
Abstract
Throughout life, bone is continuously remodelled. Bone is formed by osteoblasts, from mesenchymal origin, while osteoclasts induce bone resorption. This process is tightly regulated. During inflammation, several growth factors and cytokines are increased inducing osteoclast differentiation and activation, and chronic inflammation is a condition that initiates systemic bone loss. Rheumatoid arthritis (RA) is a chronic inflammatory auto-immune disease that is characterised by active synovitis and is associated with early peri-articular bone loss. Peri-articular bone loss precedes focal bone erosions, which may progress to bone destruction and disability. The incidence of generalised osteoporosis is associated with the severity of arthritis in RA and increased osteoporotic vertebral and hip fracture risk. In this review, we will give an overview of different animal models of inflammatory arthritis related to RA with focus on bone erosion and involvement of pro-inflammatory cytokines. In addition, a humanised endochondral ossification model will be discussed, which can be used in a translational approach to answer osteoimmunological questions.
Similar content being viewed by others


Bone Destruction in Rheumatoid Arthritis
Osteoclast Differentiation in Inflammatory Arthritis
Osteoclasts are multinucleated motile cells derived from hematopoietic stem cells during a multistep differentiation process called osteoclastogenesis through cell-cell contact or the secretion of local factors. Osteoclast differentiation is regulated by two essential cytokines: the macrophage colony-stimulating factor-1 (M-CSF) and the receptor activator of nuclear factor-kB ligand (RANKL) [1, 2]. M-CSF is considered a crucial factor responsible for the survival and proliferation of osteoclast precursor cells [3]. RANKL was cloned from activated T cells as a new member of the tumour necrosis factor (TNF) superfamily in 1997 [4]. RANKL is expressed by cells of mesenchymal origin in bone and is a direct regulator of osteoclast formation and bone turnover. In addition, cells of the immune system, such as T cells [5, 6] and B cells express RANKL [2, 7, 8]. The role of RANKL as the critical osteoclastogenic factor was demonstrated using knockout mice. These mice developed severe osteopetrosis due to complete absence of osteoclasts [9]. The receptor for RANKL is RANK, a type I transmembrane protein expressed on osteoclast precursor cells and mature osteoclasts. The binding of RANKL to RANK is inhibited by the decoy receptor osteoprotegerin (OPG). The RANKL/OPG ratio is an important determinant for the balance in osteoclastogenesis.
Osteoclasts are designed to resorb bone by generating a sealing zone, an actin ring made of densely packed podosomes, beneath their ruffled border, into which they secrete hydrochloric acid to solubilise calcium from bone and proteolytic enzymes, such as matrix metalloproteinases, cathepsin K and phosphatases (tartrate-resistant acid phosphatase (TRAP)) to degrade the remaining matrix.
Increase of osteoclast numbers and activity is a hallmark of inflammatory bone loss. The inflammatory microenvironment promotes not only precursor recruitment from bone marrow but also their subsequent differentiation into mature osteoclasts. The first indirect description of bone-resorbing cells in RA dates back to the nineteenth century [10] and was revised more than a century later by Bromley and Woolley [11] and Leisen [12]. Osteoclast in RA were recently identified and characterised in more detail by Gravallese et al. [13]. It was also demonstrated that RA patients with active disease have greater synovial RANKL expression when compared with those with lower disease activity, and this increase in RANKL is accompanied by a decrease in OPG expression [14, 15], leading to a microenvironment that favours osteoclastogenesis. Osteoclast precursor cells accumulate inside the dense inflammatory synovially derived tissue located both at the interface with bone and inside the bone erosions themselves (synovial pannus).
Similar changes are also found in animal models of inflammatory arthritis, described in detail in ‘Critical Processes in Bone Remodelling in Animal Models of Arthritis’. Studies involving these demonstrated the crucial role of osteoclasts in the pathogenesis of articular bone erosion in arthritis induced by adjuvant [16], antigen [17], collagen [18–20], serum transfer [21–23] and TNF [24–26]. By inducing arthritis in osteoclast-free mice, it was shown that these mice are completely protected from bone erosions [21, 24]. Furthermore, RANKL knockout mice are also resistant to inflammation-induced bone loss [21].
Osteoblast Differentiation in Inflammatory Arthritis
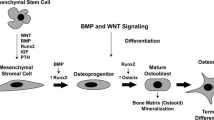
Derived from mesenchymal stem cells, osteoblast precursor cells require up-regulation of the Runx2 [27] and Osterix [28] transcription factors. Maturation to a non-proliferative matrix-producing osteoblast is marked by the expression of alkaline phosphatase, type 1 collagen along with non-collagen proteins such as osteocalcin, osteopontin and bone sialoprotein. Mature osteoblasts are capable of producing a characteristic extracellular collagenous matrix that subsequently becomes mineralised by deposition of hydroxyapatite crystals (reviewed in [29, 30]). Osteoblasts are not only responsible for the synthesis and mineralisation of bone, but they are also able to modulate osteoclast differentiation by stimulating osteoclast differentiation via production of M-CSF and RANKL [31, 32] or inhibiting osteoclast differentiation by OPG production [33, 34]. Other factors involved in osteoblast-osteoclast interaction include paracrine regulators, such as parathyroid hormone (PTH) and prostaglandin E2 (PGE2), which increase RANKL expression by osteoblasts [35, 36]. In addition, bone remodelling relies upon two other key pathways that regulate osteoblast differentiation and function, the canonical Wingless (Wnt) [37, 38] and the tumor growth factor-beta (TGF-β)/bone morphogenetic protein (BMP) [39] pathways.
As described in the previous section, osteoclasts are the principal cell type responsible for bone loss in RA [21, 40]; however, other cell types, including synovial fibroblasts and macrophages, might also contribute to bone erosion [41, 42]. Nevertheless, osteoblast differentiation and function has recently been suggested to be abnormal at sites of focal bone erosion in RA.
A murine study using the K/BxN model of serum transfer arthritis showed that the rate of bone formation is similar in arthritic and non-arthritic bone, suggesting that in RA, increased osteoclast resorption activity is not counterbalanced by osteoblast driven bone formation. Furthermore, within the arthritic bone, mineralisation of the newly formed bone in areas adjacent to sites of inflammation is reduced compared with bone surfaces adjacent to normal bone marrow. This suggests that inflammatory tissue impairs osteoblast activity [43]. Using the same K/BxN model of serum transfer arthritis, inflammation and bone erosion were induced and inflammation was then allowed to resolve completely. Proceeding inflammation resolution a significant increase in bone formation at previous inflammation-bone interfaces is observed, correlating with altered synovial expression of Wnt signalling components that favour anabolic signalling [44].
The Wnt signalling antagonist Dickkopf-1 (DKK-1) is increased in the mouse model overexpressing human TNF-α (hTNF.Tg). Prophylactic treatment with an antibody against DKK-1 prevented focal erosion, an effect also due in part to up-regulation of OPG expression in synovial tissue, suggesting that DKK-1 can act to inhibit bone formation [45]. In line with this, DKK-1 levels are higher in the sera of patients with RA than in either normal controls or patients with ankylosing spondylitis. Levels of DKK-1 correlate with disease activity in RA [46].
Other factors such as hypoxia and reduced pH in the arthritic bone microenvironment also play a role in impaired bone formation [47, 48].
Although uncommon, repair of bone erosions through formation of new bone has been described in patients undergoing conventional disease-modifying anti-rheumatic drug (DMARDs) therapy. Moreover, the repair occurred predominantly in patients with low disease activity at the time of follow-up [49].
Critical Processes in Bone Remodelling in Animal Models of Arthritis
Disruption of the balance between bone resorption and formation is observed in several rheumatic diseases. Pro-inflammatory cytokines, as IL-1, IL-6, IL-17 and TNF-α, are able to stimulate osteoclast differentiation by expansion of the osteoclast precursor pool, up-regulation of RANKL expression in osteoblasts and/or in synovial fibroblasts and by synergism with RANKL itself, normally resulting in bone loss [26, 50, 51]. Pro-inflammatory cytokines, such as IL-1, IL-6 and TNF-α, can be found in high concentrations in the synovial fluid and tissues of RA patients. Chronic inflammation in RA leads to focal articular bone erosions within inflamed joints, as well as generalised osteoporosis. This bone loss progresses throughout the disease, correlates with disease severity and if untreated, it might progress to joint deformity and fractures [52]. Pro-inflammatory cytokines, secreted predominantly by macrophages, synovial fibroblasts and lymphocytes within the inflamed synovium and pannus, mediate the erosive process by enhancing osteoclast differentiation and activity.
Animal models provide the opportunity for investigating the pathogenesis of RA, to unravel the mechanism related with bone resorption/formation, to identify drug targets and test potential therapeutics. These models include induced and genetically manipulated arthritis models. In this section, we will describe some of the most commonly used arthritis models (Table 1) and discuss the role of critical cytokines in each model (Table 2).
Collagen-Induced Arthritis
The collagen-induced arthritis (CIA) model was first described in 1977, when Trentham and his colleagues reported that immunisation of rats with an emulsion of human, chick or rat type II collagen (CII) in complete Freund’s adjuvant (CFA) or incomplete Freund’s adjuvant (IFA) resulted in the development of an erosive polyarthritis associated with an auto-immune response against cartilage [53]. Others also described similar protocols for induction of CIA in mice [71] and in non-human primates [72]. One of the aspects of the immune response in this model is the production of CII-specific antibodies [73]. As in human RA, mice immunised with CII also produce rheumatoid factor [74]. The histology of CIA resembles RA and it is possible to observe cell infiltrate in synovial tissue and destruction of bone and cartilage (Table 1).
CIA susceptibility is linked to the expression of specific MHC class II molecules, which in mice is referred to as the H-2 complex. Strains expressing H-2q or H-2r are susceptible to CIA [75]. The induction of arthritis in mice of a C57BL/6 (H-2b) background [76] has facilitated the use of gene knockout mice and more recently by the generation of the congenic C57BL/6N.Q strain, which expresses the arthritis-susceptible q haplotype of the MHC class II region [77].
The induction of CIA in mice is mediated by both auto-reactive T and B cells. Antigen-specific T cells are predominantly involved in the induction phase of the disease, supporting the activation of collagen-specific B cells and auto-antibodies. These pathogenic antibodies recognise their endogenous antigen in the joint resulting in complement activation, immune complex formation and triggering of a local inflammatory response including pro-inflammatory cytokines, whereby monocytes, granulocytes and T cells are attracted to the joint cavity (review in [78]) (Fig. 1; Table 2). Several studies demonstrated the importance of T cells in the induction of the disease in the CIA model. Holmdahl et al. showed that administration of CII-specific T cells can induce arthritis in naïve mice [79]. Moreover, mice deficient for CD4+ T cells are less susceptible to CIA than wild-type mice [80].

