
Abstract
The majority of the morbidity and mortality associated with hepatitis B virus infection is due to viral persistence and its consequences. The heterogeneity of outcomes from HBV infection suggests that both viral and host factors influence the development of chronic infection. Study of host genetic susceptibility has revealed a number of genes including MHC class II loci and cytokine receptors, which decrease the risk of persistence. On the viral side, the replication system is adapted to generate high levels of virions without stimulating the innate immune system. Secreted viral proteins (HBsAg and HBeAg) suppress innate responses through inhibition of TLR signaling, which leads to a weak adaptive immune response with an exhausted phenotype that is incapable of inducing viral elimination. However, even when the adaptive immune system begins to take effect after HBe seroconversion, the ability of the virus to mutate and evade T and B cell-mediated responses helps to sustain persistent infection. Understanding the mechanisms of persistence is important for the design of therapeutic strategies. Although there are currently no specific drugs that target the viral minichromosome (cccDNA), it is expected that in the future we will be able to use existing drugs more effectively to eliminate the infection.
Similar content being viewed by others

Introduction
Hepatitis B virus (HBV) is a member of the hepadnavirus family of small hepatotropic DNA viruses. It is a small enveloped virus with a diameter of 42–44 nm and a genome of only 3.2 kb. The small size contrasts with the biological and clinical importance of this pathogen. Worldwide it is estimated that 2 billion (one third of the human population) people have been infected with HBV. WHO reports that 350 million people are chronically infected with HBV and approximately 25 % of these will die prematurely as a result of their infection [1]. HBV is responsible for a number of disease presentations, ranging from fulminant hepatic failure to cirrhosis and hepatocellular carcinoma. At least 600,000 people a year die from hepatocellular carcinoma and a similar number from decompensated cirrhosis.
The natural history of HBV infection is associated with transmission at the time of birth or in early childhood. However, infection may be acquired later in life through parenteral exposure such as sexual intercourse or sharing of intravenous needles. Acute infection, particularly in children, may be asymptomatic or may result in a severe hepatitic illness with flu-like symptoms and jaundice that persists for several weeks. Acute infection resolves spontaneously in the majority of individuals but persistent (or chronic) infection can develop, particularly in those with asymptomatic acute infection and in the very young. Chronic infection develops in 90 % of people who acquire HBV at birth, 20–50 % of those who acquire infection in early childhood and in less than 5 % of those who acquire infection in adult life. Given the importance of chronic infection imposed on global populations in terms of morbidity and mortality, the focus of this article is to review what is currently known about the mechanisms of HBV persistence.
HBV replication
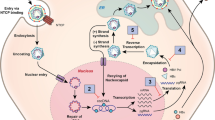
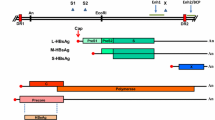
The complex mechanism of HBV replication (Fig. 1) plays an important role in viral persistence. HBV in the blood is a spherical structure surrounded by a lipid envelope containing the HBV envelope protein hepatitis B surface antigen (HBsAg) [2]. Within the envelope is a nucleocapsid made up of the hepatitis B core protein (HBcAg) and the viral genome, which is a relaxed circular, partially double-stranded DNA of 3,200 bp. The viral genome contains four overlapping open reading frames, pol, precore/core, preS/S and X. The polymerase ORF encodes a multifunctional polymerase enzyme that has reverse transcriptase, DNA polymerase and RNAseH activities. The precore/core ORF contains two translation initiation codons allowing it to encode both the nucleocapsid protein and a secreted e-antigen (HBeAg) that has immunomodulatory properties [3]. The preS/S ORF contains three translation initiation codons encoding a large, middle and small version of the HBsAg. The function of the protein encoded by the X ORF is not as well characterized as those of the other proteins. However, it has an essential role in transcriptional regulation of both viral and host proteins.

HBV replication
Recently, the cellular receptor for HBV has been identified as the sodium taurocholate transporter peptide found on the baso-lateral border of hepatocytes [4]. The mechanism of attachment, internalization of the virus and uncoating has not been well characterized. The relaxed circular DNA of the virus is released from the nucleocapsid into the host cell nucleus where host polymerases complete the positive strand DNA to form a fully double-stranded genome [5]. The HBV genome adopts a supercoiled configuration of a covalent closed circular DNA (cccDNA) that behaves as a minichromosome associating with histone and other DNA chaperone proteins [6]. The formation of the cccDNA-minichromosome within the nucleus protects the virus from innate immune sensors that would otherwise trigger responses against the genome or against the infected cell.
