3S + Na)+ 489.0263, found 489.0274.
General synthetic procedure for (12a–c)
Pyridium bromide perbromide (1.79 g, 5.60 mmol) was added to a solution of 10a–c (0.9 g, 5.08 mmol) in AcOH (100 mL) with stirring for 3 h at room temperature. The reaction mixture was poured into ice-cold water and then extracted with ethyl acetate (3 × 50 mL). The combined organic phases were washed with saturated aqueous NaHCO3, water, and brine, dried, and filtered and concentrated in vacuo to give crude 11a–c as a yellow oil (1.29 g, 98 %). The resulting crude 11a–c could be used without further purification. To a stirred solution of bromomethylketone 11a–c (1.30 g, 5.1 mmol) and aminopyridine 4 (0.82 g, 5.1 mmol) in EtOH (80 mL) was added NaHCO3 (0.43 g, 5.1 mmol) at room temperature. The reaction mixture was heated to reflux for 8 h. After removing EtOH, the residue was extracted with ethyl acetate and water. The combined organic phases were washed with water, 1 N HCl, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (12a–b, hexane/ethyl acetate = 1/1, Rf = 0.24; 12c, hexane/ethyl acetate = 4/1, Rf = 0.22).
N-(3-(6-(Trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl)acetamide(12a)
Pale yellow solid;Yield: 39 %;1H NMR (500 MHz, CDCl3): δ 2.19 (s, 3H), 7.31 (d, J = 11.6 Hz, 1H), 7.39 (t, J = 9.8 Hz, 1H), 7.51 (s, NH), 7.60 (d, J = 9.8 Hz, 1H), 7.68 (d, J = 11.1 Hz, 2H), 7.93 (s, 1H), 8.07 (s, 1H), 8.47(s, 1H);13C NMR (125 MHz, CDCl3): δ 24.7, 109.6, 117.4, 118.1, 119.9, 120.7, 122.0, 124.6, 124.7, 129.6, 133.7, 138.5, 145.2, 147.1, 168.5; EI-HRMS calculated for (C16H12F3N3O + Na)+ 342.0830, found 342.0835.
N-(3-(6-(Trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl)methanesulfonamide (12b)
Pale yellow solid;Yield: 20 %;1H NMR (500 MHz, CDCl3): δ 3.05 (s, 3H), 7.30 (dt, J = 7.0, 1.3 Hz, 1H), 7.34 (dd, J = 9.5, 1.8 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.69–7.74 (m, 3H), 7.82 (t, J = 1.9 Hz, 1H), 7.99 (s, 1H), 8.51(s, 1H);13C NMR (125 MHz, CDCl3): δ 39.4, 110.0, 118.0, 118.5, 120.7, 121.3, 123.0, 124.8, 124.9, 130.2, 130.3, 134.2, 137.6, 145.2, 146.2; EI-HRMS calculated for (C15H12F3N3O2S+H)+ 356.0681, found 356.0705.
4-Methyl-N-(3-(6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl)benzene sulfonamide (12c)
Pale yellow solid;Yield: 21 %;1H NMR (500 MHz, CDCl3): δ 2.30 (s, 3H), 7.14–7.19 (m, 3H), 7.26–7.29 (m, 2H), 7.63 (dd, J = 7.7, 0.9 Hz, 1H), 7.66–7.70 (m, 4H), 7.85 (brs, NH), 7.89 (s, 1H), 8.45(s, 1H);13C NMR (125 MHz, CDCl3): δ 21.5, 109.9, 118.0, 118.8, 120.7, 121.0, 122.7, 124.7, 127.3, 129.7, 129.9, 134.0, 136.0, 137.5, 143.9, 145.2, 146.5; EI-HRMS calculated for (C21H16ClF3N3O2S+H)+ 432.0994, found 432.1020.
3-(8-Chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenyl acetate (19)
To a solution of 18 (300 mg, 0.80 mmol) in benzene (10 mL) was added POCl3 (1.2 mL, 13.04 mmol) dropwise at room temperature. The reaction mixture was heated to reflux for 6 h. After cooling to room temperature, the mixture was poured into iced-water and then extracted with ethyl acetate (3 × 50 mL). The combined organic phases were washed with water and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 12/1, Rf = 0.25). White solid; Yield: 90 %;1H NMR (400 MHz, CDCl3): δ 2.35 (s, 3H), 6.92 (s, 1H), 7.25 (d, J = 8.6 Hz, 1H), 7.54–7.61 (m, 3H), 7.77 (s, 1H), 8.51(s, 1H); 13C NMR (100 MHz, CDCl3): δ 21.1, 113.9, 117.6, 117.9, 119.6, 121.5, 121.8, 122.5, 122.9, 124.9, 125.3, 126.5, 126.8, 129.7, 130.1, 130.5, 151.3, 169.2; EI-HRMS calculated for (C16H10ClF3N2O2+H)+ 355.0461, found 355.0474.
