
Abstract
Integrated light and electron microscopes (ILEMs) will enable a new generation of high-precision correlative imaging experiments. To fully exploit these systems, samples must contain dual-modality probes that highlight the position of macromolecules in the context of cell ultrastructure. We demonstrate that the fluorescent proteins (FPs) GFP (green), YFP (yellow) and mCherry can be used as dual-modality probes for ILEM when preserved using the in-resin fluorescence (IRF) technique, which delivers stable active fluorophores in lightly stained, resin-embedded cells and tissues. However, we found that vacuum pressure in the ILEM affects the photophysics of FPs in IRF sections. Here, we show that reducing the vacuum pressure reduces fluorescence intensity of GFP and YFP, which is a consequence of water extraction from the sample and is reversible on re-creation of partial pressure with water vapour (but not oxygen or nitrogen gas). We also find that, although fluorescence intensity is reduced at a partial pressure of 200 Pa (created using water vapour), the FP intensity is remarkably stable over time in vacuum and resistant to photobleaching during imaging. We are thus able to define imaging strategies for standard FPs acting as dual-modality probes in a single ‘multi-colour’ integrated microscope system.
Similar content being viewed by others


Introduction
From the discovery of Aequorea victoria green fluorescent protein (GFP) [1], fluorescent proteins (FPs) have developed into ubiquitous tools for localising macromolecules in biological samples [2]. The GFP molecule consists of 238 amino acids in a single chain, folded into a ϐ-barrel, at the centre of which sits the fluorescent chromophore para-hydroxybenzylidene imidazolone (PHBI). Four water molecules sit within the cavity that surrounds the chromophore, contributing to a hydrogen-bonding network that is important for the photoactive properties of GFP [3]. On excitation of GFP by light in the near-UV range, a proton-transfer reaction to a nearby amino acid group results in emission in the green region of the spectrum.
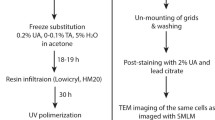
FP-protein constructs expressed in cells and tissues may be localised using diffraction-limited or super-resolution light microscopy [4]. Correlative light and electron microscopy (CLEM) is then required to place the FP in the context of cell ultrastructure. Different CLEM workflows have been developed for different applications [5, 6]. Recently, a number of in-resin fluorescence (IRF) methods have been reported that preserve the activity of GFP and other fluorophores through sample processing for electron microscopy (EM), thereby improving the accuracy of FP localisation, and promoting standard fluorophores to the role of dual-modality probes [7–11]. Generally, cells and tissues expressing FPs are immobilised using high-pressure freezing [12], after which the water in the sample is substituted at low temperature for a solvent containing a small percentage of uranyl acetate and water, before infiltration with and polymerisation in a water-tolerant resin. Maintenance of a small amount of water in the final resin block is key to preserving FP activity. Indeed, our work indicates that a quick freeze substitution (QFS) protocol [13] that minimises dehydration time leads to stable, active FPs in resin-embedded cells [10, 14].
We demonstrated our IRF protocol using the GFP-C1 construct to highlight the subcellular localisation of the lipid diacylglycerol, in combination with the mCherry-H2B (histone 2B) construct to highlight the nucleus in HeLa and COS-7 cells [10]. GFP and mCherry fluorescence was maintained through processing into several acrylic resins, and the FPs have remained stable in the polymerised resin blocks for several years. Ultrathin sections can be cut from IRF blocks and sequentially imaged in a fluorescence microscope and electron microscope for CLEM experiments. Although some membrane contrast is sacrificed due to the low concentration of heavy metal stains necessary to achieve fluorophore preservation, the result is an accurate overlay of FP localisation onto the underlying cell ultrastructure. Overlay accuracy can be further improved by imaging the IRF sections in an integrated light and scanning electron microscope (ILSEM) [15] where the sample remains in situ in a stable vacuum environment whilst imaging in both modalities [14].