Cell types and cytokines involved in bone loss and arthritis development in different arthritis mouse models. The arthritis development in the streptococcal cell wall-induced arthritis (SCW) model is mediated by synovial fibroblast and innate immune cells as macrophages, γδ T cells and polymorphonuclear cells (PMN). These cells secrete IL-1, IL-6, IL-23 and TNF-α. No bone erosion is observed in this acute joint inflammation model. In the antigen-induced arthritis (AIA) model, macrophages, B cells and T cells are responsible for disease induction. In AIA, the main cytokines involved are IL-17A, IL-23 and TNF-α. In this model, mild (1) to moderate (2) bone erosion can be observed. The AIA flare-up model is driven mainly by antigen-specific memory T cells that activate synoviocytes leading to synovial inflammation within hours followed by joint destruction. The collagen-induced arthritis (CIA) is an erosive polyarthritis model associated with an auto-immune response against cartilage. CIA is mediated by auto-reactive T and B cells directed against type II collagen. B cells can be differentiated in plasma cells that produce auto-antibodies. Immune complex-mediated immune activation and complement play a role in the progression of the disease. In addition, many pro-inflammatory cytokines such as IL-1, IL-6, IL-17A, IL-22, IL-23 and TNF-α play a role in the development and/or progression of CIA. The degree of bone erosion can vary between mild (1) and severe (3)
Antigen-Induced Arthritis
Antigen-induced arthritis (AIA) is seen after intra-articular injection of protein antigen (e.g. methylated bovine serum albumin) into the knee joints of animals that have previously been immunised with the same antigen [81]. A swelling of the injected knee appears within the following days. The histopathological appearance of AIA bears similarities to RA, including synovial lining layer hyperplasia, perivascular infiltration with lymphocytes and plasma cells, lymphoid follicles, pannus and cartilage erosions. The erosiveness is related to the ability of the antigen to bind cartilage and is also dependent of the immunisation strategy used [58] (Fig. 1). However, unlike RA, AIA is a monoarticular disease that affects only the injected joints. When induced as described above, AIA is considered to be an acute disease model where the initial inflammation subsides after approximately 1–3 weeks, depending on the immunisation strategy [58]. Moreover, by locally re-injecting a small amount of antigen, a reactivation of arthritis, called flare-up reaction can be induced. While acute AIA involves besides T and B cells also macrophages and immune complex formation, flare-up AIA is predominantly mediated by memory T cells [68] (Fig. 1; Tables 1 and 2). Susceptibility to AIA is not MHC class II restricted, and this makes the model useful for studies involving transgenic and gene knockout mice.
Streptococcal Cell Wall-Induced Arthritis
A single systemic injection of group A streptococcal cell wall (SCW) peptidoglycan-polysaccharide complexes results in the development of a chronic erosive polyarthritis in genetically susceptible female Lewis rats [82]. A variation of this model involves the local injection of SCW directly into the ankle joint in rats or knee joint in mice potentially followed by systemic challenge with SCW. The initial intra-articular application of SCW causes a local, acute TLR2/NOD2-driven inflammatory reaction. This acute phase is clinically evident within 24 h after the injection, and it is histologically characterised by neutrophil and monocyte infiltration into the synovium (Fig. 1; Tables 1 and 2). The systemic (intravenous) challenge with SCW causes pronounced reactivation of the arthritic response in the joint [55]. This model of ‘reactivation’ results in an immunologically mediated inflammatory response that mimics the exacerbation of a chronic arthritic course. The chronic erosive arthritic stage is related to effector T cell activation and the dysregulation of macrophage function, characterised by accumulation of mononuclear cells with release of pro-inflammatory cytokines and erosive destruction of subchondral and peri-articular bone and cartilage [57, 83, 84]. Local challenge with SCW leads to a reactivation of arthritis and a flare-up reaction can be induced; this results in chronic inflammation, bone erosion and cartilage damage without involvement of adjuvant such as CFA.
Spontaneous Models
K/BxN
K/BxN mice, which express both the T cell receptor transgene KRN, a TCR specific for a peptide from bovine pancreatic ribonuclease and the MHC class II molecule Ag7, develop arthritis [85]. In this model, the T cell receptor recognises the ubiquitous cytoplasmic enzyme glucose-6-phosphate isomerase (GPI) and provokes, through B cell differentiation and proliferation, high levels of anti-GPI antibodies. These antibodies are directly pathogenic upon transfer and appear to recognise endogenous cationic GPI, which seems to associate preferentially with the cartilage surface, in a complement- and FcγR-dependent manner [60, 86, 87]. This process is known as K/BxN serum transfer model.
SKG and gp130
Sakaguchi et al. [61] described a model of arthritis that occurred spontaneously (SKG). These mice harbour a mutation of the gene encoding the Src homology 2 domain of z-associated protein 70 (ZAP70), a key signalling molecule in T cells [61]. The aberrant T cell receptor signalling, as a result of aberrant ZAP-70, leads to a failure in thymic deletion and the emergence of auto-immune T cells.
Mice with a homozygous mutation in the gp130 IL-6 receptor subunit spontaneously develop arthritis due to enhanced gp130-mediated STAT3 activation and develop lymphocyte-mediated RA-like joint disease [62].
IL-1Ra−/−
Transgenic IL-1a overexpression was shown to induce chronic, destructive arthritis [88]. On the other hand, IL-1 receptor antagonist (IL-1Ra)-deficient mice spontaneously develop arthritis due to an increased sensitivity to IL-1. Elimination of IL-1Ra results in a model of arthritis dependent on T cells. When generated on a BALB/c (but not on C57BL/6), this mouse spontaneously develops a polyarthritis. These mice showed synovial and peri-articular inflammation, with invasion of granulation tissue and articular erosion. It also generates antibodies against type II collagen, IgG and (unlike the SKG mouse) double-stranded DNA but not IgM rheumatoid factor [63]. Disease is evident as early as 5 weeks of age, with morbidity exceeding 80 % by 8 weeks.
hTNF.tg
Human TNF-α transgenic (hTNF.tg) mice, which possess a modified human TNF-α gene, that resulted in pronounced TNF-α overexpression developed chronic inflammatory arthritis, from which no joints are spared, with a 100 % incidence. The histopathology in this model is characterised by hyperplasia of the synovium, inflammatory infiltrates in the joint space, pannus formation and cartilage and bone destruction. hTNF.tg mice are used as a model of RA for the inflammatory and bone destruction phases since this arthritis bypasses the adaptive initiation phase of arthritis [64].
BXD2
The BXD2 recombinant inbred mouse generated by inbreeding the intercrossed progeny of C57BL/6J and DBA/2J mice for more than 20 generations develop spontaneous erosive arthritis [65]. This progresses as the mice age. These mice show hallmarks of autoimmune disease, including increasing titers of circulating immune complexes and correlate with joint disease [66].
DNase II−/−IFN-IR−/−
A large amount of chromosomal DNA is degraded during programmed cell death and definitive erythropoiesis. DNase II is an enzyme that digests the chromosomal DNA of apoptotic cells and nuclei expelled from erythroid precursor cells after macrophages have engulfed them. DNase II−/− mice die as embryos as a result of the constitutive production of interferon-β (IFN-β), and this lethality can be rescued by a deficiency of the IFN-I receptor (IFN-IR) gene. DNaseII−/−IFN-IR−/− mice develop an inflammatory polyarthritis associated with high levels of anti-cyclic citrullinated peptide antibody and rheumatoid factor. Inadequate degradation of mammalian DNA from erythroid precursors and apoptotic cells by macrophages lead them to produce TNF-α, which activates synovial cells to produce various cytokines, leading to the development of chronic polyarthritis [67].
Pro-inflammatory Cytokines in Bone Remodelling
TNF-α
TNF-α is expressed mainly by macrophages and synovial lining cells, as well as by activated T cells, within the RA-inflamed joint [89]. Within the RA-inflamed joint, TNF-α is a dominant pro-inflammatory cytokine and is able to induce the production of other pro-inflammatory cytokines (IL-1, IL-6 and IL-8) [90].
TNF-α acts in synergy with RANKL and prompts robust osteoclastogenesis by osteoclast precursors, via TNF type 1 receptor (TNFr1) and deletion of TNFr1 abrogates this response [91]. TNF-α promotes the survival of differentiated mature osteoclasts [92]. On the other hand, interactions between TNF-α and IL-1 result in osteoclastic activity independently of RANKL [93].
TNF-α induces expression of the stromal cell product M-CSF, which maintains survival and longevity of osteoclast precursors. The fact that M-CSF plays a central role in TNF-induced osteoclastogenesis is confirmed by the capacity of an antibody directed against the M-CSF receptor, c-Fms, to completely arrest pathological osteoclastogenesis and bone resorption, inflammation in a serum-transfer arthritis model [94].
Osteoclast-associated receptor (OSCAR) is a key receptor in the process of osteoclast differentiation, expressed by osteoclasts at the erosion front and by mononuclear cells around synovial microvessels. Serum levels of soluble OSCAR were lower in RA patients than in healthy controls. Moreover, monocytes with high OSCAR expression exhibited an enhanced potential to differentiate into osteoclasts. TNF-α induces OSCAR expression in monocytes of RA patients, facilitating their differentiation into osteoclasts and bone resorption [95].
The relevance of TNF-α for arthritic bone destruction has been demonstrated in several experimental models and was finally confirmed by clinical trials. As discussed in ‘hTNF.tg’, hTNF.tg mice developed chronic inflammatory arthritis, with hyperplasia of the synovium, inflammatory infiltrates in the joint space, pannus formation and cartilage and bone destruction [64]. Blocking either TNF-α (using anti-TNF-α antibodies) or RANKL (using OPG.Fc) signalling in this murine model results in reduced osteoclastogenesis and bone erosion [25]. Low-dose TNF-α-inhibiting therapy, with adalimumab, also reduces bone erosions by reducing the number of circulating and joint-invading osteoclast precursors. This effect is uncoupled from its anti-inflammatory action [26]. On the other hand, treatment with exogenous TNF-α increase numbers of CD11b1 osteoclast precursor cells [96]. In CIA, the application of TNF-specific neutralising antibodies reduced disease activity and bone damage [97]. In mice with a FcγRIIB-deficient C57BL/6 background, TNF-α is indispensable while IL-17 is dispensable in the pathogenesis of arthritis. In this model, TNF-α mediates the increase in frequency of osteoclast precursors in circulation and their migration into the joints. TNF-α also decreases OPG expression, leading to up-regulated osteoclastogenesis associated with severe cartilage and bone destruction [98].
Although the observation in the murine models is largely mediated by the anti-inflammatory effects of TNF inhibition, direct reduction of osteoclast-mediated bone loss and augmentation of osteoblast-mediated bone formation are potential mechanisms by which TNF-α inhibition reduces structural damage in RA.
The efficacy and safety of the TNF-α antagonists infliximab, etanercept, adalimumab, golimumab and certolizumab in RA patients were demonstrated in several clinical studies, and these drugs are now frequently used in clinical practice [99] (Fig. 2).

New anti-rheumatic drugs targeting osteoclastogenesis and bone loss. RANKL is a key cytokine in osteoclastogenesis and bone resorption. Downregulation of RANKL expression by mesenchymal and T cells can be achieved by targeting IL-6, TNF and IL-1. Also, depleting B cells with an antibody targeting CD20 can result in lower RANKL expression. RANKL itself can be blocked directly by denosumab. Small-molecule tyrosine kinase inhibitors can be used to block T cell activation. Abatacept can interfere with osteoclast formation by targeting CD80 and CD86. IL-17A acts on synovial fibroblast and on osteoblast, influencing osteoclast differentiation and activity. Odanacatib inhibits cathepsin K, preventing collagen and other matrix protein degradation during bone resorption
IL-1
The pro-inflammatory cytokine IL-1 belongs to a family of cytokines that includes IL-1α, IL-1β, IL-18 and the recently called cytokine IL-36 and IL-37. In RA joints, activated macrophages and synovial fibroblasts are sources of IL-1 production [88, 100]. IL-1 signals through its cognate receptor, IL-1R1 complexed with IL-1RAcP. IL-1 receptor antagonist (IL-1Ra) is a soluble protein that competes with IL-1 for binding to the IL-1R1 receptor [101].
IL-1 stimulates bone resorption through a primary action on osteoblasts, which are induced by IL-1 to transmit a short-range signal that stimulates osteoclastic bone resorption [102]. In vitro, IL-1 promotes the fusion of osteoclast precursors [103] and prolongs the survival of mature osteoclasts [104]. IL-1 also has an important role in osteoclast activation [105]. IL-1 is able to exacerbate osteoclastogenesis by cooperating with RANKL and M-CSF, while TNF-α is not involved in this IL-1-stimulated osteoclast differentiation pathway [106].
Data from various animal models suggest an important role for IL-1 in the pathogenesis of inflammatory arthritis. Overexpression of IL-1α or IL-1β or deficiency of the soluble IL-1Ra in arthritis models resulted in the development of disease, which is associated with bone and cartilage destruction [63, 88, 107, 108]. Mice deficient of IL-1R1 do not develop arthritis after KRN serum transfer [109].