3-(8-Chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenyl cyclohexane carboxylate (21a)
To a solution of 19 (257 mg, 0.72 mmol) in THF (20 mL) was added a solution of NaOH (50 mg, 1.25 mmol) in water (10 mL) with stirring for 3 h at room temperature. After removing THF, the resulting mixture was extracted with ethyl acetate. The combined organic phases were washed with water and brine, dried, and filtered and concentrated in vacuo. The resulting crude 20 could be used without further purification. Cyclohexanecarboxylic chloride (28 mg, 0.19 mmol) was added to a solution of 20 (50 mg, 0.16 mmol), TEA (19 mg, 0.19 mmol), and DMAP (4 mg, 0.03 mmol) in anhydrous CH2Cl2 (20 mL) slowly in an ice bath. After stirring for 3 h at room temperature, the reaction mixture was poured into ice water and then extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with 1 N HCl, water, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 20/1, Rf = 0.23) to afford 21a as a pale yellow solid (64 mg, 94 %). 1H NMR (400 MHz, CDCl3): δ 1.25–1.39 (m, 4H), 1.57–1.65 (m, 2H), 1.81–1.84 (m, 2H), 2.06–2.09 (m, 2H), 2.58 (t, J = 10.1 Hz, 1H), 6.92 (s, 1H), 7.24 (m, 1H), 7.52–7.59 (m, 3H), 7.78 (s, 1H), 8.52(s, 1H);13C NMR (100 MHz, CDCl3): δ 25.3, 25.6, 28.8, 43.1, 113.9, 117.7, 119.6, 121.4; EI-HRMS calculated for (C21H18ClF3N2O2 + Na)+ 445.0907, found 445.0907.
1-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenoxy)propan-2-one (21b)
Using the same method as for the preparation of 8a, starting with 20 (73 mg, 0.23 mmol), 1-chloroacetone (0.5 mL, 17.41 mmol) and K2CO3 (161 mg, 1.17 mmol), 21b was generated as a pale yellow solid (30 mg, 35 %). 1H NMR (400 MHz, CDCl3): δ 2.30 (s, 3H), 4.66 (s, 2H), 6.91 (s, 1H), 7.06 (d, J = 7.9 Hz, 1H), 7.28 (s, 1H), 7.35 (d, J = 7.5 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.76 (s, 1H), 8.48 (s, 1H);13C NMR (100 MHz, CDCl3): δ 26.6, 73.0, 113.9, 114.6, 116.5, 117.5, 117.9, 119.6, 120.9, 121.4, 122.1, 124.1, 126.5, 129.6, 130.2, 130.6, 141.3, 158.5, 204.6; EI-HRMS calculated for (C17H12ClF3N3O2+H)+ 369.0618, found 369.0670.
General synthetic procedure for (27a–d)
POCl3 (0.3 mL, 3.40 mmol) was added to a mixture of 24 or 26a–c (0.17 mmol) and pyridine (0.93 mL, 11.60 mmol) in anhydrous dichloroethane (14 mL) at room temperature. The reaction mixture was heated to reflux for 7 h. After cooling to room temperature, the reaction mixture was concentrated, filtered, and extracted with ethyl acetate. The combined organic phases were washed with 1 N HCl, water, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 2/1, Rf = 0.24).
N-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenyl)acetamide (27b)
Pale yellow solid; Yield: 96 %;1H NMR (400 MHz, CDCl3): δ 2.17 (s, 3H), 6.90 (s, 1H), 7.44–7.48 (m, 2H), 7.63 (d, J = 6.7 Hz, 1H), 7.74 (s, 1H), 7.96 (s, 1H), 8.16 (s, 1H), 8.53(s, 1H);13C NMR (100 MHz, CDCl3): δ 24.5, 113.9, 117.5, 117.9, 119.7, 119.8, 121.1, 121.5, 122.1, 123.6, 124.2, 126.4, 129.2, 129.6, 130.0, 139.1, 141.5, 168.9; EI-HRMS calculated for (C16H11ClF3N3O + Na)+ 376.0440, found 376.0447.
N-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenyl)cyclohexane carboxamide (27c)
Pale yellow solid; Yield: 32 %;1H NMR (400 MHz, CDCl3): δ 1.28–1.35 (m, 2H), 1.50–1.59 (m, 2H), 1.71 (m, 2H), 1.83–1.85 (m, 2H), 1.94–1.97 (m, 2H), 2.26 (t, J = 11.6 Hz, 1H), 6.90 (s, 1H), 7.44–7.52 (m, 2H), 7.55 (s, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.75 (s, 1H), 7.95 (s, 1H), 8.54 (s, 1H);13C NMR (100 MHz, CDCl3): δ 25.6, 29.6, 46.5, 113.8, 117.4, 117.8, 119.7, 119.8, 121.0, 122.2, 123.3, 126.3, 129.4, 129.6, 130.0, 139.1, 141.5, 174.6; EI-HRMS calculated for (C21H19ClF3N3O + Na)+ 444.1066, found 444.1075.