In the process of establishing that GFP is active and visible in IRF sections in the vacuum of the ILSEM, we noted that alterations in vacuum pressure caused unexpected changes in the intensity of the emitted fluorescence. As the vacuum pressure decreased from 200 to 10−3 Pa, so the fluorescence intensity decreased, yet this effect could not be attributed solely to photobleaching, as the majority of the signal was recovered on return to 200 Pa and then to atmospheric pressure [10]. Since it is known that removal of the hydration shell from the chromophore can affect GFP dynamics [16], we hypothesised that the reduction in FP intensity with reducing vacuum pressure was a result of water extraction from the sample.
We now further investigate the effects of vacuum pressure on FPs in IRF sections. By manipulating the ILSEM to create partial pressures with dry nitrogen and oxygen gases, we confirm that water is essential for maintaining FP activity in vacuum. Using a vacuum test chamber, we were able to measure the effect of vacuum pressure on fluorescence intensity with greater precision, demonstrating quantitatively that repeated exposure to vacuum does not adversely influence FP activity. These findings confirm and extend our previous work showing that FPs can act as ‘dual-modality’ probes within the integrated light and electron microscope, and add mechanistic insights which will have significant implications for future experimental design.
Results
Standard fluorescent proteins are active in cells in resin in the electron microscope
In order to test the relationship between vacuum pressure and fluorescence intensity, we examined HeLa cells transfected with an expression construct encoding GFP-C1 or infected with a recombinant vaccinia virus encoding YFP-A3 [17]. As previously observed [10, 18], GFP-C1, which binds to the lipid diacylglycerol in cellular membranes, localised to the nuclear envelope, nucleoplasmic reticulum, Golgi apparatus, ER and vesicles (Fig. 1a, d–f); mCherry-H2B localised to the nucleus. Imaging the alternative constructs mCherry-C1 and GFP-H2B in the ILSEM highlights that the preservation and vacuum stability of the FPs in resin are independent of the specific construct used (Fig. 1b). Imaging IRF sections of HeLa cells infected with vaccinia virus YFP-A3 confirmed that YFP can also be preserved in resin (Fig. 1c) and imaged in vacuo (Fig. 1g–k). Furthermore, CLEM experiments performed using an ILSEM show that YFP-A3 localises to both immature (Fig. 1i) and mature virions inside the cell (Fig. 1h, j) and at the plasma membrane (Fig.1k), consistent with it being an inner core viral protein.

Preservation of FPs in IRF sections and imaging in the ILSEM. a–c Light microscopy of 200 nm sections taken from resin blocks containing HeLa cells expressing the fluorescently tagged constructs GFP-C1/mCherry-H2B (a), GFP-H2B/mCherry-C1 (b) and HeLa cells infected with vaccinia virus YFP-A3 (c). Images were acquired using a standard widefield epifluorescence light microscope and 100×/0.75 NA air objective. d Composite image showing GFP-C1 overlaid onto the corresponding electron micrograph; images were acquired sequentially using an ILSEM. e, f Boxed detail from (d) showing GFP-C1 (which binds to the lipid diacylglycerol) at Golgi stacks and endoplasmic reticulum. g Composite image showing YFP-A3 overlaid onto the corresponding electron micrograph; images were acquired sequentially using the ILSEM. YFP-A3 localises to viral particles within the cytoplasm and at the cell surface. h–k Boxed detail from G showing YFP-A3 localised to immature (i), mature (h, j) and extracellular (k) viral particles. ER endoplasmic reticulum, G Golgi stack, N nucleus, M mitochondrion. Scale bars = 10 μm (a–c), 5 μm (d, g), 500 nm (e, f, h–k)
FP activity in vacuo recovers with water vapour but not with nitrogen or oxygen
The intensity of GFP-C1 fluorescence in cells in IRF sections reduced as the vacuum pressure in the ILSEM decreased from atmospheric pressure (~105 Pa) to high vacuum (~10−3 Pa). GFP intensity was optimal at 200 Pa, visible from 200 to 40 Pa, but too dim below 40 Pa for effective imaging using an sCMOS detector. This effect could not be attributed solely to photobleaching since FP intensity recovered when the vacuum pressure was cycled back to 200 Pa using water vapour to create partial pressure, and then to atmosphere (Fig. 2a). To test the hypothesis that the effect of vacuum pressure on FP activity in IRF sections was caused by extraction of water, we instead created partial pressure in the ILSEM with oxygen gas (Fig. 2b) or nitrogen gas (Fig. 2c). In both cases, fluorescence intensity was too low for effective imaging from 200 Pa to high vacuum. Fluorescence intensity was not recovered on cycling back to 200 Pa in the presence of oxygen or nitrogen. However, on returning to atmospheric pressure by venting the system and opening the chamber door to air (~70 % humidity), the GFP signal was recovered. Furthermore, following GFP signal loss in oxygen or nitrogen partial pressure conditions, fluorophore activity could be recovered without returning to atmosphere by re-creating partial pressure at 200 Pa with water vapour (Fig. 2d, e). The same FP response to partial pressures created with water vapour, oxygen and nitrogen was observed for YFP-A3 (Fig. 3).