The cccDNA uses host cellular transcriptional machinery to transcribe a full-length pregenomic RNA that is part of viral replication as well as mRNA, which encodes viral polypeptides [5]. Pregenomic RNA is packaged with the HBV polymerase protein into the nucleocapsid where first the negative strand of the new HBV genome and then the positive strand are synthesized. Processing of the reverse transcription and DNA polymerization within the nucleocapsid is also presumed to assist in evasion of innate immune sensors. Completed nucleocapsids associate with large, middle and small HBsAg proteins at the cell membrane and bud out into the serum [7]. As cccDNA lacks a centromere and other components of mitotic regulation, cell division may lead to loss or dilution of cccDNA. However, some of the nucleocapsids may re-enter the nucleus to maintain the levels of cccDNA within the cell rather than budding out into the blood.
Host factors
Natural history studies demonstrate that the outcome of HBV infection is heterogeneous with different individuals developing acute or chronic infection from the same viral strain. To explain the variation in disease outcomes, a number of groups have sought host genetic variants that influence the infection [8]. Twin studies from Taiwan confirm that concordance for HBsAg status (as a marker for HBV chronicity) is significantly higher among monozygotic twins compared to dizygotic twin pairs, indicating that host genetics may be responsible for around 50 % of the variation in disease outcome [9].
Sequence diversity in the major histocompatibility (MHC) loci on chromosome 6 has been recognized as a source of variation in disease outcome in many infectious and autoimmune diseases. For this reason, a number of investigators have looked at the distribution of alleles in the MHC class I and II loci in subjects with either acute (self-limiting) or chronic (persistent) HBV infection [10]. Consistent associations with the MHC class II allele HLA-DRB1*1302 have been found across a wide range of ethnic groups. HLA-DRB1*1302 is found more frequently in patients with self-limiting infection, but no specific mechanism has yet been described to explain how this association is linked to viral elimination [11]. Some reports have suggested that peptide binding by this allele is more promiscuous than other alleles in the DRB1 locus, which would allow a wider range of viral epitopes to be presented and reduce the opportunity for virus to evade T cell-mediated immunity. In support of this theory, it has also been demonstrated that individuals who are homozygous at the MHC class II loci are less likely to eliminate the virus than those who are heterozygous [12]. Presumably in this case the increased number of HLA alleles is capable of binding and presenting a wider diversity of viral epitopes.
Recently, genome-wide association studies have been used to identify single nucleotide polymorphisms (SNPs) associated with HBV persistence. Consistent associations with SNPS in the MHC class II region have again been reported even when this hypothesis-free approach has been employed. SNPs in both the HLA-DPA and HLA-DPB regions have been associated with the outcome of HBV, and although these loci are in close proximity to the HLA-DRB loci, there does not appear to be any linkage disequilibrium to explain these associations [13]. One study looking at the cell surface display of HLA-DP protein appears to show that SNPs associated with viral elimination are also associated with high levels of HLA-DP display, suggesting that antigen presentation is critical to mounting an effective adaptive immune response to the virus [14].
Using a genome-wide scan in West African sibling pairs affected by chronic HBV infection, we used microsatellite genotyping to identify chromosomal regions linked to HBV outcome. A region of chromosome 21 containing a cluster of cytokine receptor II genes was linked to chronic HBV infection [15]. Resequencing and gene association studies across this region identified SNPS in the IL-10RB and IFNAR2 genes that conferred susceptibility to chronic infection. IL-10RB encodes a cell membrane receptor that partners with a number of ligand-specific receptor subunits allowing it to participate in signal transduction from interleukin 10 (IL-10), interleukin 22 and interleukins 28a, 28b and 29, which are now known as type III interferons or interferon lambda (IFN-λ). The variant of the IL-10RB associated with viral elimination was found to be a more effective signal transducer for IFN-λ.
Viral factors
Undoubtedly the small size of the HBV genome and the mechanism of replication contribute to the ability of HBV to evade innate immune sensing and therefore to persist. However, there are other features in the viral replication that favor viral persistence. The HBV DNA polymerase enzyme is error prone, leading to nucleotide and amino acid substitutions in viral progeny. Inevitably the random generation of variants is likely to produce progeny with impaired viability particularly when taking into account the overlapping reading frames of HBV. However, the high rate of nucleoside substitutions guarantees that some of the progeny are replication competent, and when selection pressures are applied those species with sequence-encoded advantages will become dominant. This principal has been demonstrated effectively by the use of low potency antiviral drugs such as lamivudine where amino acid substitutions in the polymerase protein confer resistance to the drug [16]. It is believed that sequence variation, particularly in B and T cell epitopes, allows the virus to persist even in the face of virus-specific adaptive immune responses.