1-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenyl)pyrrolidine-2,5-dione (27d)
Pale yellow solid; Yield: 28 %;1H NMR (400 MHz, CDCl3): δ 2.95 (s, 4H), 6.93 (s, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.76–7.82 (m, 3H), 8.63 (s, 1H);13C NMR (100 MHz, CDCl3): δ 28.4, 114.0, 117.7, 118.0, 119.7, 121.5, 122.6, 124.2, 125.7, 126.4, 127.1, 128.2, 129.8, 130.3, 132.7, 140.7, 175.8; EI-HRMS calculated for (C18H11ClF3N3O2+H)+ 394.0570, found 394.0608.
tert-Butyl (3-(8-chloro-6-(trifluoromethyl)imidazo[1,5-a]pyridin-3-yl)phenyl) carbamate (27a)
Pale yellow solid;Yield: 13 %;1H NMR (400 MHz, CDCl3): δ 1.53 (s, 9H), 6.71 (s, 1H), 6.90 (s, 1H), 7.41–7.48 (m, 3H), 7.76 (s, 1H), 7.88 (s, 1H), 8.58 (s, 1H);13C NMR (100 MHz, CDCl3): δ 28.2, 113.7, 117.3, 117.6, 118.3, 119.7, 119.9, 122.3, 122.5, 124.2, 126.3, 129.5, 130.0, 139.4, 141.6, 152.6; EI-HRMS calculated for (C19H17ClF3N3O2 + Na)+ 434.0859, found 434.0865.
N-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl)acetamide (33a)
Acetic anhydride (20 mg, 0.20 mmol) was added to a mixture of 32 (50 mg, 0.16 mmol) and DMAP (3 mg, 0.02 mmol) in anhydrous CH2Cl2 (10 mL) with stirring for 1 h at room temperature. After removing the solvent, the residue was extracted with ethyl acetate and water. The combined organic phases were washed with 1 N HCl, saturated Na2CO3, water, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/hexane = 33–50 %, Rf = 0.23). Pale yellow solid; Yield: 50 %;1H NMR (400 MHz, CDCl3): δ 2.19 (s, 3H), 7.35–7.39 (m, 2H), 7.62–7.66 (m, 2H), 7.72 (s, 1H), 7.94 (s, 1H), 8.06 (s, 1H), 8.39 (s, 1H);13C NMR (100 MHz, CDCl3): δ 24.6, 111.3, 116.6, 117.6, 119.6, 120.3, 121.5, 122.2, 123.4, 124.2, 129.5, 133.1, 138.5, 142.6, 147.5, 168.8; EI-HRMS calculated for (C16H11ClF3N3O + Na)+ 376.0440, found 376.0453.
General synthetic procedure for (33b–c)
Cyclohexanecarboxylic chloride (28 mg, 0.19 mmol) or toluenesulfonyl chloride (39 mg, 0.21 mmol) was added to a solution of 32 (50 mg, 0.16 mmol), TEA (19 mg, 0.19 mmol), and DMAP (4 mg, 0.03 mmol) in anhydrous CH2Cl2 (10 mL) slowly in an ice bath. After stirring for 3 h at room temperature, the reaction mixture was poured into ice water and then extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with 1 N HCl, water, and brine, dried, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 9/1, Rf = 0.22).
N-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-yl)phenyl)cyclohexane carboxamide (33b)
Pale yellow solid; Yield: 54 %; 1H NMR (400 MHz, CDCl3): δ 1.28–1.33 (m, 2H), 1.51–1.60 (m, 2H), 1.72 (m, 2H), 1.84–1.86 (m, 2H), 1.96–1.99 (m, 2H), 2.25 (m, 1H), 7.35–7.39 (m, 2H), 7.48 (s, 1H), 7.66 (d, J = 7.0 Hz, 2H),7.98 (s, 1H), 8.13 (s, 1H), 8.40 (s, 1H);13C NMR (100 MHz, CDCl3): δ 25.6, 25.7, 29.7, 46.6, 111.3, 116.6, 116.9, 117.5, 119.6, 120.2, 122.0, 123.3, 124.2, 129.5, 133.0, 138.7, 142.7, 147.6, 174.7; EI-HRMS calculated for (C21H19ClF3N3O+H)+ 422.12470, found 422.12446.
N-(3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridin-2-y l)phenyl)-4-toluenesulfonamide (33c)
Pale yellow solid;Yield: 68 %;1H NMR (400 MHz, CDCl3): δ 2.46 (s, 3H), 7.00 (d, J = 7.5 Hz, 1H), 7.33–7.35 (m, 3H), 7.40 (s, 1H), 7.58 (s, 1H), 7.83–7.87 (m, 4H), 8.10 (d, J = 7.4 Hz, 1H), 8.40 (s, 1H);13C NMR (100 MHz, CDCl3): δ 21.7, 111.3, 116.6, 117.0, 119.7, 123.3, 124.4, 128.2, 128.5, 128.6, 129.4, 129.6, 129.7, 129.8, 131.5, 133.9, 134.9, 136.5, 142.7, 145.1, 146.6; EI-HRMS calculated for (C21H15ClF3N3O2S+H)+ 466.06038, found 466.07034.