Response of GFP in IRF sections to partial pressure created using water vapour, oxygen or nitrogen. a–c Images were collected at atmospheric pressure (Atm), and across increments of decreasing vacuum pressure from 200 Pa to high vacuum (~10−3 Pa), before cycling back to 200 Pa and then Atm. a Fluorescent signal was retained in the presence of water vapour at 200 Pa, declined in intensity as the chamber pressure reduced to high vacuum, and recovered as the pressure was cycled back to 200 Pa and then Atm. Conversely, the signal degraded immediately in the presence of oxygen (b) or nitrogen (c) and remained unchanged until the chamber returned to Atm. d, e Switching from partial pressure created by either oxygen or nitrogen gas to water vapour resulted in the recovery of a significant proportion of the original fluorescent intensity. Scale bars = 10 μm

Response of YFP in IRF sections to partial pressure created using water vapour, oxygen or nitrogen. a–c Images were collected at atmospheric pressure (Atm), and across increments of decreasing vacuum pressure from 200 Pa to high vacuum (~10−3 Pa), before cycling back to 200 Pa and then Atm. a Fluorescent signal was retained in the presence of water vapour at 200 Pa, declined in intensity as the chamber pressure reduced to high vacuum, and recovered as the pressure was cycled back to 200 Pa and then Atm. Conversely, the signal degraded immediately in the presence of oxygen (b) or nitrogen (c) and remained unchanged until the chamber returned to Atm. d, e Switching from partial pressure created by either oxygen or nitrogen gas to water vapour resulted in the recovery of a significant proportion of the original fluorescent intensity. Scale bars = 10 μm
Quantification of the effect of vacuum pressure on GFP fluorescence intensity
To quantify the response of FPs to vacuum pressure in IRF sections, we built an offline vacuum chamber with pumping system, vacuum gauge, ‘up to air valve’ for rapid venting, and high precision leak valve for accurate pressure control. The system included an imaging window to hold a glass coverslip, with the IRF sections exposed to vacuum, and the light microscope optics outside the chamber (Fig. 4a). Vacuum-response quantification studies were performed using cells expressing the GFP-C1 construct. As the QFS protocol preserves fluorophore activity across a wide range of expression levels, we chose to exclude bright cells over-expressing the GFP-C1 construct as they are not physiologically relevant, and dim cells that would be difficult to quantify (Fig. 4b, red circles).

Response of GFP fluorescence intensity to changing vacuum pressure was quantified using a custom-built vacuum test chamber. a Components of the vacuum test chamber. b Inset showing the glass coverslip holding the IRF sections, mounted for an imaging experiment. c Image showing part of a typical field of view analysed during vacuum tests. Cells expressing either very high or very low levels of the GFP-C1 construct were excluded from the analysis (red circles). Cells expressing the construct at more ‘physiological’ levels (assessed by membrane morphology) were included in the analysis (green circles). Scale bar = 100 μm
Previously, we demonstrated that IRF sections mounted on glass slides in buffered glycerol were more resistant to photobleaching than fixed whole cells [10]. Here, we performed similar photobleaching experiments on dry mounted 200 nm-thick IRF sections at atmospheric pressure and under vacuum (Fig. 5). Time-lapse recordings of 30 min duration were carried out (300 ms exposure every 30 s) at atmospheric pressure (~105 Pa) and at 200 Pa. At atmospheric pressure, fluorescence intensity reduced over the first 30 min by 6.7 %. In comparison, at 200 Pa, the baseline fluorescence intensity was lower but the intensity dropped by only 1.4 % over the first 30 min. Both sets of IRF sections were then left at atmospheric pressure for 30 min without imaging, prior to performing a second time-lapse recording. At atmospheric pressure, no recovery of signal was noted after the rest period, and over the second time-lapse recording, a further drop in intensity of only 1.1 % was measured, indicating that a proportion of the fluorophores had been irreversibly photobleached (Fig. 5). In the vacuum experiments, on returning to 200 Pa after the rest period at atmosphere, the baseline fluorescence intensity was similar to the first time-lapse experiment and the intensity again dropped by only 1.8 % over the second 30-min recording.