For many years after the discovery of HBV, the presence of HBeAg in serum was associated with high viral replication and its absence was thought to be associated with asymptomatic carriage, a low replication state with normal liver biochemistry and resolution of histological liver injury. However, it is now clear that many patients who have lost HBeAg and who have acquired antibody to HBeAg (anti-HBe) may progress to a high viral replication phase with rapid progression to cirrhosis. The molecular explanation for the HBeAg-negative viral replication is that it is the result of mutations in the preCore region of the viral genome, which allows transcription and translation of the nucleocapsid antigen while preventing the production of the HBeAg [17]. This form of the virus is therefore still able to persist even in the presence of antibodies. HBeAg-negative hepatitis is associated with increased T cell responses, but variation in T cell epitopes in the viral proteins may help to avoid adaptive immune responses [18].
Although vertical transmission of HBV leads to chronic infection in a high proportion of children, transmission can be interrupted successfully by using a combination of active and passive immunization. Administering the HBsAg vaccine and HBV hyperimmune immunoglobulin at the time of birth reduces the risk of infection by 90 %. However, in a small number of children chronic infection develops even in the face of vaccine-induced antibody to HBsAg (anti-HBs). This immune escape is the result of viral amino acid variation in the key ‘a’ determinant of the HBsAg. Although the original reports identified a mutation at position 145 in the HBsAg protein, subsequently a number of other variants have been described that are not effectively bound by anti-HBs and therefore allow the virus to persist even in the face of a strong humoral immune response.
Host–viral interactions
One of the most important determinants of HBV persistence is the age at which the individual acquires the infection. Mother-to-baby (vertical) transmission is responsible for approximately 40 % of infections in Asia and around 10 % of infections in sub-Saharan Africa. Transmission normally occurs at the time of birth and results in chronic infection in over 90 % of cases in the absence of active and passive immunization. In contrast, acquisition of infection during early childhood results in chronic infection in 20–50 % of cases, and acquisition in adult life only results in chronic infection in 5 % of cases. The mechanism of chronicity in neonates and children is not fully characterized but a number of mechanisms have been implicated. Most important of these is neonatal tolerization. The HBeAg is a small polypeptide, which is capable of crossing the placenta and can be found in the serum of neonates born to HBeAg-positive mothers [19]. It is thought that neonatal exposure to HBeAg establishes immune tolerance to the HBeAg and to the nucleocapsid protein, which is known to be the target of both CD4+ and CD8+ T cell responses.
It is widely recognized that chronic HBV infection progresses through four stages beginning with the ‘immune tolerant’ phase [20]. During this phase infected individuals have extremely high viral loads in the absence of biochemical or histological liver damage. During this phase both innate and adaptive immune responses to the virus appear to be absent.
Gene expression studies during early infection in chimpanzees suggest that HBV is a stealth virus inducing minimal responses in the liver [21]. Specifically in this model there appeared to be an absence of an interferon response, which would be expected in a viral infection. It is not clear how the virus avoids interferon induction but the usual innate immune triggers such as double-stranded RNA or other replication intermediates are hidden from the immune system. Furthermore, HBV is not cytopathic so it does not trigger the release of damage-associated molecular patterns such as high mobility group B1 protein.
Adaptive immune responses to HBV antigens are impaired in individuals with chronic HBV infection when compared to individuals with acute self-limiting infection. Since the 1990s it has been known that both CD4+ and CD8+ T cell responses are affected in terms of the magnitude of the response and in terms of the breadth of the viral T cell epitopes that are recognised [22]. More recent immunological techniques have shown that HBV-specific T cells have an anergic or exhausted phenotype [22]. In addition, some studies suggest the regulatory T cells are also present that inhibit effector immune responses [23].
It is now thought that the soluble antigens secreted by HBV-infected cells play an important role in modulating adaptive immune responses [24]. Priming of adaptive immune responses requires both the presence of non-self antigens and a ‘danger’ signal. Danger signals are normal transduced through Toll- like receptors, which recognize components of bacterial, viral or fungal cell walls or genomes. HBeAg and HBsAg have now been shown to interfere with the expression or signal transduction from TLR2, TLR7 and TLR9 [25–27].
New treatment options
The principal goal of treatment is to cure the infection before the patient progresses to a life-threatening complication such as decompensated cirrhosis or hepatocellular carcinoma. Serologically, a cure is marked by the absence of HBV DNA in serum and by the loss of HBsAg. In reality, loss of HBsAg occurs in only a minority of treated patients. In patients who start therapy during the HBeAg-positive phase of infection, treatment with interferon-based therapy may induce HBsAg loss in around 20 % of those who manage to seroconvert from HBeAg to anti-HBe during a 2-year follow-up. This group of patients can also achieve reasonable levels of HBsAg loss when treated with nucleoside analogs over a 5-year period. Unfortunately, this is a minority of patients, and HBsAg loss is extremely rare among the patients who form the majority in most clinics who are HBeAg negative when treatment commences.