General synthetic procedure for (37a–c)
A mixture of 5-bromopyrimidin-2-amine 35–36 (222 mg, 1.27 mmol) and bromoacetone 3 (257 mg, 1.0 mmol) in dioxane (10 mL) with or without NaHCO3 (84 mg, 1.00 mmol) was stirred until reflux for 7 h. After cooling to room temperature, ethyl acetate was added, washed with water and brine, dried over calcium oxide, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (gradient eluent: ethyl acetate/hexane = 20–50 %, Rf = 0.21).
3-(6-Bromoimidazo[1,2-a]pyrimidin-2-yl)phenyl acetate (37a)
White solid;Yield: 11 %; 1H NMR (400 MHz, CDCl3): δ 2.33 (s, 3H), 7.10 (d, J = 7.9 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.75 (s, 1H), 7.77 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 8.51 (s, 1H), 8.55 (s, 1H);13C NMR (100 MHz, CDCl3): δ 21.2, 104.8, 106.6, 119.6, 122.1, 123.7, 129.8, 132.5, 134.2, 150.7, 151.2, 158.6, 169.5; EI-HRMS calculated for (C14H10BrN3O2+H)+ 332.00346, found 332.00372.
3-(Imidazo[1,2-a]pyrimidin-2-yl)phenyl acetate (37b)
Pale yellow solid; Yield: 21 %;1H NMR (400 MHz, CDCl3): δ 2.32 (s, 3H), 6.82 (t, J = 5.6 Hz, 1H), 7.07 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.76 (m, 2H), 7.83 (d, J = 7.7 Hz, 1H), 8.39 (d, J = 6.3 Hz, 1H), 8.49 (s, 1H);13C NMR (100 MHz, CDCl3): δ 21.2, 106.5, 108.9, 119.5, 121.7, 123.6, 129.7, 133.1, 134.7, 146.2, 148.6, 150.1, 151.2, 169.5; EI-HRMS calculated for (C14H11 N3O2+H)+ 254.09295, found 254.09322.
3-(Imidazo[1,2-a]pyrazin-2-yl)phenyl acetate (37c)
Pale yellow solid;Yield: 11 %;1H NMR (400 MHz, CDCl3): δ 2.34 (s, 3H), 7.12 (d, J = 7.6 Hz, 1H), 7.47 (t, J = 7.5 Hz, 1H), 7.74 (s, 1H), 7.83 (d, J = 7.3 Hz, 1H), 7.89 (s, 1H), 7.95 (s, 1H), 8.07 (s, 1H), 9.10 (s, 1H);13C NMR (100 MHz, CDCl3): δ 21.1, 109.4, 118.6, 119.6, 122.0, 123.7, 129.8, 129.9, 134.4, 140.9, 143.8, 146.8, 151.2, 169.4; EI-HRMS calculated for (C14H11 N3O2+H)+ 254.09295, found 254.09288.
2-(3-Nitrophenyl)imidazo[1,2-a]pyrimidine (37d)
A mixture of 2-aminopyrazine 34 (195 mg, 2.05 mmol) and bromoacetone 29 (660 mg, 2.70 mmol) in ethanol (20 mL) was stirred until reflux for 3 h, After cooling to room temperature, the mixture was concentrated. The residue was dissolved in ethyl acetate, washed with 1 N HCl, water, and brine, dried over calcium oxide, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (methanol/methylene chloride = 0–1 %, Rf = 0.21) to afford 37d as a yellow solid (24 mg, 5 %). 1H NMR (400 MHz, CDCl3): δ 6.95 (t, J = 1.7 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.97 (s, 1H), 8.21 (d, J = 7.9 Hz, 1H), 8.45 (d, J = 7.6 Hz, 1H), 8.50 (d, J = 5.9 Hz, 1H), 8.61 (d, J = 1.2 Hz, 1H), 8.80 (s, 1H);13C NMR (100 MHz, CDCl3): δ 107.1, 108.8, 109.4, 113.6, 113.9, 120.9, 123.1, 129.9, 132.2, 133.3, 134.9, 150.8; EI-HRMS calculated for (C12H8N4O2+H)+ 241.07255, found 241.07288.