Time-lapse recordings of fluorescence intensity from GFP in 200 nm-thick IRF sections at atmosphere and 200 Pa. Consecutive recordings of 30 min duration were acquired (30 ms exposure every 30 s), with a 30 min pause at atmosphere in between first and second recordings. At atmospheric pressure (~105 Pa; black line), fluorescence intensity declined by 6.7 % during the first acquisition period, reaching a plateau by the end of the recording period. The signal did not recover during the waiting phase, indicating that the initial reduction was caused predominantly by non-reversible photobleaching. Under vacuum conditions (200 Pa; red line), photobleaching of the signal remaining at this pressure was minimal, as shown by the similarity in intensity during first and second acquisition periods
The vacuum chamber was also used to quantify the effects of vacuum on fluorescence intensity observed in the ILSEM shown in Fig. 2. Fluorescence intensity in 200 nm IRF sections decreased as the vacuum pressure decreased, from a maximum at atmospheric pressure to a minimum at high vacuum (~10−3 Pa). On increasing vacuum pressure back to atmosphere, a slight reduction in intensity was noted over the pressure range, suggesting a lag in rehydration of the sections and thus fluorescence recovery when leaking the vacuum to air (~70 % humidity) rather than water vapour (Fig. 6a). Over repeated cycles of pumping to vacuum and venting to atmosphere (Fig. 6b), the fluorescence intensity averaged across multiple regions of interest reduced by only 0.1 % at atmosphere. This analysis leads us to propose a strategy for imaging standard fluorophores in IRF sections of cells or tissues inside the ILSEM (Fig. 7).

Quantification of vacuum pressure effects on GFP fluorescence intensity. Atmospheric pressure (Atm) and 200 Pa boundaries are marked by dashed vertical lines. a Graph showing intensity of fluorescence normalised to the initial fluorescence level. Each data point is averaged over five ROIs and three pressure cycles. As vacuum pressure decreased, the FP intensity decreased, and recovered on return to atmospheric pressure. b Scatter plot of the raw data (fluorescence intensity, arbitrary units) for each of five ROIs (i.e. five cells) acquired over three pressure cycles showing that GFP fluorescence intensity was stable over multiple cycles of vacuum exposure

Suggested workflow for imaging standard fluorescent proteins in IRF sections in the ILSEM
Discussion
The integration of light and electron microscopes heralds a new era for high-precision correlative microscopy, where both light and electron signals are recorded in situ from a single specimen. We have demonstrated that standard FPs (GFP, YFP, mCherry) can be preserved in-resin in a state that is active in vacuum. However, we found that the vacuum affects the photophysics of GFP in several ways. First, we observed a drop in fluorescence intensity as the vacuum pressure decreases due to extraction of water from the sample, and which can be reversed by re-introducing water into the system at partial vacuum pressure or atmospheric pressure. Second, we show that although fluorescence intensity is reduced at a partial pressure of 200 Pa (created using water vapour), the FP intensity is remarkably stable and resistant to photobleaching during imaging. Finally, we show that holding IRF sections in vacuum leads to very minor losses in fluorescence over time.