Blocking TNF-α and IL-1 in the hTNF.tg mouse model leads to an almost-complete remission of disease, suppression of osteoclast differentiation, synovial inflammation, bone erosion and cartilage. Articular changes caused by chronic overexpression of TNF-α are not completely neutralised by monotherapies that target TNF-α, IL-1, or RANKL. Blocking TNF-α alone reduces the numbers of osteoclasts and bone erosion; however, recombinant IL-1Ra treatment is not as effective as TNF-α treatment [110].
In hTNF.tg mice that are deficient for IL-1, cartilage destruction was completely blocked and bone erosion and osteoclast formation partly reduced despite the presence of synovial inflammation. Protection of cartilage is based on the loss of IL-1 on hematopoietic cells. This study suggested that TNF-mediated cartilage damage is completely and TNF-mediated bone damage is partially dependent on IL-1, suggesting that IL-1 is a crucial mediator for inflammatory cartilage and bone degradation [111]. Moreover, lack of IL-1 completely reversed the increase osteoclast formation and bone resorption in hTNF.tg mice and the increase levels of RANKL in these mice. Structural parameters and osteoclast and osteoblast numbers were indistinguishable from wild-type mice [112].
IL-1Ra exposure or genetic ablation of IL-1RI ablated TNF-stimulated osteoclastogenesis, in vivo and in vitro. IL-1 directly targeted osteoclast precursors and promoted the osteoclast phenotype in a TNF-independent manner in the presence of permissive levels of RANKL. IL-1 is able to induce RANKL expression by stromal cells and directly stimulate osteoclast precursor differentiation. Thus, IL-1 mediates the osteoclastogenic effect of TNF by enhancing stromal cell expression of RANKL and directly by stimulating differentiation of osteoclast precursors [113].
In contrast to these in vivo and in vitro data, targeting IL-1 has not yet provided powerful therapeutics for the treatment of RA [114] (Fig. 2).
IL-6
IL-6 belongs to the family of cytokines that includes IL-11, leukemia inhibitory factor (LIF) and oncostatin M (OSM). The signalling pathway of IL-6 includes two molecules, a specific receptor for IL-6 and a cell-surface glycoprotein called gp130 as a signal transducer [115]. IL-6 can signal via its membrane bound IL-6 receptor but also via trans-signalling, meaning that IL-6 forms a complex with soluble IL-6 receptor. This complex can bind to gp130 on the cell membrane of cells trigging IL-6 signalling. IL-6 singling is possible then on cells lacking IL-6 receptor on the membrane. IL-6 is a pro-inflammatory cytokine with pleiotropic biological activities. This cytokine is produced by a variety of cell types in the inflamed RA bone microenvironment including macrophages, fibroblast-like synoviocytes and chondrocytes [116]. IL-6 contributes to the induction and maintenance of the autoimmune process through B cell modulation and Th17 cell differentiation. In patients with RA, synovial fluid levels [117, 118] and circulating levels of IL-6 [119] are increased. Moreover, in synovial fluid IL-6 soluble receptor levels correlate with progressive joint damage in RA [120].
Several studies tried to elucidate the role of IL-6 in bone formation/resorption. Kurihara et al. demonstrated that IL-6 enhances osteoclast-like multinucleated cell formation in long-term human bone marrow cultures by inducing IL-1β release [121]. IL-6 soluble receptor triggers osteoclast-like multinucleated cell formation by IL-6, suggesting that locally or systemically produced soluble IL-6 receptor is involved in IL-6-mediated osteoclast recruitment and osteoclastic bone resorption [122]. More recently, it was demonstrated that IL-6 regulates osteoclast differentiation by modulating its interaction with the soluble IL-6 receptor complex expressed by osteoblast lineage cells, leading to an up-regulation of cyclooxygenase (COX)-2-dependent prostaglandin E2 (PGE2) synthesis. This, in turn, up-regulates RANKL expression while down-regulating OPG expression, resulting in enhanced osteoclastogenesis [36]. In vitro, blocking of IL-6 receptor reduced osteoclast formation in mouse monocyte cells stimulated with either RANKL or RANKL plus TNF-α [123]. IL-6 receptor expression increases during in vitro osteoblast differentiation and IL-6 function as a differentiation regulator of preosteoblast cells but as an apoptosis initiator in more mature cells [124].
Mice overexpressing IL-6 develop osteopenia with severe alterations in cortical and trabecular bone microarchitecture, as well as uncoupling of bone formation from resorption, with decreased osteoblast and increased osteoclast number and activity [125]. On the other hand, mice deficient in IL-6 are protected against inflammation and bone destruction in an AIA model [126, 127]. Moreover, IL-6 is an important mediator of bone destruction in AIA once it regulates T cell production of key osteoclastogenic cytokines and inflammation-induced bone marrow osteoclast differentiation [128]. Mice lacking IL-6 on the DBA/1J are completely protected from CIA, accompanied by a reduced antibody response to type II collagen and the absence of inflammatory cells and tissue damage in knee joints. However, removal of IL-6 in hTNF.tg mice does not affect the development of the disease, showing that IL-6 plays a crucial role in the development of autoimmune CIA but not in the TNF-α-dependent inflammatory arthritis (Table 2) [129]. Blocking the IL-6 receptor in a murine CIA model delayed the onset of inflammation and reduced joint destruction. In addition, administration of IL-6 receptor neutralising antibodies at the time of CIA induction completely abolished the inflammatory response indicating that IL-6 plays an important role in the initiation of arthritis [130]. Furthermore, administration of blocking antibodies against the IL-6 receptor in hTNF.tg mice did not inhibit inflammation as described by Alonzi et al. However, it significantly reduced osteoclast formation and bone erosion [123], by reducing the number of osteoclast precursors in bone marrow [131]. In several studies, performed in different arthritis mouse models, blocking IL-6 receptor was very successful. As a result of these studies, a humanised anti-IL-6 receptor monoclonal antibody, tocilizumab [132], entered into clinical trials and it has been shown to be an effective treatment in several large phase III clinical trials in RA with rapid and sustained improvement in disease activity, reducing radiographic joint damage and improving physical function [133]. Since then several agents binding either IL-6 receptor including sarilumab [134], ALX-0061 [135] or IL-6 sirukumab [136], clazakizumab [137], olokizumab [138] or MEDI5117 [139] were developed and are now in clinical trials (Fig. 2).
IL-17
IL-17 family of cytokines consists of six members, IL-17A-IL-17F. IL-17A is not only produced by Th17 but also by other cells such as, CD8+ T cells, CD4−CD8− γδ T cells, natural killer (NK) cells, innate lymphoid cells (ILC3) and potentially by mast cells and neutrophils.
The presence of IL-17-producing cells, in particular CD4+IL-17A+ cells, in blood and synovial fluid of RA patients has been correlated with inflammatory activity [140]. IL-17A levels in synovial fluids are significantly higher in RA patients compared with osteoarthritis patients [141].
Th17 cells besides IL-17A are also able to produce other cytokines such as IL-17F, IL-21, IL-22 and GM-CSF. Th17 cells can be differentiated from naïve CD4+ T cells by TGF-β and IL-6 or IL-21. The pro-inflammatory cytokine IL-23 is not required for the polarisation of Th17 cells but is crucial in their maturation and stability [142]. The retinoic acid-related orphan nuclear hormone receptor (ROR)γt acts as a lineage-specific transcription factor [143]. Th17 cells express the chemokine receptor CCR6. Inflamed synovial cells in both SKG arthritis and human RA also produce CCL20, the ligand for CCR6, facilitating the migration of arthritogenic Th17 cells to inflamed joints [144]. As mentioned above, Th17 cells are not the only cell type able to produce IL-17. Also, γδ T cells and potentially neutrophils are able to produce IL-17A. Equal numbers of CD4+ Th17 and IL-17 producing γδ T cells are found in the joints of CIA mice, and in vitro, both populations similarly induce osteoclastogenesis. However, individual depletion and adoptive transfer studies revealed that, in vivo, Th17 cells dominated with regard to bone destruction [50].
In mouse, Th17 cells directly contribute to bone damage as they express receptor activator of RANKL and have the ability to activate RANK expression on bone-resorbing osteoclasts [144]. IL-17 induces differentiation of osteoclast progenitors into mature osteoclasts in vitro, by first acting on osteoblasts, which stimulates both COX-2-dependent PGE2 synthesis and osteoclast differentiation factor gene expression [141]. Treatment of human monocytes with only IL-17 induces osteoclastogenesis. This effect could be inhibited by adding OPG and infliximab, suggesting that TNF-α and RANKL are, at least in part, responsible for the IL-17-induced osteoclastogenesis [145]. IL-17A gene transfer, using minicircle DNA, induced the expansion of IL-17RA+CD11b+Gr1low osteoclast precursors. The expansion preceded noticeable joint inflammation [146]. In CIA mice overexpression of IL-17, using a viral vector, in the knee joint promotes osteoclastic bone destruction by enhancing RANKL expression and up-regulating the RANKL/OPG ratio in the synovium. Systemic OPG treatment prevents joint damage induced by local overexpression of IL-17 [147].
Interestingly, in mice lacking IL-17, CIA was markedly suppressed. In this model IL-17 is responsible for the priming of collagen-specific T cells and collagen-specific IgG2a production [148]. Lubberts et al. used anti-IL-17 antibody therapy to demonstrate that IL-17 plays a role not only in early stages of CIA but also during disease progression. Anti-IL-17 antibodies therapy suppress joint inflammation and prevent cartilage and bone destruction. Furthermore, treatment reduces IL-6 levels and the number of synovial IL-1-positive and RANKL-positive cells [149]. Neutralisation of IL-17, in a T cell-mediated model of AIA, prevents joint swelling and significantly suppress joint inflammation, cartilage proteoglycan depletion and reduces bone erosions. Synovial expression of cathepsin K, RANKL, IL-1β and TNF-α was suppressed by blocking IL-17 [59]. Transfer of IL-17-deficient donor bone marrow into CIA DBA/1J mice inhibits development and severity of clinical arthritis, due to reduction in the secretion of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6 [150]. Mice lacking IL17-RA were similarly to IL-23-deficient mice completely protected against CIA [151] and from bone pathology. Mice deficient in IL-17 or IL-23 present less bone destruction and osteoclast formation in a lipopolysaccharide (LPS)-induced model of inflammatory bone destruction, which is not induced by an autoantigen but is T cell dependent. This suggests that IL-23-stimulated proliferation of Th17 cells plays a pivotal role in inflammatory bone destruction by inducing RANKL through an IL-17 effect on mesenchymal cells [152].
The development of spontaneous arthritis is completely suppressed in the progeny of IL-1Ra-deficient mice when crossed with IL-17-deficient mice, indicating that IL-17 and IL-1 are necessary for this spontaneous development of arthritis [153].
Several blockers for IL-17 pathway are been evaluated in clinical trials as secukinumab, a fully human IgG1k anti-IL-17A monoclonal antibody [154, 155], ixekizumab (a humanised IgG4 anti-IL-17A monoclonal antibody) [156, 157] or brodalumab, a fully human IgG2 anti-IL-17RA monoclonal antibodies [158, 159] (Fig. 2). However, none of these are yet approved for the treatment of RA.
IL-23
Interleukin-23 is a heterodimeric cytokines belonging to the IL-12 family. IL-23 is a pro-inflammatory cytokine composed of two subunits, p19 and p40. The p40 subunit is shared with IL-12. IL-23 is mainly expressed by macrophages, monocytes and DCs. The IL-23 receptor (IL-23R) is composed of IL-12Rβ1 combined with a specific chain, IL-23R and is found on T cells, NK cells, macrophages and DCs (reviewed in [160]).
Several studies suggested a role of IL-23 in osteoclastogenesis. Chen et al. demonstrated that IL-23 drives osteoclast development by acting directly on myeloid precursor cells and indirectly by stimulating RANKL production in osteoblasts [161].