2-(3-Nitrophenyl)imidazo[1,2-a]pyrazine (37e)
A mixture of 2-aminopyrazine 36 (95 mg, 1.0 mmol) and bromoacetone 29 (488 mg, 2.0 mmol) in ethanol (10 mL) was stirred until reflux for 3 h. After cooling to room temperature, the mixture was concentrated. Then the residue was dissolved in ethyl acetate and washed with water. The combined aqueous phase was extracted with ethyl acetate. The combined organic phase was washed with brine, dried over calcium oxide, and filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (methylene chloride/methanol = 100/1, Rf = 0.21) to afford 37e as a yellow solid (40 mg, 17 %). 1H NMR (400 MHz, CDCl3): δ 7.68 (t, J = 7.6 Hz, 1H), 7.97 (s, 1H), 8.11 (s, 1H), 8.13 (s, 1H), 8.24 (d, J = 7.3 Hz, 1H), 8.37 (d, J = 6.7 Hz, 1H), 8.80 (s, 1H), 9.18 (s, 1H);13C NMR (100 MHz,CDCl3): δ 102.6, 110.0, 114.3, 117.1, 118.9, 121.2, 123.4, 129.7, 130.1, 132.2, 134.6, 144.1; EI-HRMS calculated for (C12H8N4O2+H)+ 241.07255, found 241.07260.
Biology
In vitro GLP-1R activation assay (Chen et al. 2007)
CHO-K1 cells (4 × 106/100 mm dish) were transiently transfected with the pCMV6-GLP-1R (Origene #SC124060) and pCRE-Luc (Promega #631911) plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). After 24 h incubation at 37 °C, cells were seeded into 96-well culture plates (2 × 104/well), and further incubated at 37 °C overnight. At the time of assay, GLP-1 (7–37) (Sigma, St. Louis, MO) or test compounds in DMSO were added to the plate. After 8 h incubation, cells were lysed and luciferase activity quantified using the Steady-Glo luciferase assay system (Promega #E2550).
Data were analyzed in Excel and EC50 values were determined graphically from dose–response curves in OriginPro.
Results and discussion
Chemistry
The general synthetic pathway yielding the novel derivatives 6a, 6b, and 8a–8f is outlined in Scheme 1. 3-Hydroxy acetophenone 1 was converted into 3-acetoxy acetophenone 2 by acetylation. Treatment of substituted acetophenone 2 with bromine in the presence of AlCl3 in Et2O (Bunders et al. 2010) afforded á-bromomethylketone 3. Subsequently, 3 and the substituted 2-aminopyridines 4, 5 were allowed to react in the presence of sodium bicarbonate in refluxing ethanol (Fookes et al. 2008), resulting in the generation of compounds 6a and 6b. The deacetylation of compounds 6a and 6b with sodium hydroxide afforded compounds 7a and 7b, respectively. The akylation or acylation of 7a and 7b furnished the target compounds 8a–8f.

Reagents and conditions: (a) acetic anhydride, DMAP, anhydrous CH2Cl2, RT; (b) Br2, AlCl3, Et2O, ice bath; (c) NaHCO3, EtOH, reflux; (d) NaOH, THF/H2O, RT; (e) R1Cl, K2CO3, acetone, reflux or R2Cl, pyridine, 0 °C or RT
The derivatives 12a–12c were readily prepared in three steps, as illustrated in Scheme 2. 3-Aminoacetophenone 9 was first acylated with acyl chloride or sulfonyl chloride, as appropriate, to produce compounds 10a–10c. Subsequent bromination of 10a–10c with PBB (pyridinium bromide perbromide) in acetic acid (Yu et al. 2008) afforded compounds 11a–11c, which were then cyclized with 5-trifluoro-2-aminopyridine in refluxing ethanol to give the desired derivatives 12a–12c.

Reagents and conditions: (a) acetic anhydride, DMAP, anhydrous CH2Cl2, RT or R3Cl, pyridine, 0 °C or RT; (b) pyridium bromide perbromide, AcOH, RT; (c) NaHCO3, EtOH, reflux
Next we synthesized the imidazo[1,5-α]pyridine derivatives 19, 21a–21b, and 27a–27d, as detailed in Scheme 3. The intermediate 2-aminomethylpyridine 15, prepared from 2,3-dichloro-5-triflouropyridine 14 in two steps using a method reported elsewhere (Stolting 2004), was treated with 3-acetoxybenzoinc acid 17, followed by cyclization in the presence of POCl3 in refluxing benzene (Bower and Ramage 1955) to give intermediate 19. Then, deacylation of 19 and subsequent acylation or alkylation of the resulting compound 20 with appropriate acyl chloride or 1-chloroacetone resulted in the generation of the desired compounds 21a–21b. For the synthesis of derivatives 27a–27d, the first step is the amidation of intermediate 15 with benzoic acid 23 in the presence of DCC and DMAP in dichloromethane, resulting in the generation of compound 24. Then, deprotection of 24 with trifluoroacetic acid (Mu 2001) followed by acylation of the resulting compound 25 afforded the compounds 26a–26c. Finally, the amides 24 and 26a–26c were cyclized in the presence of POCl3 and pyridine in refluxing dichloroethane (Cookson et al. 1986), resulting in the generation of the target derivatives 27a–27d, respectively.