On a practical level, our experiments define a strategy for correlative imaging in the integrated microscope. Fluorescence imaging should be performed at ILSEM chamber pressures of 40 to 200 Pa, with partial pressure created by water vapour rather than nitrogen gas. Electron imaging should be performed in high vacuum to minimise scattering of electrons and maximise contrast from the IRF sections. FP intensity recovers when the vacuum pressure is increased to 200 Pa in the presence of water vapour, enabling repeated cycles of fluorescence microscopy and electron imaging on the same sample. Using this workflow, it is possible to sequentially image an area of interest for fluorescence and electron signals, before moving the specimen to acquire light and electron images from a new region of interest. Since the effect of time under vacuum on fluorescence intensity is minor, the same sample can be left in situ for several days for complex or large area imaging experiments.
Although we have shown that the extraction of water affects FP activity, it is difficult to determine the exact molecular mechanism underlying the reduction in FP intensity with reducing vacuum pressure. It is not likely to be caused by unfolding of the FP itself since the effect is reversible, and it is known that gas phase rGFP ions remain folded and stable under vacuum [19]. The reduction in FP intensity may be directly caused by the removal of the resident water molecules surrounding the chromophore, which has been shown to inactivate gas phase rGFP under vacuum [19]. It is also possible that the effect is indirectly caused by alteration of pH upon removal of water molecules from the FP microenvironment, to which FPs are highly sensitive [20]. The situation may be further complicated by the removal of reactive oxygen species from the chromophore by the vacuum, thus reducing damage to the chromophore over time [21], which could explain the observed stability and resistance to photobleaching.
Dissecting the molecular mechanism of the FP response to vacuum in cells in IRF samples will be inherently problematic. In comparison to photophysical studies in vacuum of isolated GFP molecules as crystals [22] or in the gas phase [19, 23–25], GFP in resin-embedded cells will be affected by a complex unquantifiable microenvironment created by the interplay of protein folding, the biochemistry of the cellular location, aldehyde cross-linking of proteins, uranyl acetate binding, solvent extraction of water and macromolecules, and resin-macromolecule interactions. For now, our approach has been driven by the need to understand in vacuo FP imaging on a ‘systems’ level, and the practical implications for integrated imaging using FPs as dual-modality probes. Indeed, it will be interesting in the future to examine the vacuum response of new iterations of FPs that withstand fixation, leading to improved electron contrast whilst maintaining FP activity [26].
Finally, it is interesting to consider that modulation of FP emission is the core concept of localisation-based ‘super-resolution’ light microscopy techniques including PALM and STORM [27]. It would be a logical step to investigate whether the modulation of FP emission by vacuum pressure could be used to incorporate subdiffraction light microscopy into the ILSEM, which could lead to correlation of FP-labelled proteins to cellular structures with a precision in the order of 10 nm. This resolution would be sufficient to localise proteins to trafficking vesicles and membrane domains without the need for antibodies, in a single ‘multi-colour’ integrated microscope system.
Materials and methods
Cell culture, constructs and transfection
Human cervical cancer epithelial HeLa cells were obtained from the American Type Culture Collection (ATCC, CCL-2). Cells were maintained in DMEM supplemented with 10 % fetal bovine serum in 10- or 15-cm tissue culture dishes. PKCε was kindly provided by Peter J. Parker (London Research Institute, UK). EGFP-PKCε was made as previously described [18]. Cells were transfected with 0.5 μg DNA (for 3.5 cm MatTek dishes) of each construct using Lipofectamine LTX and PLUS reagent (Invitrogen, Life Technologies Ltd, Paisley) in OPTIMEM medium (Gibco, Life Technologies Ltd., Paisley). The transfection mix was added to the cells in antibiotic-free medium. Microscopy and/or fixation was performed 18–24 h after transfection. Alternatively, HeLa cells were infected with vaccinia virus encoding YFP-A3 [17] at a multiplicity of infection (MOI) of 2 in serum-free DMEM. Infected cells were fixed at 18 h post-infection for downstream processing.