Nucleoside analogs are highly successful in suppressing viral replication and preventing the progression of disease to cirrhosis or decompensation. Now that these clinical goals have been achieved, the target of therapy has been raised to achieve HBsAg loss. Two years ago, Hadziyiannis et al. [28] reported an intriguing trial in which HBeAg-negative patients who had been successfully treated for 4 years or more with adefovir underwent treatment withdrawal. As would be anticipated, many of these patients suffered flares of hepatitis requiring reinstatement of their antiviral therapy. However, 25 % of the patients lost HBsAg during 4 years of follow-up. Attempts to replicate the findings in this small study are now underway.
An alternative therapeutic strategy for patients who have been treated for prolonged periods with nucleoside analogs is to exploit the immunomodulatory properties of interferon once some degree of antiviral adaptive immune response has been reinstated. A number of studies provide evidence that viral suppression with nucleoside analogs is associated with some degree of ‘immune restitution’ with improvement of CD4+ and CD8+ T cell responses [29]. Interferon has pleotropic effects stimulating both antiviral and immunomodulatory responses. T cell immunity is stimulated by upregulation of proteins in the antigen presentation pathways and has been shown to increase HBV-specific CD4 responses. Interferon alpha stimulates the activity of APOBEC, a cytosine deaminase that has recently been shown to decrease cccDNA in infected cells [30, 31]. Previous studies combining interferon with nucleoside analogs have administered both therapeutic modalities simultaneously, but it has now been proposed to evaluate interferon therapy in patients who have been treated with nucleoside analogs for prolonged periods in whom the cccDNA levels may already be reduced and the viral suppression may have resulted in enhanced adaptive immunity.
A logical intervention to overcome the effects of HBeAg and HBsAg is to use direct TLR agonists to stimulate adaptive responses to HBV. There is a risk in using TLR agonists that might induce non-specific inflammatory responses or autoimmunity when administered systemically. However, there is now an oral TLR7 agonist in clinical development that, given its route of delivery, should primarily target the liver. To date, there are no trials of TLR stimulation in chronic HBV infection [32].
Finally, there is now renewed interest in the use of therapeutic vaccination in chronic HBV infection. The purpose of this intervention is to drive T cell responses against the virus to eliminate infected cells. Although vaccination with the HBsAg is highly successful in prophylactic vaccines, there is a major immunological challenge to stimulate responses in patients who already have high circulating levels of viral antigens. There are now a number of novel vaccine adjuvants that could be used to stimulate immune responses to viral antigens presented in the skin that might theoretically overcome the anergy in existing T cell responses. The ideal target patient group is likely to be those with HBeAg-negative disease who have been on nucleoside analogs for prolonged periods.
Summary
The success of HBV as a global pathogen is dependent on its ability to generate persistent infection. Viral factors, host factors and host-viral interactions all contribute to viral persistence. However, once established, persistent infection is exceptionally difficult to terminate therapeutically. A number of strategies are available to challenge this situation but will need to be evaluated in carefully designed clinical trials. In the meantime, treatment with nucleoside analogs, which remain the backbone of therapy, is highly successful in reducing most of the morbidity and mortality from HBV.