Reagents and conditions: (a) CH3NO2, KOH, DMSO, RT; (b) SnCl2, conc. HCl, EtOH, reflux; (c) acetic anhydride, reflux; (d) DCC, DMAP, anhydrous CH2Cl2, RT; (e) POCl3, benzene, reflux; (f) NaOH, THF/H2O, RT; (g) R4Cl, TEA, DMAP, anhydrous CH2Cl2, RT, or R5Cl, K2CO3, acetone, reflux. (h) di-tert-butyl dicarbonate, TEA, dioxane/H2O; (i) DCC, DMAP, anhydrous CH2Cl2, RT; (j) CF3COOH, anhydrous CH2Cl2, RT; (k) acetic anhydride, DMAP, anhydrous CH2Cl2, RT or R6Cl, TEA, DMAP, anhydrous CH2Cl2, RT or succimyl chloride, K2CO3, CH3CN, reflux; (l) POCl3, pyridine, dichloroethane, reflux
Scheme 4 describes the synthesis of derivatives 33a–33c. The formation of á-diazoketone intermediate 30 was achieved from á-bromomethylketone 28 by treatment with N,N′-ditosylhydrazine and DBU (Toma et al. 2007). Subsequent coupling of á-diazoketone with 3-chloro-5-trifluoro-2-aminopyridine in the presence of 10 mol % Cu(OTf)2 in dichloroethane (DCE) (Yadav et al. 2007) afforded substituted 2-arylimidazo[1,2-α]pyridine 31. Reduction of 31 with stannous chloride in a refluxing mixture of ethanol and concentrated hydrochloride (Denora et al. 2008) resulted in the generation of compound 32. Finally, the acylation of 32 with the corresponding acyl chloride afforded derivatives 33a–33c.

Reagents and conditions: (a) pyridium bromide perbromide, AcOH, reflux; (b) TsNHNHTs, DBU, THF, RT; (c) 10 mol %Cu(OTf)2, dichloroethane, 80 °C; (d) SnCl2, conc. HCl, EtOH, reflux; (e) acetic anhydride, DMAP, anhydrous CH2Cl2, RT or R8Cl, TEA, DMAP, anhydrous CH2Cl2, RT
The syntheses of compounds 37a–37e are detailed in Scheme 5. The preparation of intermediate 35 was achieved by bromination of 2-aminopyrimidine 34 with NBS in refluxing acetonitrile. The subsequent cyclization of 35, 2-anmonoprimidine 34, and 2-aminopyrazine 36 with intermediate 3 yielded the target derivatives 37a–37c, respectively. The cyclization of 2-anmonoprimidine 34 and 2-aminopyrazine 36 with intermediate 29 yielded the target derivatives 37d–37e, respectively.

Reagents and conditions: (a) NBS, CH3CN, reflux; (b) dioxane, reflux or NaHCO3, dioxane, reflux; (c) EtOH, reflux
Biology
The compounds prepared in this study were evaluated in terms of GLP-1R agonist activity using an in vitro activation efficacy assay in CHO-K1 cells (Chen et al. 2007), and the magnitude of the responses have been compared at two concentrations of compounds used. GLP-1 (7–37) was used as the positive control and DMSO (0.1 %) was used as the negative control. Induction values represent luciferase activities driven by CRE (cAMP response element). Compounds were grouped into three series according to fused-heterocyclic ring type.
In general, the first series of compounds, 6a–6b, 8a–8f, 12a–12c, and 33a–33c (Fig. 3), based on the imidazo[1,2-α]pyridine structure and containing various substituted groups, generated higher responses than those of the second series. Compound 6b is the model compound, in which replacement of the acetyl group with propanyl-2-one 8d, mesyl 8e, or tosyl 8f resulted in a significant increase in magnitude of the response, suggesting that the hydrogen-bond donor is preferred to be this region and that the length of linker affects binding to the ago-allosteric binding site of GLP-1R. To determine whether the chlorine in imidazo[1,2-α]pyridine is essential for its activity, compounds 6a and 8a–8c were synthesized. Compounds 8a and 8b showed good responses similar to that of compounds 8d and 8e at 10 μM. However, a loss of response was observed for compounds 6a and 8c. The bioisosteric replacement of the ester group (6a, 8b–8c) with an amide group (12a–12c) resulted in comparable activities; in particular, compound 12a exhibited a two fold enhanced response. On the contrary, compounds 33a and 33c exhibited decreased responses. However, compound 33b, which contains acyclohexanecarboxamide group, exhibited a moderate response. Further increases in concentration resulted in a significant drop in the responses of compounds 8a, 6a–6b, 8f, and 12a–12b to an about <1-fold increase, likely due to cytotoxicity at a high concentration (100 μM). Surprisingly, compound 8e, which generated the highest response at 10 μM, also exhibited the greatest response at a high concentration (100 μM).