IRF preparation
IRF preparation was performed as described previously [10, 14]. Note that virus-infected cells were fixed with 4 % formaldehyde in 0.1 M phosphate buffer pH 7.4 prior to freezing. Briefly, cells were spun in a 1.5 ml Eppendorf tube to form a pellet, resuspended in an equal volume of media containing 20 % BSA and maintained at 37 °C. Fifty microliters of the cell suspension was pelleted in a blocked 200 μl pipette tip, from which the cells could be transferred to membrane carriers after removal of the blockage and loaded into the EMPACT2 high-pressure freezer using the rapid transfer system (Leica Microsystems, Vienna). Carriers containing frozen cells were stored under liquid nitrogen. Quick freeze substitution (QFS) was performed using a modified version of the method described by McDonald and Webb [13]. The substitution medium was 5 % H2O in acetone, with 0.1 % uranyl acetate diluted from a 20 % stock in methanol. After freeze substitution, the cells were infiltrated with HM20 resin at −50 °C in an automated freeze substitution unit (AFS2; Leica Microsystems, Vienna) and polymerised under 360 nm UV light over 48 h, before warming to room temperature.
Polymerised blocks were trimmed from the moulds and stored at room temperature in the dark. Prior to sectioning, the membrane carriers were carefully trimmed away by hand, and the blocks cut and trimmed perpendicular to the cell layer to allow examination of the full depth of the layer. For vacuum testing, ultrathin sections of 200 nm were cut using a UC6 ultramicrotome (Leica Microsystems, Vienna), collected on 22 × 22 mm glass coverslips, and stored for a maximum of 48 h in the dark at 4 °C. Widefield epifluorescence microscopy was performed on 200 nm sections using a standard benchtop microscope (Axio Scope.A1; Zeiss, Cambridge) and EC Epiplan-Neofluar 100×/0.75 NA air objective. Where sections were to be used for ILSEM, indium tin oxide (ITO) coated glass coverslips were used (Delmic B.V., Delft) to collect 200 nm sections, and the coverslip attached to a specimen holder [14].
ILSEM
ILSEM was performed on the same day as sectioning using a SECOM light microscope platform (Delmic B.V., Delft) with Nikon Plan Apo 100×/1.4 NA objective and vacuum compatible immersion oil, mounted on a Quanta 250 FEG SEM (FEI Company, Eindhoven). GFP fluorescence was stimulated by excitation of GFP using a 488 nm laser light source and multi-band filters (Di01-R405/488/594 dichroic, FF01-446/532/646-25 emission; Semrock, Rochester, NY). Images were collected using an sCMOS camera (Zyla 5.5; Andor Technology, Belfast) with a laser power of 1.1 mW at the sample level and exposure time of 8 s, at atmospheric pressure (ambient pressure with chamber door closed only for imaging), and across chamber pressures from 200 Pa to high vacuum (approximately 10−3 Pa). During vacuum series acquisition, short pauses were introduced after each target pressure was achieved (30 min when changing from atmosphere to 200 Pa, 20 Pa to high vacuum, high vacuum to 200 Pa, and 200 Pa to atmosphere; 5 min at all other points; and 10 min resting with chamber door open after returning to atmosphere) to allow for stabilisation of the system and specimens. Partial pressure conditions were created either using water vapour as standard for a Quanta FEG SEM, or using oxygen or nitrogen as auxiliary gases.
When directly comparing fluorescence intensity across a range of vacuum pressures, the power of illumination and time for each exposure were kept constant, and adjustments in brightness, contrast and RGB levels were made post-acquisition to closely match the levels of background fluorescence between images. For SEM imaging, the vCD backscatter detector (FEI Company, Eindhoven) was used at a working distance of 5.6 mm, and inverted contrast images were acquired in high vacuum (2.5 keV, spot size 3.5, 30 μm aperture, and pixel dwell time of 60 μs for a 3072*1536 image frame). The electron micrographs were adjusted to enhance contrast, and composite images of dual signals were generated as described previously [10, 14].
Vacuum test chamber for IRF sections
A chamber was constructed to allow imaging of GFP fluorescence intensity across a range of vacuum pressures, based on a five-way cross with DN50KF flanges. One opening was connected to a reducing T (LewVac, FL-RT63LF-50KF) that attached to the pumping system (Turbovac 50, Oerlikon and RV3, Edwards) on one side, and a vacuum gauge (Ionivac ITR90, Oerlikon) on the other. An up to air valve (UTAV-50KF, LewVac) for rapidly venting the chamber, and a high precision leak valve (215010, Oerlikon) for accurate pressure control were also attached.