References
Goldstein ST, Zhou F, Hadler SC, Bell BP, Mast EE, Margolis HS. A mathematical model to estimate global hepatitis B disease burden and vaccination impact. Int J Epidemiol 2005;34(6):1329–1339
Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1970;1(7649):695–698
Dandri M, Locarnini S. New insight in the pathobiology of hepatitis B virus infection. Gut 2012;61(Suppl 1):6–17. doi:10.1136/gutjnl-2012-302056.:i6-17
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012;1:e00049. doi:10.7554/eLife.00049
Locarnini S, Zoulim F. Molecular genetics of HBV infection. Antivir Ther 2010;15(Suppl 3):3–14. doi:10.3851/IMP1619.:3-14
Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol 2009;51(3):581–592
Urban S, Schulze A, Dandri M, Petersen J. The replication cycle of hepatitis B virus. J Hepatol 2010;52(2):282–284
Yee LJ, Thursz MR. Hepatitis B and C. In Kaslow R, McNichol J, Hill AV, editors. Genetic Susceptibility in Infectious Disease. Oxford: OUP; 2007
Lin TM, Chen CJ, Wu MM, Yang CS, Chen JS, Lin CC, et al. Hepatitis B virus markers in Chinese twins. Anticancer Res 1989;9:737
Thursz M, Yee L, Khakoo S. Understanding the host genetics of chronic hepatitis B and C. Semin Liver Dis 2011;31(2):115–127
Thursz MR, Kwiatkowski D, Allsopp CE, Greenwood BM, Thomas HC, Hill AV. Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N Engl J Med 1995;332(16):1065–1069
Thursz MR, Thomas HC, Greenwood BM, Hill AV. Heterozygote advantage for HLA class-II type in hepatitis B virus infection. Nat Genet 1997;17(1):11–12
Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet 2009;41(5):591–595
Wu TW, Chu CC, Ho TY, Chang Liao HW, Lin SK, Lin M, et al. Responses to booster hepatitis B vaccination are significantly correlated with genotypes of human leukocyte antigen (HLA)-DPB1 in neonatally vaccinated adolescents. Hum Genet 2013;132(10):1131–1139
Frodsham AJ, Zhang L, Dumpis U, Taib NA, Best S, Durham A, et al. Class II cytokine receptor gene cluster is a major locus for hepatitis B persistence. Proc Natl Acad Sci USA 2006;103(24):9148–9153
Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351(15):1521–1531
Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, et al. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 1989;2(8663):588–591
Fattovich G, McIntyre G, Thursz M, Colman K, Giuliano G, Alberti A, et al. Hepatitis B virus precore/core variation and interferon therapy. Hepatology 1995;22(5):1355–1362
Lin HH, Ohto H, Etoh T, Yoneyama T, Kawana T, Mizuno M. Studies on the risk factors of intrauterine infection of hepatitis B virus. Nihon Sanka Fujinka Gakkai Zasshi 1985;37(11):2393–2400
Thursz MR, Thomas HC. Pathogenesis of chronic hepatitis B. In Thomas HC, Lemon S, Zuckerman AJ, editors. Viral Hepatitis. 3rd ed. Oxford: Blackwell; 2005. p 308–321
Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol 2005;79(15):9369–9380
Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Postgrad Med J 2013;89(1051):294–304
Franzese O, Kennedy PT, Gehring AJ, Gotto J, Williams R, Maini MK, et al. Modulation of the CD8+ -T-cell response by CD4+ CD25+ regulatory T cells in patients with hepatitis B virus infection. J Virol 2005;79(6):3322–3328
Vincent IE, Zannetti C, Lucifora J, Norder H, Protzer U, Hainaut P, et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One 2011;6(10):e26315
Vincent IE, Zannetti C, Lucifora J, Norder H, Protzer U, Hainaut P, et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One 2011;6(10):e26315
Wang S, Chen Z, Hu C, Qian F, Cheng Y, Wu M, et al. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol 2013;190(10):5142–5151
Xu N, Yao HP, Lv GC, Chen Z. Downregulation of TLR7/9 leads to deficient production of IFN-alpha from plasmacytoid dendritic cells in chronic hepatitis B. Inflamm Res 2012;61(9):997–1004
Hadziyannis SJ, Sevastianos V, Rapti I, Vassilopoulos D, Hadziyannis E. Sustained responses and loss of HBsAg in HBeAg-negative patients with chronic hepatitis B who stop long-term treatment with adefovir. Gastroenterology 2012;143(3):629–636
Boni C, Penna A, Bertoletti A, Lamonaca V, Rapti I, Missale G, et al. Transient restoration of anti-viral T cell responses induced by lamivudine therapy in chronic hepatitis B. J Hepatol 2003;39(4):595–605
Abe H, Ochi H, Maekawa T, Hatakeyama T, Tsuge M, Kitamura S, et al. Effects of structural variations of APOBEC3A and APOBEC3B genes in chronic hepatitis B virus infection. Hepatol Res 2009;39(12):1159–1168
Chowdhury S, Kitamura K, Simadu M, Koura M, Muramatsu M. Concerted action of activation-induced cytidine deaminase and uracil-DNA glycosylase reduces covalently closed circular DNA of duck hepatitis B virus. FEBS Lett 2013;587(18):3148–3152
Zhang X, Kraft A, Broering R, Schlaak JF, Dittmer U, Lu M. Preclinical development of TLR ligands as drugs for the treatment of chronic viral infections. Expert Opin Drug Discov 2012;7(7):597–611
Compliance with ethical requirements and Conflict of interest
This article does not contain any studies with human or animal subjects. The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thursz, M. Basis of HBV persistence and new treatment options. Hepatol Int 8 (Suppl 2), 486–491 (2014). https://doi.org/10.1007/s12072-013-9504-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-013-9504-6