In vitro responses of compounds 6a–6b, 8a–8f, 12a–12c and 33a–33c on CHO-K1 cells at 10 and 100 μM. The cells were transfected with the pCMV6-GLP-1R and pCRE-Luc plasmids. The transfected and cultured cells were incubated with different compounds for 8 h, and luciferase activity quantified using the Steady-Glo luciferase assay system. Vertical and horizontal axes show the fold increases compared to the control and the synthesized compounds, respectively. Values shown are mean ± SD of three independent experiments. Significant difference from 0.1 % DMSO treated group: *p ≤ 0.05, **p ≤ 0.01 and ***p < 0.001
In the second series of compounds, 19, 21a–21b, and 27a–27d (Fig. 4), the imidazo[1,2-α]pyridine structure was changed to an imidazo[1,5-α]pyridine structure. Unfortunately, all derivatives exhibited low responses at a low concentration (10 μM). Compounds 21a–21b and 27b–27d showed moderate responses at 100 μM. Generally, both first- and second-series compounds with substituted ester groups showed higher responses than those with a substituted amide group.

In vitro responses of compounds 19, 21a–21b, 27a–27d, and 37a–37e on CHO-K1 cells at 10 and 100 μM. Experimental details are described in Fig. 3. Vertical and horizontal axes show the fold increases compared to the control and the synthesized compounds, respectively. Values shown are mean ± SD of three independent experiments. Significant difference from 0.1 % DMSO treated group: *p ≤ 0.05, **p ≤ 0.01, and ***p < 0.001
Finally, a nitrogen atom was introduced into the six-membered fused-heterocyclic ring to evaluate the effect of electron density on activity (Fig. 4). The majority of the compounds 37a–37e thus generated showed loss of responses compared to the first series. We speculated that the loss of responses might be attributable to a decreased interaction between the π-electron and the receptor.
Over the half of the synthesized compounds, the effects did not appear concentration-dependent. However the effects of the compounds on coupling the GLP-1R to the signaling way may well be concentration-dependent, but the responses measured did not appear concentration-dependent due to cytotoxicity.
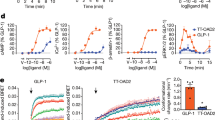
In addition, selected compounds 8a, 8b and 8e which showed >2.5-fold increases at 10 μM were assayed further to determine concentration–response curves (Fig. 5) and calculate EC50 values (Table 1). Compound 8e, bearing chlorine substitution imidazo[1,2-α]pyridine ring and mesyl group of benzene ring, was found to be a potent GLP-1R agonist exhibiting an EC50 of 7.89 μM. Compounds 8a and 8b, without chlorine substitution of imidazo[1,2-α]pyridine ring, were about threefold less potent, with EC50 values of 20 μM and 17 μM, respectively (Table 1). Concentration–response curves of selected compounds are shown in Fig. 5. The concentration are in a range from 1 μM to 100 μM. Compounds 8b and 8e showed above 50 % response (8b, 52 %; 8e, 58 %) at their EC50 values, while compound 8a showed lower response 43 % at EC50 value (Fig. 5). Thus, compound 8e may serve as a GLP-1R agonist with potential for application.


In conclusion, these new compounds, synthetic methodology developed and preliminary biological evaluation results could be helpful in further design and discovery of more potent GLP-1R agonists for the treatment of DM2.
References
Bower, J. D., and Ramage, G. R. 1955. Heterocyclic systems related to pyrrocoline. Part I. 2: 3a-Diazaindene. Journal of Chemical Society, 2834:2836.
Bunders, C.A., J.J. Richards, and C. Melander. 2010. Identification of aryl 2-aminoimidazoles as biofilm inhibitors in Gram-negative bacteria. Bioorganic and Medicinal Chemistry Letters 20: 3797–3800.
Buse, J.B., R.R. Henry, J. Han, D.D. Kim, M.S. Fineman, and A.D. Baron. 2004. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 27: 2628–2635.
Chen, D., J. Liao, N. Li, C. Zhou, Q. Liu, G. Wang, R. Zhang, S. Zhang, L. Lin, K. Chen, X. Xie, F. Nan, A.A. Young, and M.W. Wang. 2007. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proceedings of the National Academy of Sciences of the United States of America 104: 943–948.
Cookson, R.C., P.J. Dudfield, and D.I.C. Scopes. 1986. Synthesis of carbocyclic C-nucleoside analogues from 8,9,10-trinorborn-5-en-2-ol. Journal of the Chemical Society, Perkin Transactions 1: 393–398.
Deacon, C.F., M.A. Nauck, M. Toft-Nielsen, L. Pridal, B. Willms, and J.J. Holst. 1995. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 44: 1126–1131.
Denora, N., V. Laquintana, M.G. Pisu, R. Dore, L. Murru, A. Latrofa, G. Trapani, and E. Sanna. 2008. 2-Phenyl-imidazo[1,2-a]pyridine compounds containing hydrophilic groups as potent and selective ligands for peripheral benzodiazepine receptors: synthesis, binding affinity and electrophysiological studies. Journal of Medicinal Chemistry 51: 6876–6888.