IL-23 induced osteoclastogenesis in cultures of human peripheral blood mononuclear cells (PBMCs) in the absence of osteoblasts or exogenous RANKL. This process was inhibited by osteoprotegerin, anti-IL-17 antibody or TNF inhibition, indicating the involvement of RANKL, IL-17 and TNF in IL-23-induced osteoclastogenesis [162]. Recently, a novel pathway involving IL-23 in myeloid cells was identified as a major mechanism for the stimulation of osteoclastogenesis in inflammatory arthritis. In human PBMCs, IL-23 induces the expression of MDL-1, a PU.1 transcriptional target during myeloid differentiation, which orchestrates osteoclast differentiation through activation of DNAX activating protein of 12 kDa and its ITAMs. IL-23-elicited osteoclastogenesis is independent of the RANKL pathway and uses a unique myeloid DNAX activating protein of 12-kDa-associated lectin-1+/DNAX activating protein of 12 kDa+ cell subset [163].
IL-23 induced RANKL expression by CD4+ T cells; however, the effects of IL-23 on osteoclastogenesis via T cells are less clear because both stimulatory and inhibitory effects have been described [164–166].
IL-23p19-deficient mice are protected from CIA. IL-23 gene-targeted mice do not develop clinical signs of disease and are completely resistant to the development of joint and bone pathology [58]. In vivo blockade of endogenous IL-23 activity by treatment with anti-IL-23 antibody attenuates CIA in rats by preventing both inflammation and bone destruction [162]. In mouse, administration of anti-IL-23p19 before clinical CIA onset results in a milder disease [58].
Systemic IL-23 exposure using hydrodynamic delivery of IL-23 minicircle DNA in vivo induced chronic arthritis, severe bone loss and myelopoiesis in the bone marrow and spleen, which resulted in increased osteoclast differentiation and systemic bone loss [167].
Clinical studies performed with antibodies targeting the p40 subunit of IL-12 and IL-23 (ustekinumab and briakinumab) or the p19 subunit of IL-23 (tildrakizumab and guselkumab) have been performed in patients with psoriasis, ankylosing spondylitis and psoriatic arthritis [168]. However, no studies were yet described for the application of these in the treatment of rheumatoid arthritis (Fig. 2).
Bone Repair: New Tools, Possible Interventions and Perspectives
Endochondral Ossification
In addition to animal models of inflammatory arthritis, there is a high interest in humanised in vivo models to study the link between the immune system and bone remodelling. Besides bone erosion, two of the most important factors common to many arthropathies are insufficient bone repair and excessive bone formation. Interestingly, both of these processes involve the process of endochondral ossification [169, 170], which is insufficient in one instance and undesirable in the other. In order to study the roles of specific cytokines on the various stages of bone erosion, insufficient bone formation and osteophyte formation new in vivo models are needed. In the field of regenerative medicine, much interest is now focused on the harnessing of this process to generate new bone and repair large bone defects, often mediated by mesenchymal stem cells (MSCs) [171–176]. This model could be harnessed to determine the effects of specific cytokines on all stages of bone formation and erosion in a controlled humanised model of bone formation. Indeed, the important role of TNF-α in endochondral bone formation using fracture healing models has already been established [177–179]. In this new model of endochondral ossification, MSCs can be chondrogenically primed in vitro in various biomaterials or as scaffold-free pellets. The early stages of condensation and chondrogenic differentiation in the presence of various cytokines can thus be assessed in vitro. Upon subcutaneous implantation of these constructs, they undergo complete endochondral ossification, becoming vascularised and forming marrow-containing bone. To date, this model has been used to demonstrate the effects of IL-1β on the various in vitro and in vivo stages of bone formation [180, 181]. We propose that the cytokines discussed in this review can also be combined in such a system to reproducibly determine their role in early chondrogenesis, bone formation, cell migration, vascularisation, marrow formation and ultimately, bone remodelling and erosion. This model represents an excellent system to analyse the specific effects of cytokines implicated in the sub-optimal repair of bone erosions in RA and the unwanted formation of osteophytes in spondyloarthritis (SpA) and osteoarthritis (OA).
Novel Therapeutic Targets
Current treatment strategies for RA include nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, disease-modifying anti-rheumatic drugs (DMARDs), such as methotrexate, hydroxychloroquine, leflunomide or sulfasalazine, and biologic response modifiers (‘biologicals’). The latest have specific mechanisms of action, including inhibiting the action of individual cytokines, blocking cell-cell interactions and depleting certain cell types. The therapeutic goals of all these treatments are the clinical remission as well as structural joint protection, prevention of erosion formation, articular cartilage loss and peri-articular bone loss/osteoporosis in and around the affected joints. The suppression of inflammation and prevention of bone loss, are closely associated with each other, since as discussed in previous sections there are clear evidences linking both. Inflammatory RA is associated with reduced bone density and increased risk of fragility fractures/osteoporosis. Furthermore, suppression of inflammation halts generalised bone loss, thereby preventing increased fracture risk in RA patients [182].
In ‘Pro-inflammatory Cytokines in Bone Remodelling’, we mentioned the biologicals available that target TNF-α, IL-1, IL-6, IL-17 and IL-23 cytokine pathways. Now, we would like to discuss new treatment options that have as a target: B cells (rituximab), T cell costimulation (abatacept), JAK (tofacitinib), RANKL (denosumab) and cathepsin K (odanacatib).
B Cell Depletion (Rituximab)
Rituximab is a monoclonal chimeric anti-CD20 antibody that recognises a determinant expressed on intra-medullary pre-B- to B-memory stage lymphocytes, this antibody is used as a B cell depletive therapy [183] (Fig. 2).
The efficiency of rituximab was demonstrated in patients who had responded inadequately to treatment with a TNF-α antagonist in combination with background methotrexate therapy [184] and in those who had responded inadequately to methotrexate therapy [183]. Rituximab improved the signs and symptoms of RA 24 weeks after the start of the treatment. Among patients that do not respond to TNF-α antagonist therapy, progression of bone erosion and joint space narrowing on plain radiographs was slower in patients treated with combination treatment with rituximab compared with methotrexate alone in [185, 186].
In RA patients treated with rituximab, there was a decrease of RANKL expression in the synovium and a decrease in RANK-positive pre-osteoclasts [187] (Fig. 2). Moreover, rituximab treatment reduced levels of sera markers of bone resorption and bone formation. This reduction correlates with the decrease in disease activity [188].
Concerning the safety profile despite the fact that rituximab does not deplete fully mature plasma cells, repeated administration of the biologic agent frequently induces a reduction of immunoglobulins, particularly IgG, which may carry an increased risk of infection. Reactivation of occult hepatitis B infection has been reported in patients with RA treated with Rituximab. Moreover, rare occurrence of a usually fatal central nervous system demyelinating disease, progressive multifocal leukoencephalopathy has also been reported [189].
Inhibition of T Cell Costimulation (Abatacept)
Abatacept is a recombinant fusion protein comprising the extracellular domain of human cytotoxic T lymphocyte antigen 4 (CTLA4) and a fragment of the Fc domain of human IgG1 that selectively modulates the CD80/CD86:CD28 co-stimulatory signal required for full T cell activation. CTLA4 (CD152) is a surface protein on T lymphocytes, which negatively regulates T cell activity [190].
In rats with CIA, abatacept treatment results in reduced synovitis and osteoclasts number in the synovium [191]. In a hTNF.tg model, CTLA4 directly inhibit the formation of osteoclast through binding to CD80/86 on monocytes and thereby preventing these cells to develop to osteoclasts [192].
In patients with active RA inadequately responsive to a TNF antagonist, abatacept in combination with background DMARD therapy was more effective than background DMARD therapy alone in reducing the signs and symptoms of RA and improving physical function (Fig. 2). [193]. The combination of Abatacept and methotrexate was also more effective than methotrexate monotherapy in controlling the signs and symptoms of RA and in improving physical function among patients with RA active despite methotrexate therapy [194]. Moreover, in combination with background methotrexate therapy, abatacept slowed significantly the progression of bone erosion and joint cartilage space narrowing on plain radiographs [194, 195]. Abatacept treatment of inadequate responders to methotrexate reduces the progression of erosions in RA patients compared with patients treated with placebo [196].
The safety profile of abatacept is favourable, particularly in relation to serious (including opportunistic) infections in comparison with anti-TNF agents [197]. The most common infections reported are pneumonia, urinary tract infection and cellulitis. Antibodies to abatacept developed in ≤3 % of patients, with no association between immunogenicity and adverse events [198].
JAK Inhibitors (Tofacitinib)
The binding of cytokines, as for example IL-1, IL-2, IL-6, IL-8, GM-CSF and IL-10 to their receptors activates the Janus kinase (JAK)-signal transducer and activator of transcription (STAT), which further translocate to the nucleus and regulate gene expression. The JAK family comprises four members, namely JAK1-3 and tyrosine kinase 2 (TYK2) [199].
Studies performed by Changelian P. team demonstrated that administration of tofacitinib (CP-690,550), an inhibitor JAK3 and JAK1, in the two arthritis animal models CIA and adjuvant-induced arthritis, reduced the clinical and histological manifestations of joint inflammation including bone and cartilage damage [200]. Moreover, similar experiments performed by La Branche et al., using an adjuvant-induced arthritis model showed that tocacitinib decreases edema, inflammation and suppress osteoclast-mediated structural damage to arthritic joints, being this secondary to a decrease in RANKL production [201]. Tofacitinib reduces metalloproteinase and interferon-regulated gene expression in rheumatoid synovial biopsies, and clinical improvement correlates with reductions in STAT1 and STAT3 phosphorylation [202]. Others suggest a role for JAKs in the differentiation of human dendritic cells, once tofacitinib decreases CD80/CD86 expression and T cell stimulatory capability through suppression of type I IFN signalling [203].
In humans, the efficiency of tofacitinib was demonstrated in different clinical trials and is the first small molecule approved by the US Food and Drug Administration (FDA) for the treatment of RA (Fig. 2). Clinical trials demonstrated that tofacitinib has comparable efficacy with TNF-α inhibitor treatment in patients with RA under methotrexate treatment [204], and that tofacitinib in combination with methotrexate is also an effective treatment for TNF non-responders [205]. In patients who had not previously received methotrexate, tofacitinib monotherapy was superior to methotrexate in reducing signs and symptoms of RA and inhibiting the progression of structural joint damage [206]. Moreover, several studies demonstrated that tofacitinib has a sustained efficacy in the treatment of RA patients who have an inadequate response to methotrexate, etanercept, infliximab or adalimumab [69, 207, 208].
Relative to the safety profile of tofacitinib, treatment with this inhibitor has been associated with an increased risk of serious infections, infestations, malignancy and lymphoma. Frequently, patients had an increase in both low-density and high-density lipoprotein cholesterol, alanine aminotransferase, aspartate aminotransferase and serum creatinine levels and an reduction in neutrophil counts [199].
Several other JAK inhibitors are being developed with the aim to treat RA, among them some are in preclinical, as filgotinib (a JAK1/2 inhibitor, GLPG0634) [209], AG490 (a JAK2 inhibitor) [210], CEP-33779 (a JAK2 inhibitor) [211] or already in clinical development, as baricitinib (a JAK1/2 inhibitor, LY-3009104 or INCB028050) [212, 213] and decernotinib (a selective JAK3 inhibitor, VX-509) [214].
Anti-RANKL (Denosumab)
Denosumab (formerly known as AMG162) is a humanised IgG2 monoclonal antibody that inhibits RANKL activity and has been studied in clinical trials in patients with osteoporosis resulting in strongly suppressed bone resorption, by inhibiting the activation, proliferation and survival of osteoclasts [215].
In a study in RA patients, there was no evidence of an effect of denosumab on joint space narrowing or on disease activity. Addition of twice-yearly injections of denosumab to ongoing methotrexate treatment inhibited structural damage evident on magnetic resonance imaging, produced a sustained decrease in markers of bone turnover and resulted in increased bone mineral density, with no increase in the rates of adverse events as compared with placebo [216] (Fig. 2).