The glass coverslips upon which IRF sections were collected were mounted using a custom flange (polished outer surface, central 5 mm-diameter hole) with the sections facing into the chamber and sealed onto the flange using vacuum grease. A customised upright epifluorescence microscope built from Thorlabs 30 mm cage components was used to image the sections. Images were obtained using an Olympus UPLFLN 20×/0.5 NA objective, a fluorescence filter set (MF469-35, MD498, MF535-22; Thorlabs), an f = 150 mm tube lens, and an sCMOS camera (Flash 4.0 V2, Hamamatsu). Illumination was provided using an LED (M490L2, Thorlabs). Image data was acquired using Micromanager [28] with 2 × 2 binning and an exposure time of 300 ms. The power of illumination at the sample level was recorded as 3.5 mW. The pressure measurements from the ion gauge were also recorded in Micromanager by using the analogue input of an Arduino Mega 2560 microcontroller board.
Data analysis
Individual images were imported as a sequence into Fiji [29] and the individual frames aligned using the template matching plugin (https://sites.google.com/site/qingzongtseng/template-matching-ij-plugin). Circular regions of interest (ROIs) were then drawn around a selection of fluorescent cells in the image, and their average intensity extracted. Cells exhibiting very intense fluorescence were excluded from analysis as they were considered to be non-physiological. Cells exhibiting very weak fluorescence were also excluded as they would have been difficult to quantify accurately. To assess FP response to multiple rounds of vacuum exposure, the pressure in the chamber was cyclically reduced and increased with images obtained at pre-determined settings of the pumping system and leak valve. Three pressure cycles were recorded sequentially. Each data point was averaged over the three pressure cycles and five ROIs selected.
References
Shimomura O, Johnson FH, Saiga Y (1962) Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan. Aequorea J Cell Comp Physiol 59:223–239
Giepmans BN, Adams SR, Ellisman MH, Tsien RY (2006) The fluorescent toolbox for assessing protein location and function. Science (New York, NY) 312:217–224. doi:10.1126/science.1124618
Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science (New York, NY) 273:1392–1395
Shcherbakova DM, Sengupta P, Lippincott-Schwartz J, Verkhusha VV (2014) Photocontrollable fluorescent proteins for superresolution imaging. Annu Rev Biophys 43:303–329. doi:10.1146/annurev-biophys-051013-022836
Muller-Reichert T, Verkade P (2012) Introduction to correlative light and electron microscopy. Methods Cell Biol 111:xvii–xix. doi:10.1016/b978-0-12-416026-2.03001-6
Muller-Reichert T, Verkade P (2014) Preface. Correlative light and electron microscopy II. Methods Cell Biol 124:xvii–xviii. doi:10.1016/b978-0-12-801075-4.09983-3
Bell K, Mitchell S, Paultre D, Posch M, Oparka K (2013) Correlative imaging of fluorescent proteins in resin-embedded plant material. Plant Physiol 161:1595–1603. doi:10.1104/pp. 112.212365
Kukulski W, Schorb M, Welsch S, Picco A, Kaksonen M, Briggs JA (2011) Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J Cell Biol 192:111–119. doi:10.1083/jcb.201009037
Nixon SJ, Webb RI, Floetenmeyer M, Schieber N, Lo HP, Parton RG (2009) A single method for cryofixation and correlative light, electron microscopy and tomography of zebrafish embryos. Traffic (Copenhagen, Denmark) 10:131–136. doi:10.1111/j.1600-0854.2008.00859.x
Peddie CJ et al (2014) Correlative and integrated light and electron microscopy of in-resin GFP fluorescence, used to localise diacylglycerol in mammalian cells. Ultramicroscopy. doi:10.1016/j.ultramic.2014.02.001
Watanabe S et al (2011) Protein localization in electron micrographs using fluorescence nanoscopy. Nat Methods 8:80–84. doi:10.1038/nmeth.1537