Fookes, C.J.R., T.Q. Pham, F. Mattner, I. Greguric, C. Loc’h, X. Liu, P. Berghofer, R. Shepherd, M.C. Gregoire, and A. Katsifis. 2008. Synthesis and biological evaluation of substituted[18F]imidazo[1,2-a]pyridines and [18F]pyrazolo[1,5-a]pyrimidines for the study of the peripheral benzodiazepine receptor using positron emission tomography. Journal of Medicinal Chemistry 51: 3700–3712.
Giugliano, D., E. Standl, T. Vilsbøll, J. Betteridge, R. Bonadonna, I.W. Campbell, G.-H. Schernthaner, B. Staels, A. Trichopoulou, and E. Farinaro. 2009. Is the current therapeutic armamentarium in diabetes enough to control the epidemic and its consequences? What are the current shortcomings? Acta Diabetologica 46: 173–181.
Gong, Y.-D., H.-G. Cheon, T.-H. Lee, and N.-S. Kang. 2010. A novel 3-(8-Chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridine-2-yl)phenyl acetate skeleton and pharmacophore model as glucagon-like peptide 1 receptor agonists. Bulletin of the Korean Chemical Society 31: 3760–3764.
Hribal, M.L., and G. Sesti. 2010. Liraglutide, the once-daily human GLP-1 analog, in the treatment of Type 2 diabetes. Expert Review of Endocrinology and Metabolism 5: 495–505.
Kopin, A. S., Beinborn, M. 2004. Methods and compositions for the treatment of metabolic disorders. WO2004103310A2.
Kwak, M.K., and H. Ha. 2013. Where are we now in diabetic research? Archives of Pharmacal Research 36: 142–144.
Mu, F. 2001. Design, synthesis, and biological evaluation of a series of lavendustin aanalogues that inhibit EGFR and syk tyrosine kinases, as well as tubulin polymerization. Journal of Medicinal Chemistry 44: 441–452.
Murphy, K.G., and S.R. Bloom. 2007. Nonpeptidic glucagon-like peptide 1 receptor agonists: A magic bullet for diabetes? Proceedings of the National Academy of Sciences of the United States of America 104: 689–690.
Sennik, D., F. Ahmed, and D. Russell-Jones. 2011. Exenatide, a GLP-1 agonist in the treatment of Type 2 diabetes. Expert Review of Endocrinology and Metabolism 7: 15–26.
Sjöholm, Åke. 2010. Liraglutide therapy for type 2 diabetes: Overcoming unmet needs. Pharmaceuticals 3: 764–781.
Stolting, J., Burton, B. 2004. Novel process for the preparation of 2-aminomethylpyridine derivatives. WO 2004096772 A1.
Teng, M., M.D. Johnson, C. Thomas, D. Kiel, J.N. Lakis, T. Kercher, S. Aytes, J. Kostrowicki, D. Bhumralkar, L. Truesdale, J. May, U. Sidelman, J.T. Kodra, A.S. Jørgensen, P.H. Olesen, J.C. De Jong, P. Madsen, C. Behrens, I. Pettersson, L.B. Knudsen, J.J. Holst, and J. Lau. 2007. Small molecule ago-allosteric modulators of the human glucagon-like peptide-1 (hGLP-1) receptor. Bioorganic and Medicinal Chemistry Letters 17: 5472–5478.
Teng, M., Truesdale, L. K., Bhuumralkar, D., Kiel, D., Johnson, M. D., Thomas, C. 2000. Non-peptide GLP-1 agonists. WO2000042026.
Toma, T., J. Shimokawa, and T. Fukuyama. 2007. N, N′-ditosylhydrazine: aconvenient reagent for facile synthesis of diazoacetates. Organic Letters 9: 3195–3197.
Wang, M. W., Yuan, Y. Y., Zhou, L. 2009. A kind of receptor signaling transolution positive modulators, preparation methods and uses thereof. WO 2009129696A1.
Whitehouse, F.W. 1997. Insulin therapy and its shortcomings—the need for new approaches. Diabetic Medicine 14: S5–S8.
Yadav, J.S., B.V. Subba Reddy, Y. Gopal Rao, M. Srinivas, and A.V. Narsaiah. 2007. Cu(OTf)2-catalyzed synthesis of imidazo[1,2-a]pyridines from α-diazoketones and 2-aminopyridines. Tetrahedron Letters 48: 7717–7720.
Yu, G.J., C.L. Yoo, B. Yang, M.W. Lodewyk, L. Meng, T.T. El-Idreesy, J.C. Fettinger, D.J. Tantillo, A.S. Verkman, and M.J. Kurth. 2008. Potent s-cis-locked bithiazole correctors of ΔF508 cystic fibrosis transmembrane conductance regulator cellular processing for cystic fibrosis therapy. Journal of Medicinal Chemistry 51: 6044–6054.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2011-000-7061).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhang, YJ., Shen, LL., Cheon, HG. et al. Synthesis and biological evaluation of glucagon-like peptide-1 receptor agonists. Arch. Pharm. Res. 37, 588–599 (2014). https://doi.org/10.1007/s12272-013-0253-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-013-0253-9