Patients receiving methotrexate for erosive RA treated in combination with denosumab were protected against erosion, and not only prevented bone loss but increased hand bone mineral density [217].
The rate of hospitalised infection among RA patients receiving denosumab concurrently with biologic agents for RA was not increased compared with those receiving zoledronate [218].
Cathepsin K Inhibitor (Odanacatib)
Cathepsin K is selectively expressed in osteoclasts and synovial fibroblasts and is secreted to degrade collagen and other matrix proteins during bone resorption.
Mutations in the cathepsin K gene are linked to pycnodysostosis, an autosomal recessive osteochondrodysplasia characterised by osteosclerosis, bone fragility and decreased bone turnover [219].
In RA patients, serum levels of cathepsin K were elevated and correlated with radiological destruction [220]. Moreover, rheumatoid factor positivity is associated with increased joint destruction and up-regulation of cathepsin k gene expression in the peripheral blood in patients treated with methotrexate [221].
The rationale for cathepsin K inhibition is that it will inhibit a protease and affect matrix degradation rather than osteoclast differentiation or apoptosis. This means that the number of osteoclasts and their function should not be reduced. This may allow osteoclast to osteoblast communication that contributes to maintaining bone formation, while suppressing bone resorption.
Indeed, animal studies demonstrated that cathepsin K inhibition reduces bone resorption without inhibiting bone formation [222, 223]. Deletion of cathepsin K in a hTNF.tg model results in reduced area of bone erosion although cathepsin K did not completely protect against inflammatory bone lesions [224]. In CIA mice, inhibition of cathepsin K delays the onset and reduces the disease severity, bone erosion and cartilage degradation [225].
Odanacatib is designed to avoid uptake by lysosomes (Fig. 2). The efficiency of odanacatib in osteoporosis treatment has been evaluated. Both phases 2 and 3 studies have been completed and fracture reduction has been demonstrated [226–228]. The report of the pivotal phase 3 fracture trial called The Long-Term Odanacatib Fracture Trial (LOFT), with the background and study design of fracture end point trial and baseline characteristics of its participants was recently described [226]. The results were presented last year but are still not fully published [229]. In the LOFT trial, odanacatib significantly reduced the risk of three types of osteoporotic fractures compared with placebo in the primary efficacy analysis, and also reduced the risk of the secondary endpoint of clinical vertebral fractures. The rates of adverse events overall in LOFT were generally balanced between patients taking odanacatib and placebo; however, morphea-like skin lesions and atypical femoral fractures occurred more often in the Odanacatib group than in the placebo group.
To our knowledge, there are no clinical studies about the possible application and efficacy of odanacatib for the treatment of RA; however, based in preclinical studies in animals and in the data obtained in the clinical trials for osteoporosis is reasonable to speculate about a positive outcome of the use of this inhibitor in the treatment of RA.
Concluding Remarks
Osteoclasts have been recognised as the critical bone-resorbing cells. Studies in arthritis models have revealed the importance of many pro-inflammatory cytokines that can influence the activity of these cells directly or indirectly. The RANKL-RANK-OPG system is critical in the differentiation and activation of osteoclasts and many of the pro-inflammatory cytokines produced during persistent joint inflammation act via this pathway in the bone-resorbing process. Moreover, new data may indicate also RANKL-independent osteoclastogenesis for TNF-α and IL-23. The different animal models reflect different aspect of the pathogenesis of arthritis also in terms of cellular interactions and mediators involved in osteoclast activation and bone erosion. From these in vivo studies, the list of potential therapeutic options to treat RA inflammation including the bone-resorbing process is growing but the real proof needs to come from clinical trials. Humanised models to study bone formation and erosion might be useful to fulfil the gap between mouse and human studies. A real challenge for the future is how to repair damaged bone after controlling the inflammatory destructive process. New options via modulation of the Wnt signalling and the TGF-β/BMP pathway and perhaps the balance between IL-23, IL-17 and IL-22 might be a promising way to go; however, further research is needed on how to stimulate osteoblast activity after resolving inflammation.
Abbreviations
- AIA:
-
Antigen-induced arthritis
- BMP:
-
Bone morphogenetic protein
- CFA:
-
Complete Freund’s adjuvant
- CIA:
-
Collagen-induced arthritis
- CII:
-
Type II collagen
- COX2:
-
Cyclooxygenase 2
- CTLA4:
-
Cytotoxic T lymphocyte antigen 4
- DKK-1:
-
Dickkopf-1
- DMARDs:
-
Disease-modifying anti-rheumatic drugs
- FDA:
-
Food and drug administration
- GPI:
-
Glucose-6-phosphate isomerase
- hTNF.tg:
-
Human TNF-α transgenic
- IFA:
-
Incomplete Freund’s adjuvant
- IFN-β:
-
Interferon β
- IFN-IR:
-
Interferon I receptor
- JAK:
-
Janus kinase
- LIF:
-
Leukemia inhibitor factor
- LOFT:
-
The long-term odanacatib facture trial
- LPS:
-
Lipopolysaccharide
- M-CSF:
-
Macrophage colony-stimulating factor-1
- MSCs:
-
Mesenchymal stem cells
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- OA:
-
Osteoarthritis
- OPG:
-
Osteoprotegerin
- OSCAR:
-
Osteoclast-associated receptor
- OSM:
-
Oncostatin M
- PBMCs:
-
Peripheral blood mononuclear cell
- PGE2:
-
Prostaglandin E2
- PsA:
-
Psoriatic arthritis
- PTH:
-
Parathyroid hormone
- RA:
-
Rheumatoid arthritis
- RANKL:
-
Receptor activator of nuclear factor-kB ligand
- RORγt:
-
Retinoic acid-related orphan nuclear hormone receptor γt
- SCW:
-
Streptococcal cell wall
- SpA:
-
Spondyloarthritis
- STAT:
-
Signal transducer and activator of transcription
- TNF:
-
Tumour necrosis factor
- TNFr1:
-
TNF type 1 receptor
- TRAP:
-
Tartate-resistant acid phosphatase
- TYK2:
-
Tyrosine kinase 2
- Wnt:
-
Wingless
- ZAP70:
-
z-associated protein 70
References
Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, San Martin J, Dansey R (2012) Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov 11(5):401–419
Adamopoulos IE, Bowman EP (2008) Immune regulation of bone loss by Th17 cells. Arthritis Res Ther 10(5):225
Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW Jr, Ahmed-Ansari A, Sell KW, Pollard JW, Stanley ER (1990) Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A 87(12):4828–4832
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A et al (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 95(7):3597–3602
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20(1):62–68
Kamiya S, Okumura M, Chiba Y, Fukawa T, Nakamura C, Nimura N, Mizuguchi J, Wada S, Yoshimoto T (2011) IL-27 suppresses RANKL expression in CD4+ T cells in part through STAT3. Immunol Lett 138(1):47–53
Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y (2000) Prostaglandin E2 induces expression of receptor activator of nuclear factor-kappa B ligand/osteoprotegrin ligand on pre-B cells: implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res 15(7):1321–1329
Titanji K, Vunnava A, Sheth AN, Delille C, Lennox JL, Sanford SE, Foster A, Knezevic A, Easley KA, Weitzmann MN et al (2014) Dysregulated B cell expression of RANKL and OPG correlates with loss of bone mineral density in HIV infection. PLoS Pathog 10(10):e1004497
Theill LE, Boyle WJ, Penninger JM (2002) RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol 20:795–823
Weichselbaum A (1878) Die feineren veränderungen des gelenkknorpels bei fungöser synovitis und karies der gelenkenden. Arch Pathol Anat Physiol Klin Med 73(4):461–475
Bromley M, Woolley DE (1984) Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum 27(9):968–975
Leisen JC, Duncan H, Riddle JM, Pitchford WC (1988) The erosive front: a topographic study of the junction between the pannus and the subchondral plate in the macerated rheumatoid metacarpal head. J Rheumatol 15(1):17–22
Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR (1998) Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol 152(4):943–951
Crotti TN, Smith MD, Weedon H, Ahern MJ, Findlay DM, Kraan M, Tak PP, Haynes DR (2002) Receptor activator NF-kappaB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Ann Rheum Dis 61(12):1047–1054
Ellabban AS, Kamel SR, Ahmed SS, Osman AM (2012) Receptor activator of nuclear factor kappa B ligand serum and synovial fluid level. A comparative study between rheumatoid arthritis and osteoarthritis. Rheumatol Int 32(6):1589–1596
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S et al (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402(6759):304–309
Engdahl C, Lindholm C, Stubelius A, Ohlsson C, Carlsten H, Lagerquist MK (2013) Periarticular bone loss in antigen-induced arthritis. Arthritis Rheum 65(11):2857–2865
Romas E, Sims NA, Hards DK, Lindsay M, Quinn JW, Ryan PF, Dunstan CR, Martin TJ, Gillespie MT (2002) Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am J Pathol 161(4):1419–1427
Lubberts E, Oppers-Walgreen B, Pettit AR, Van Den Bersselaar L, Joosten LA, Goldring SR, Gravallese EM, Van Den Berg WB (2002) Increase in expression of receptor activator of nuclear factor kappaB at sites of bone erosion correlates with progression of inflammation in evolving collagen-induced arthritis. Arthritis Rheum 46(11):3055–3064
Sims NA, Green JR, Glatt M, Schlict S, Martin TJ, Gillespie MT, Romas E (2004) Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis. Arthritis Rheum 50(7):2338–2346
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM (2001) TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol 159(5):1689–1699
Yang C, McCoy K, Davis JL, Schmidt-Supprian M, Sasaki Y, Faccio R, Novack DV (2010) NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PLoS One 5(11):e15383
Orosa B, Garcia S, Martinez P, Gonzalez A, Gomez-Reino JJ, Conde C (2014) Lysophosphatidic acid receptor inhibition as a new multipronged treatment for rheumatoid arthritis. Ann Rheum Dis 73(1):298–305
Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, Steiner G, Smolen JS, Wagner EF, Schett G (2002) Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest 110(10):1419–1427
Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Turk B, Pietschmann P, Woloszczuk W, Haralambous S et al (2002) Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum 46(3):785–792
Binder NB, Puchner A, Niederreiter B, Hayer S, Leiss H, Bluml S, Kreindl R, Smolen JS, Redlich K (2013) Tumor necrosis factor-inhibiting therapy preferentially targets bone destruction but not synovial inflammation in a tumor necrosis factor-driven model of rheumatoid arthritis. Arthritis Rheum 65(3):608–617
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M et al (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89(5):755–764
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108(1):17–29
Walsh NC, Gravallese EM (2010) Bone remodeling in rheumatic disease: a question of balance. Immunol Rev 233(1):301–312
Baum R, Gravallese EM (2014) Impact of inflammation on the osteoblast in rheumatic diseases. Curr Osteoporos Rep 12(1):9–16
Balani D, Aeberli D, Hofstetter W, Seitz M (2013) Interleukin-17A stimulates granulocyte-macrophage colony-stimulating factor release by murine osteoblasts in the presence of 1,25-dihydroxyvitamin D(3) and inhibits murine osteoclast development in vitro. Arthritis Rheum 65(2):436–446
Gori F, Hofbauer LC, Dunstan CR, Spelsberg TC, Khosla S, Riggs BL (2000) The expression of osteoprotegerin and RANK ligand and the support of osteoclast formation by stromal-osteoblast lineage cells is developmentally regulated. Endocrinology 141(12):4768–4776
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M et al (1998) Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 139(3):1329–1337