McDonald KL, Morphew M, Verkade P, Muller-Reichert T (2007) Recent advances in high-pressure freezing: equipment- and specimen-loading methods. Methods Mol Biol (Clifton, NJ) 369:143–173
McDonald KL, Webb RI (2011) Freeze substitution in 3 hours or less. J Microsc 243:227–233. doi:10.1111/j.1365-2818.2011.03526.x
Peddie CJ, Liv N, Hoogenboom JP, Collinson LM (2014) Integrated Light and Scanning Electron Microscopy of GFP-Expressing. Cell Method Cell Biol 124:363–389. doi:10.1016/b978-0-12-801075-4.00017-3
Liv N et al (2013) Simultaneous correlative scanning electron and high-NA fluorescence microscopy. PLoS One 8:e55707. doi:10.1371/journal.pone.0055707
Nickels JD et al (2012) Dynamics of Protein and its Hydration Water: Neutron Scattering Studies on Fully Deuterated GFP. Biophys J 103:1566–1575
Arakawa Y, Cordeiro JV, Schleich S, Newsome TP, Way M (2007) The release of vaccinia virus from infected cells requires RhoA-mDia modulation of cortical actin. Cell Host Microbe 1:227–240. doi:10.1016/j.chom.2007.04.006
Domart MC et al (2012) Acute manipulation of diacylglycerol reveals roles in nuclear envelope assembly & endoplasmic reticulum morphology. PLoS One 7:e51150. doi:10.1371/journal.pone.0051150
Frankevich V, Barylyuk K, Chingin K, Nieckarz R, Zenobi R (2013) Native biomolecules in the gas phase? The case of green fluorescent protein Chemphyschem. Eur J Chem Phys Phys Chem 14:929–935. doi:10.1002/cphc.201200959
Zimmer M (2002) Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem Rev 102:759–781
Bourgeois D, Regis-Faro A, Adam V (2012) Photoactivated structural dynamics of fluorescent proteins. Biochem Soc Trans 40:531–538. doi:10.1042/bst20120002
Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, Ormo M, Remington SJ (1997) Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc Natl Acad Sci U S A 94:2306–2311
Lammich L, Petersen MA, Nielsen MB, Andersen LH (2007) The gas-phase absorption spectrum of a neutral GFP model chromophore. Biophys J 92:201–207. doi:10.1529/biophysj.106.093674
Petrone A, Caruso P, Tenuta S, Rega N (2013) On the optical absorption of the anionic GFP chromophore in vacuum, solution, and protein. Phys Chem Chem Phys: PCCP 15:20536–20544. doi:10.1039/c3cp52820k
Rajput J et al (2009) Photoabsorption studies of neutral green fluorescent protein model chromophores in vacuo. Phys Chem Chem Phys: PCCP 11:9996–10002. doi:10.1039/b914276b
Paez-Segala MG et al (2015) Fixation-resistant photoactivatable fluorescent proteins for CLEM. Nat Methods 12:215–218. doi:10.1038/nmeth.3225, 214 p following 218
Requejo-Isidro J (2013) Fluorescence nanoscopy. Methods Appl J Chem Biol 6:97–120. doi:10.1007/s12154-013-0096-3
Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N (2010) Computer control of microscopes using microManager Current protocols in molecular biology / edited by Frederick M Ausubel [et al] Chapter 14:Unit14.20 doi:10.1002/0471142727.mb1420s92
Schindelin J et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi:10.1038/nmeth.2019
Acknowledgments
The authors would like to acknowledge funding from Cancer Research UK, and from the MRC, BBSRC and EPSRC under grant award MR/K01580X/1 to LC and Peter O’Toole (York University). We would like to thank Peter O’Toole and Sander den Hoedt (DELMIC B.V.) for useful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Elisabeth Brama and Christopher J. Peddie are joint first authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brama, E., Peddie, C.J., Jones, M.L. et al. Standard fluorescent proteins as dual-modality probes for correlative experiments in an integrated light and electron microscope. J Chem Biol 8, 179–188 (2015). https://doi.org/10.1007/s12154-015-0143-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12154-015-0143-3