Moverare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Borjesson AE, Sjogren K, Windahl SH, Farman H, Kindlund B et al (2014) Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med 20(11):1279–1288
Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, Chandrasekhar S, Martin TJ, Onyia JE (2001) Catabolic effects of continuous human PTH (1--38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology 142(9):4047–4054
Liu XH, Kirschenbaum A, Yao S, Levine AC (2005) Cross-talk between the interleukin-6 and prostaglandin E(2) signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-{kappa}B (RANK) ligand/RANK system. Endocrinology 146(4):1991–1998
Byun MR, Hwang JH, Kim AR, Kim KM, Hwang ES, Yaffe MB, Hong JH (2014) Canonical Wnt signalling activates TAZ through PP1A during osteogenic differentiation. Cell Death Differ 21(6):854–863
Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG (2006) Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J Cell Sci 119(Pt 7):1283–1296
Tang N, Song WX, Luo J, Luo X, Chen J, Sharff KA, Bi Y, He BC, Huang JY, Zhu GH et al (2009) BMP-9-induced osteogenic differentiation of mesenchymal progenitors requires functional canonical Wnt/beta-catenin signalling. J Cell Mol Med 13(8B):2448–2464
Shealy DJ, Wooley PH, Emmell E, Volk A, Rosenberg A, Treacy G, Wagner CL, Mayton L, Griswold DE, Song XY (2002) Anti-TNF-alpha antibody allows healing of joint damage in polyarthritic transgenic mice. Arthritis Res 4(5):R7
Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D (2007) Macrophages in rheumatoid arthritis. Histol Histopathol 22(5):581–586
Pap T, Muller-Ladner U, Gay RE, Gay S (2000) Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res 2(5):361–367
Walsh NC, Reinwald S, Manning CA, Condon KW, Iwata K, Burr DB, Gravallese EM (2009) Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J Bone Miner Res 24(9):1572–1585
Matzelle MM, Gallant MA, Condon KW, Walsh NC, Manning CA, Stein GS, Lian JB, Burr DB, Gravallese EM (2012) Resolution of inflammation induces osteoblast function and regulates the Wnt signaling pathway. Arthritis Rheum 64(5):1540–1550
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C et al (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13(2):156–163
Wang SY, Liu YY, Ye H, Guo JP, Li R, Liu X, Li ZG (2011) Circulating Dickkopf-1 is correlated with bone erosion and inflammation in rheumatoid arthritis. J Rheumatol 38(5):821–827
Brandao-Burch A, Utting JC, Orriss IR, Arnett TR (2005) Acidosis inhibits bone formation by osteoblasts in vitro by preventing mineralization. Calcif Tissue Int 77(3):167–174
Utting JC, Robins SP, Brandao-Burch A, Orriss IR, Behar J, Arnett TR (2006) Hypoxia inhibits the growth, differentiation and bone-forming capacity of rat osteoblasts. Exp Cell Res 312(10):1693–1702
Ideguchi H, Ohno S, Hattori H, Senuma A, Ishigatsubo Y (2006) Bone erosions in rheumatoid arthritis can be repaired through reduction in disease activity with conventional disease-modifying antirheumatic drugs. Arthritis Res Ther 8(3):R76
Pollinger B, Junt T, Metzler B, Walker UA, Tyndall A, Allard C, Bay S, Keller R, Raulf F, Di Padova F et al (2011) Th17 cells, not IL-17+ gammadelta T cells, drive arthritic bone destruction in mice and humans. J Immunol 186(4):2602–2612
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL (2000) TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 106(12):1481–1488
Scott DL (2000) Prognostic factors in early rheumatoid arthritis. Rheumatology (Oxford) 39(Suppl 1):24–29
Trentham DE, Townes AS, Kang AH (1977) Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med 146(3):857–868
Holmdahl R, Mo J, Nordling C, Larsson P, Jansson L, Goldschmidt T, Andersson M, Klareskog L (1989) Collagen induced arthritis: an experimental model for rheumatoid arthritis with involvement of both DTH and immune complex mediated mechanisms. Clin Exp Rheumatol 7(Suppl 3):S51–55
Esser RE, Stimpson SA, Cromartie WJ, Schwab JH (1985) Reactivation of streptococcal cell wall-induced arthritis by homologous and heterologous cell wall polymers. Arthritis Rheum 28(12):1402–1411
Joosten LA, Abdollahi-Roodsaz S, Heuvelmans-Jacobs M, Helsen MM, van den Bersselaar LA, Oppers-Walgreen B, Koenders MI, van den Berg WB (2008) T cell dependence of chronic destructive murine arthritis induced by repeated local activation of Toll-like receptor-driven pathways: crucial role of both interleukin-1beta and interleukin-17. Arthritis Rheum 58(1):98–108
Lens JW, van den Berg WB, van de Putte LB, Berden JH, Lems SP (1984) Flare-up of antigen-induced arthritis in mice after challenge with intravenous antigen: effects of pre-treatment with cobra venom factor and anti-lymphocyte serum. Clin Exp Immunol 57(3):520–528
Cornelissen F, Asmawidjaja PS, Mus AM, Corneth O, Kikly K, Lubberts E (2013) IL-23 dependent and independent stages of experimental arthritis: no clinical effect of therapeutic IL-23p19 inhibition in collagen-induced arthritis. PLoS One 8(2):e57553
Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, van den Berg WB (2005) Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol 167(1):141–149
Matsumoto I, Staub A, Benoist C, Mathis D (1999) Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 286(5445):1732–1735
Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S et al (2003) Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 426(6965):454–460
Sawa S, Kamimura D, Jin GH, Morikawa H, Kamon H, Nishihara M, Ishihara K, Murakami M, Hirano T (2006) Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J Exp Med 203(6):1459–1470
Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y (2000) Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med 191(2):313–320
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G (1991) Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J 10(13):4025–4031
Taylor BA, Wnek C, Kotlus BS, Roemer N, MacTaggart T, Phillips SJ (1999) Genotyping new BXD recombinant inbred mouse strains and comparison of BXD and consensus maps. Mamm Genome 10(4):335–348
Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL et al (2008) Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9(2):166–175
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443(7114):998–1002
Buchner E, Brauer R, Schmidt C, Emmrich F, Kinne RW (1995) Induction of flare-up reactions in rat antigen-induced arthritis. J Autoimmun 8(1):61–74
Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, Krishnaswami S, Burgos-Vargas R, Wilkinson B, Zerbini CA et al (2009) The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum 60(7):1895–1905
Zwerina K, Koenders M, Hueber A, Marijnissen RJ, Baum W, Heiland GR, Zaiss M, McLnnes I, Joosten L, van den Berg W et al (2012) Anti IL-17A therapy inhibits bone loss in TNF-alpha-mediated murine arthritis by modulation of the T-cell balance. Eur J Immunol 42(2):413–423
Courtenay JS, Dallman MJ, Dayan AD, Martin A, Mosedale B (1980) Immunisation against heterologous type II collagen induces arthritis in mice. Nature 283(5748):666–668
Cathcart ES, Hayes KC, Gonnerman WA, Lazzari AA, Franzblau C (1986) Experimental arthritis in a nonhuman primate. I. Induction by bovine type II collagen. Lab Investig 54(1):26–31
Nandakumar KS (2010) Pathogenic antibody recognition of cartilage. Cell Tissue Res 339(1):213–220
Tarkowski A, Holmdahl R, Klareskog L (1989) Rheumatoid factors in mice. Monogr Allergy 26:214–229
Nabozny GH, Bull MJ, Hanson J, Griffiths MM, Luthra HS, David CS (1994) Collagen-induced arthritis in T cell receptor V beta congenic B10.Q mice. J Exp Med 180(2):517–524
Campbell IK, Hamilton JA, Wicks IP (2000) Collagen-induced arthritis in C57BL/6 (H-2b) mice: new insights into an important disease model of rheumatoid arthritis. Eur J Immunol 30(6):1568–1575
Backlund J, Li C, Jansson E, Carlsen S, Merky P, Nandakumar KS, Haag S, Ytterberg J, Zubarev RA, Holmdahl R (2013) C57BL/6 mice need MHC class II Aq to develop collagen-induced arthritis dependent on autoreactive T cells. Ann Rheum Dis 72(7):1225–1232
Pollinger B (2012) IL-17 producing T cells in mouse models of multiple sclerosis and rheumatoid arthritis. J Mol Med (Berl) 90(6):613–624
Holmdahl R, Klareskog L, Rubin K, Larsson E, Wigzell H (1985) T lymphocytes in collagen II-induced arthritis in mice. Characterization of arthritogenic collagen II-specific T-cell lines and clones. Scand J Immunol 22(3):295–306
Ehinger M, Vestberg M, Johansson AC, Johannesson M, Svensson A, Holmdahl R (2001) Influence of CD4 or CD8 deficiency on collagen-induced arthritis. Immunology 103(3):291–300
Brackertz D, Mitchell GF, Mackay IR (1977) Antigen-induced arthritis in mice. I. Induction of arthritis in various strains of mice. Arthritis Rheum 20(3):841–850
Cromartie WJ, Craddock JG, Schwab JH, Anderle SK, Yang CH (1977) Arthritis in rats after systemic injection of streptococcal cells or cell walls. J Exp Med 146(6):1585–1602
van den Broek MF, van Bruggen MC, Stimpson SA, Severijnen AJ, van de Putte LB, van den Berg WB (1990) Flare-up reaction of streptococcal cell wall induced arthritis in Lewis and F344 rats: the role of T lymphocytes. Clin Exp Immunol 79(2):297–306
Allen JB, Wong HL, Costa GL, Bienkowski MJ, Wahl SM (1993) Suppression of monocyte function and differential regulation of IL-1 and IL-1ra by IL-4 contribute to resolution of experimental arthritis. J Immunol 151(8):4344–4351
Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL et al (1999) From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 10(4):451–461
Corr M, Crain B (2002) The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J Immunol 169(11):6604–6609
Solomon S, Kolb C, Mohanty S, Jeisy-Walder E, Preyer R, Schollhorn V, Illges H (2002) Transmission of antibody-induced arthritis is independent of complement component 4 (C4) and the complement receptors 1 and 2 (CD21/35). Eur J Immunol 32(3):644–651
Niki Y, Yamada H, Seki S, Kikuchi T, Takaishi H, Toyama Y, Fujikawa K, Tada N (2001) Macrophage- and neutrophil-dominant arthritis in human IL-1 alpha transgenic mice. J Clin Invest 107(9):1127–1135
Choy EH, Panayi GS (2001) Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med 344(12):907–916
Butler DM, Maini RN, Feldmann M, Brennan FM (1995) Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur Cytokine Netw 6(4):225–230
Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y (2001) Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem 276(1):563–568
Lee SE, Chung WJ, Kwak HB, Chung CH, Kwack KB, Lee ZH, Kim HH (2001) Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of Akt and ERK. J Biol Chem 276(52):49343–49349
O’Gradaigh D, Ireland D, Bord S, Compston JE (2004) Joint erosion in rheumatoid arthritis: interactions between tumour necrosis factor alpha, interleukin 1, and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclasts. Ann Rheum Dis 63(4):354–359
Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL (2005) M-CSF mediates TNF-induced inflammatory osteolysis. J Clin Invest 115(12):3418–3427
Herman S, Muller RB, Kronke G, Zwerina J, Redlich K, Hueber AJ, Gelse H, Neumann E, Muller-Ladner U, Schett G (2008) Induction of osteoclast-associated receptor, a key osteoclast costimulation molecule, in rheumatoid arthritis. Arthritis Rheum 58(10):3041–3050
Li P, Schwarz EM, O’Keefe RJ, Ma L, Boyce BF, Xing L (2004) RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res 19(2):207–213
Williams RO, Feldmann M, Maini RN (1992) Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A 89(20):9784–9788
Okazaki H, Lin Q, Nishikawa K, Ohtsuji N, Tsurui H, Ohtsuji M, Amano H, Tada N, Sudo K, Nishimura H et al (2014) TNFalpha but not IL-17 is critical in the pathogenesis of rheumatoid arthritis spontaneously occurring in a unique FcgammaRIIB-deficient mouse model. Mod Rheumatol 24(6):931–938
Furst DE (2010) Development of TNF inhibitor therapies for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 28(3 Suppl 59):S5–12
Ritchlin C (2000) Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis. Arthritis Res 2(5):356–360
Seckinger P, Klein-Nulend J, Alander C, Thompson RC, Dayer JM, Raisz LG (1990) Natural and recombinant human IL-1 receptor antagonists block the effects of IL-1 on bone resorption and prostaglandin production. J Immunol 145(12):4181–4184
Thomson BM, Saklatvala J, Chambers TJ (1986) Osteoblasts mediate interleukin 1 stimulation of bone resorption by rat osteoclasts. J Exp Med 164(1):104–112
Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, Suda T (1999) Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res 247(1):84–93
Jimi E, Shuto T, Koga T (1995) Macrophage colony-stimulating factor and interleukin-1 alpha maintain the survival of osteoclast-like cells. Endocrinology 136(2):808–811
Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N et al (2000) Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med 191(2):275–286
Ma T, Miyanishi K, Suen A, Epstein NJ, Tomita T, Smith RL, Goodman SB (2004) Human interleukin-1-induced murine osteoclastogenesis is dependent on RANKL, but independent of TNF-alpha. Cytokine 26(3):138–144
Niki Y, Yamada H, Kikuchi T, Toyama Y, Matsumoto H, Fujikawa K, Tada N (2004) Membrane-associated IL-1 contributes to chronic synovitis and cartilage destruction in human IL-1 alpha transgenic mice. J Immunol 172(1):577–584
Ghivizzani SC, Kang R, Georgescu HI, Lechman ER, Jaffurs D, Engle JM, Watkins SC, Tindal MH, Suchanek MK, McKenzie LR et al (1997) Constitutive intra-articular expression of human IL-1 beta following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J Immunol 159(7):3604–3612
Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C (2002) Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med 196(1):77–85
Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, Dunstan C, Kollias G, Steiner G, Smolen J et al (2004) Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum 50(1):277–290
Zwerina J, Redlich K, Polzer K, Joosten L, Kronke G, Distler J, Hess A, Pundt N, Pap T, Hoffmann O et al (2007) TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci U S A 104(28):11742–11747
Polzer K, Joosten L, Gasser J, Distler JH, Ruiz G, Baum W, Redlich K, Bobacz K, Smolen JS, van den Berg W et al (2010) Interleukin-1 is essential for systemic inflammatory bone loss. Ann Rheum Dis 69(1):284–290
Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL (2005) IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest 115(2):282–290
Smolen JS, Landewe R, Breedveld FC, Dougados M, Emery P, Gaujoux-Viala C, Gorter S, Knevel R, Nam J, Schoels M et al (2010) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis 69(6):964–975
Garbers C, Aparicio-Siegmund S, Rose-John S (2015) The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol 34:75–82
Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, Ota Z (1997) The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum 40(6):1096–1105
Waage A, Kaufmann C, Espevik T, Husby G (1989) Interleukin-6 in synovial fluid from patients with arthritis. Clin Immunol Immunopathol 50(3):394–398
Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J (1988) Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum 31(6):784–788
Dasgupta B, Corkill M, Kirkham B, Gibson T, Panayi G (1992) Serial estimation of interleukin 6 as a measure of systemic disease in rheumatoid arthritis. J Rheumatol 19(1):22–25
Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, Nakamura I, Yamaguchi A, Kishimoto T, Suda T, Kashiwazaki S (1996) Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res 11(1):88–95
Kurihara N, Bertolini D, Suda T, Akiyama Y, Roodman GD (1990) IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol 144(11):4226–4230
Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, Koishihara Y, Ohsugi Y, Kumaki K, Taga T et al (1993) Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci U S A 90(24):11924–11928
Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G (2009) Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum 60(9):2747–2756
Li Y, Backesjo CM, Haldosen LA, Lindgren U (2008) IL-6 receptor expression and IL-6 effects change during osteoblast differentiation. Cytokine 43(2):165–173
De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, Vivarelli M, Muratori F, Berni S, Ballanti P et al (2006) Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum 54(11):3551–3563
Boe A, Baiocchi M, Carbonatto M, Papoian R, Serlupi-Crescenzi O (1999) Interleukin 6 knock-out mice are resistant to antigen-induced experimental arthritis. Cytokine 11(12):1057–1064
Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M et al (1998) Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci U S A 95(14):8222–8226
Wong PK, Quinn JM, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP (2006) Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum 54(1):158–168
Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G (1998) Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med 187(4):461–468
Takagi N, Mihara M, Moriya Y, Nishimoto N, Yoshizaki K, Kishimoto T, Takeda Y, Ohsugi Y (1998) Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum 41(12):2117–2121
Tanaka K, Hashizume M, Mihara M, Yoshida H, Suzuki M, Matsumoto Y (2014) Anti-interleukin-6 receptor antibody prevents systemic bone mass loss via reducing the number of osteoclast precursors in bone marrow in a collagen-induced arthritis model. Clin Exp Immunol 175(2):172–180
Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, Matsumoto Y, Ohsugi Y (2005) Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol 5(12):1731–1740
Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Murata N, van der Heijde D, Kishimoto T (2007) Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 66(9):1162–1167
Huizinga TW, Fleischmann RM, Jasson M, Radin AR, van Adelsberg J, Fiore S, Huang X, Yancopoulos GD, Stahl N, Genovese MC (2014) Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis 73(9):1626–1634
Van Roy M, Ververken C, Beirnaert E, Hoefman S, Kolkman J, Vierboom M, Breedveld E, t Hart B, Poelmans S, Bontinck L et al (2015) The preclinical pharmacology of the high affinity anti-IL-6R Nanobody ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther 17(1):135
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B (2014) Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 73(9):1616–1625
Mease P, Strand V, Shalamberidze L, Dimic A, Raskina T, Xu LA, Liu Y, Smith J (2012) A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Ann Rheum Dis 71(7):1183–1189
Shaw S, Bourne T, Meier C, Carrington B, Gelinas R, Henry A, Popplewell A, Adams R, Baker T, Rapecki S et al (2014) Discovery and characterization of olokizumab: a humanized antibody targeting interleukin-6 and neutralizing gp130-signaling. MAbs 6(3):774–782
Finch DK, Sleeman MA, Moisan J, Ferraro F, Botterell S, Campbell J, Cochrane D, Cruwys S, England E, Lane S et al (2011) Whole-molecule antibody engineering: generation of a high-affinity anti-IL-6 antibody with extended pharmacokinetics. J Mol Biol 411(4):791–807
Zizzo G, De Santis M, Bosello SL, Fedele AL, Peluso G, Gremese E, Tolusso B, Ferraccioli G (2011) Synovial fluid-derived T helper 17 cells correlate with inflammatory activity in arthritis, irrespectively of diagnosis. Clin Immunol 138(1):107–116
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT et al (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9):1345–1352
Lubberts E (2015) The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol 11(10):562
Paulissen SM, van Hamburg JP, Dankers W, Lubberts E (2015) The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine 74(1):43–53
Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T et al (2007) Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 204(12):2803–2812
Yago T, Nanke Y, Ichikawa N, Kobashigawa T, Mogi M, Kamatani N, Kotake S (2009) IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: a novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem 108(4):947–955
Adamopoulos IE, Suzuki E, Chao CC, Gorman D, Adda S, Maverakis E, Zarbalis K, Geissler R, Asio A, Blumenschein WM et al (2015) IL-17A gene transfer induces bone loss and epidermal hyperplasia associated with psoriatic arthritis. Ann Rheum Dis 74(6):1284–1292
Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, van den Berg WB (2003) IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol 170(5):2655–2662
Nakae S, Nambu A, Sudo K, Iwakura Y (2003) Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171(11):6173–6177
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB (2004) Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50(2):650–659
Park MJ, Park HS, Oh HJ, Lim JY, Yoon BY, Kim HY, Cho ML, Cho SG (2012) IL-17-deficient allogeneic bone marrow transplantation prevents the induction of collagen-induced arthritis in DBA/1J mice. Exp Mol Med 44(11):694–705
Corneth OB, Mus AM, Asmawidjaja PS, Klein Wolterink RG, van Nimwegen M, Brem MD, Hofman Y, Hendriks RW, Lubberts E (2014) Absence of interleukin-17 receptor a signaling prevents autoimmune inflammation of the joint and leads to a Th2-like phenotype in collagen-induced arthritis. Arthritis Rheumatol 66(2):340–349
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y et al (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203(12):2673–2682
Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y (2003) IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A 100(10):5986–5990
Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH, Psoriasis Study G et al (2010) Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2(52):52ra72
Genovese MC, Greenwald MW, Cho CS, Berman A, Jin L, Cameron G, Xie L, Braun D, Banerjee S, Warner L (2011) A phase 2 study of multiple subcutaneous doses of LY2439821, an anti-IL-17 monoclonal antibody, in patients with rheumatoid arthritis in two populations: naive to biologic therapy or inadequate responders to tumor necrosis factor alpha inhibitors. Arthritis Rheum-Us 63(10):S1017–S1017
Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, Braun D, Banerjee S (2012) Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 366(13):1190–1199
Genovese MC, Greenwald M, Cho CS, Berman A, Jin L, Cameron GS, Benichou O, Xie L, Braun D, Berclaz PY et al (2014) A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol 66(7):1693–1704
Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, Aras G, Li J, Russell CB, Thompson EH et al (2012) Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 366(13):1181–1189
Pavelka K, Chon Y, Newmark R, Erondu N, Lin SL (2012) A randomized, double-blind, placebo-controlled, multiple-dose study to evaluate the safety, tolerability, and efficacy of brodalumab (AMG 827) in subjects with rheumatoid arthritis and an inadequate response to Methotrexate. Arthritis Rheum-Us 64(10):S362–S362
Berer A, Stockl J, Majdic O, Wagner T, Kollars M, Lechner K, Geissler K, Oehler L (2000) 1,25-Dihydroxyvitamin D(3) inhibits dendritic cell differentiation and maturation in vitro. Exp Hematol 28(5):575–583
Chen L, Wei XQ, Evans B, Jiang W, Aeschlimann D (2008) IL-23 promotes osteoclast formation by up-regulation of receptor activator of NF-kappaB (RANK) expression in myeloid precursor cells. Eur J Immunol 38(10):2845–2854
Yago T, Nanke Y, Kawamoto M, Furuya T, Kobashigawa T, Kamatani N, Kotake S (2007) IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther 9(5):R96
Shin HS, Sarin R, Dixit N, Wu J, Gershwin E, Bowman EP, Adamopoulos IE (2015) Crosstalk among IL-23 and DNAX activating protein of 12 kDa-dependent pathways promotes osteoclastogenesis. J Immunol 194(1):316–324
Quinn JM, Sims NA, Saleh H, Mirosa D, Thompson K, Bouralexis S, Walker EC, Martin TJ, Gillespie MT (2008) IL-23 inhibits osteoclastogenesis indirectly through lymphocytes and is required for the maintenance of bone mass in mice. J Immunol 181(8):5720–5729
Kamiya S, Nakamura C, Fukawa T, Ono K, Ohwaki T, Yoshimoto T, Wada S (2007) Effects of IL-23 and IL-27 on osteoblasts and osteoclasts: inhibitory effects on osteoclast differentiation. J Bone Miner Metab 25(5):277–285
Ju JH, Cho ML, Moon YM, Oh HJ, Park JS, Jhun JY, Min SY, Cho YG, Park KS, Yoon CH et al (2008) IL-23 induces receptor activator of NF-kappaB ligand expression on CD4+ T cells and promotes osteoclastogenesis in an autoimmune arthritis model. J Immunol 181(2):1507–1518
Adamopoulos IE, Tessmer M, Chao CC, Adda S, Gorman D, Petro M, Chou CC, Pierce RH, Yao W, Lane NE et al (2011) IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. J Immunol 187(2):951–959
Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14(9):585–600
Shapiro F (2008) Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cell Mater 15:53–76
Kawaguchi H (2008) Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol Cell 25(1):1–6
Thompson EM, Matsiko A, Farrell E, Kelly DJ, O’Brien FJ (2014) Recapitulating endochondral ossification: a promising route to in vivo bone regeneration. J Tissue Eng Regen Med 9(8):889–902
Farrell E, van der Jagt OP, Koevoet W, Kops N, van Manen CJ, Hellingman CA, Jahr H, O’Brien FJ, Verhaar JA, Weinans H et al (2009) Chondrogenic priming of human bone marrow stromal cells: a better route to bone repair? Tissue Eng Part C Methods 15(2):285–295
Farrell E, Both SK, Odorfer KI, Koevoet W, Kops N, O’Brien FJ, de Jong RJ B, Verhaar JA, Cuijpers V, Jansen J et al (2011) In-vivo generation of bone via endochondral ossification by in-vitro chondrogenic priming of adult human and rat mesenchymal stem cells. BMC Musculoskelet Disord 12:31
Huang JI, Durbhakula MM, Angele P, Johnstone B, Yoo JU (2006) Lunate arthroplasty with autologous mesenchymal stem cells in a rabbit model. J Bone Joint Surg Am 88(4):744–752
Janicki P, Kasten P, Kleinschmidt K, Luginbuehl R, Richter W (2010) Chondrogenic pre-induction of human mesenchymal stem cells on beta-TCP: enhanced bone quality by endochondral heterotopic bone formation. Acta Biomater 6(8):3292–3301
Jukes JM, Both SK, Leusink A, Sterk LM, van Blitterswijk CA, de Boer J (2008) Endochondral bone tissue engineering using embryonic stem cells. Proc Natl Acad Sci U S A 105(19):6840–6845
Aizawa T, Kon T, Einhorn TA, Gerstenfeld LC (2001) Induction of apoptosis in chondrocytes by tumor necrosis factor-alpha. J Orthop Res 19(5):785–796
Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, Barnes GL, Graves DT, Einhorn TA (2003) Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res 18(9):1584–1592
Grimes R, Jepsen KJ, Fitch JL, Einhorn TA, Gerstenfeld LC (2011) The transcriptome of fracture healing defines mechanisms of coordination of skeletal and vascular development during endochondral bone formation. J Bone Miner Res 26(11):2597–2609
Scotti C, Piccinini E, Takizawa H, Todorov A, Bourgine P, Papadimitropoulos A, Barbero A, Manz MG, Martin I (2013) Engineering of a functional bone organ through endochondral ossification. Proc Natl Acad Sci U S A 110(10):3997–4002
Mumme M, Scotti C, Papadimitropoulos A, Todorov A, Hoffmann W, Bocelli-Tyndall C, Jakob M, Wendt D, Martin I, Barbero A (2012) Interleukin-1beta modulates endochondral ossification by human adult bone marrow stromal cells. Eur Cell Mater 24:224–236
Hauser B, Riches PL, Wilson JF, Horne AE, Ralston SH (2014) Prevalence and clinical prediction of osteoporosis in a contemporary cohort of patients with rheumatoid arthritis. Rheumatology (Oxford) 53(10):1759–1766
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, Racewicz AJ, van Vollenhoven RF, Li NF, Agarwal S et al (2006) The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum 54(5):1390–1400
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW et al (2006) Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 54(9):2793–2806
Keystone E, Emery P, Peterfy CG, Tak PP, Cohen S, Genovese MC, Dougados M, Burmester GR, Greenwald M, Kvien TK et al (2009) Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis 68(2):216–221
Tak PP, Rigby WF, Rubbert-Roth A, Peterfy CG, van Vollenhoven RF, Stohl W, Hessey E, Chen A, Tyrrell H, Shaw TM et al (2011) Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis 70(1):39–46
Boumans MJ, Thurlings RM, Yeo L, Scheel-Toellner D, Vos K, Gerlag DM, Tak PP (2012) Rituximab abrogates joint destruction in rheumatoid arthritis by inhibiting osteoclastogenesis. Ann Rheum Dis 71(1):108–113
Wheater G, Hogan VE, Teng YK, Tekstra J, Lafeber FP, Huizinga TW, Bijlsma JW, Francis RM, Tuck SP, Datta HK et al (2011) Suppression of bone turnover by B-cell depletion in patients with rheumatoid arthritis. Osteoporos Int 22(12):3067–3072
Fleischmann RM (2009) Progressive multifocal leukoencephalopathy following rituximab treatment in a patient with rheumatoid arthritis. Arthritis Rheum 60(11):3225–3228
Emery P (2003) The therapeutic potential of costimulatory blockade with CTLA4Ig in rheumatoid arthritis. Expert Opin Investig Drugs 12(4):673–681
Kliwinski C, Kukral D, Postelnek J, Krishnan B, Killar L, Lewin A, Nadler S, Townsend R (2005) Prophylactic administration of abatacept prevents disease and bone destruction in a rat model of collagen-induced arthritis. J Autoimmun 25(3):165–171
Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, Herrmann M, Smolen J, Schett G (2008) CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis 67(11):1603–1609
Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, Birbara C, Box J, Natarajan K, Nuamah I et al (2005) Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med 353(11):1114–1123
Kremer JM, Genant HK, Moreland LW, Russell AS, Emery P, Abud-Mendoza C, Szechinski J, Li T, Ge Z, Becker JC et al (2006) Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: a randomized trial. Ann Intern Med 144(12):865–876
Kremer JM, Genant HK, Moreland LW, Russell AS, Emery P, Abud-Mendoza C, Szechinski J, Li T, Teng J, Becker JC et al (2008) Results of a two-year followup study of patients with rheumatoid arthritis who received a combination of abatacept and methotrexate. Arthritis Rheum 58(4):953–963
Genant HK, Peterfy CG, Westhovens R, Becker JC, Aranda R, Vratsanos G, Teng J, Kremer JM (2008) Abatacept inhibits progression of structural damage in rheumatoid arthritis: results from the long-term extension of the AIM trial. Ann Rheum Dis 67(8):1084–1089
Atzeni F, Sarzi-Puttini P, Botsios C, Carletto A, Cipriani P, Favalli EG, Frati E, Foschi V, Gasparini S, Giardina A et al (2012) Long-term anti-TNF therapy and the risk of serious infections in a cohort of patients with rheumatoid arthritis: comparison of adalimumab, etanercept and infliximab in the GISEA registry. Autoimmun Rev 12(2):225–229
Khraishi M, Russell A, Olszynski WP (2010) Safety profile of abatacept in rheumatoid arthritis: a review. Clin Ther 32(11):1855–1870
Meier FM, McInnes IB (2014) Small-molecule therapeutics in rheumatoid arthritis: scientific rationale, efficacy and safety. Best Pract Res Clin Rheumatol 28(4):605–624
Milici AJ, Kudlacz EM, Audoly L, Zwillich S, Changelian P (2008) Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther 10(1):R14
LaBranche TP, Jesson MI, Radi ZA, Storer CE, Guzova JA, Bonar SL, Thompson JM, Happa FA, Stewart ZS, Zhan Y et al (2012) JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum 64(11):3531–3542
Boyle DL, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, Shurmur R, Singhal AK, Wei N, Rosengren S et al (2015) The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis 74(6):1311–1316
Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, Tanaka Y (2014) The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheum Dis 73(12):2192–2198
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, Garcia Meijide JA, Wagner S, Forejtova S, Zwillich SH, Gruben D, Koncz T et al (2012) Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 367(6):508–519
Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, Gruben D, Wallenstein G, Krishnaswami S, Zwillich SH et al (2013) Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 381(9865):451–460
Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, Koncz T, Krishnaswami S, Wallenstein GV, Zang C et al (2014) Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 370(25):2377–2386
Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, Gruben D, Kanik KS, Krishnaswami S, Pascual-Ramos V et al (2012) A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum 64(4):970–981
Coombs JH, Bloom BJ, Breedveld FC, Fletcher MP, Gruben D, Kremer JM, Burgos-Vargas R, Wilkinson B, Zerbini CA, Zwillich SH (2010) Improved pain, physical functioning and health status in patients with rheumatoid arthritis treated with CP-690,550, an orally active Janus kinase (JAK) inhibitor: results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 69(2):413–416
Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, Lepescheux L, Christophe T, Conrath K, Vandeghinste N et al (2013) Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol 191(7):3568–3577
Park JS, Lee J, Lim MA, Kim EK, Kim SM, Ryu JG, Lee JH, Kwok SK, Park KS, Kim HY et al (2014) JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J Immunol 192(9):4417–4424
Stump KL, Lu LD, Dobrzanski P, Serdikoff C, Gingrich DE, Dugan BJ, Angeles TS, Albom MS, Ator MA, Dorsey BD et al (2011) A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res Ther 13(2):R68
Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF et al (2010) Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 184(9):5298–5307
Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, Punwani N, Williams WV, Yeleswaram S (2014) The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol 54(12):1354–1361
Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N (2015) A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheum 67(2):334–343
Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, Holmes GB, Dunstan CR, DePaoli AM (2004) A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 19(7):1059–1066
Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R et al (2008) Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 58(5):1299–1309
Deodhar A, Dore RK, Mandel D, Schechtman J, Shergy W, Trapp R, Ory PA, Peterfy CG, Fuerst T, Wang H et al (2010) Denosumab-mediated increase in hand bone mineral density associated with decreased progression of bone erosion in rheumatoid arthritis patients. Arthritis Care Res (Hoboken) 62(4):569–574
Curtis JR, Xie F, Yun H, Saag KG, Chen L, Delzell E (2015) Risk of hospitalized infection among rheumatoid arthritis patients concurrently treated with a biologic agent and denosumab. Arthritis Rheumatol 67(6):1456–1464
Gelb BD, Shi GP, Chapman HA, Desnick RJ (1996) Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 273(5279):1236–1238
Skoumal M, Haberhauer G, Kolarz G, Hawa G, Woloszczuk W, Klingler A, Varga F, Klaushofer K (2008) The imbalance between osteoprotegerin and cathepsin K in the serum of patients with longstanding rheumatoid arthritis. Rheumatol Int 28(7):637–641
Tchetina EV, Demidova NV, Karateev DE, Nasonov EL (2013) Rheumatoid factor positivity is associated with increased joint destruction and upregulation of matrix metalloproteinase 9 and cathepsin k gene expression in the peripheral blood in rheumatoid arthritic patients treated with methotrexate. Int J Rheumatol 2013:457876
Lotinun S, Kiviranta R, Matsubara T, Alzate JA, Neff L, Luth A, Koskivirta I, Kleuser B, Vacher J, Vuorio E et al (2013) Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 123(2):666–681
Pennypacker BL, le Duong T, Cusick TE, Masarachia PJ, Gentile MA, Gauthier JY, Black WC, Scott BB, Samadfam R, Smith SY et al (2011) Cathepsin K inhibitors prevent bone loss in estrogen-deficient rabbits. J Bone Miner Res 26(2):252–262
Schurigt U, Hummel KM, Petrow PK, Gajda M, Stockigt R, Middel P, Zwerina J, Janik T, Bernhardt R, Schuler S et al (2008) Cathepsin K deficiency partially inhibits, but does not prevent, bone destruction in human tumor necrosis factor-transgenic mice. Arthritis Rheum 58(2):422–434
Svelander L, Erlandsson-Harris H, Astner L, Grabowska U, Klareskog L, Lindstrom E, Hewitt E (2009) Inhibition of cathepsin K reduces bone erosion, cartilage degradation and inflammation evoked by collagen-induced arthritis in mice. Eur J Pharmacol 613(1-3):155–162
Bone HG, Dempster DW, Eisman JA, Greenspan SL, McClung MR, Nakamura T, Papapoulos S, Shih WJ, Rybak-Feiglin A, Santora AC et al (2015) Odanacatib for the treatment of postmenopausal osteoporosis: development history and design and participant characteristics of LOFT, the Long-Term Odanacatib Fracture Trial. Osteoporos Int 26(2):699–712
Engelke K, Fuerst T, Dardzinski B, Kornak J, Ather S, Genant HK, de Papp A (2015) Odanacatib treatment affects trabecular and cortical bone in the femur of postmenopausal women: results of a two-year placebo-controlled trial. J Bone Miner Res 30(1):30–38
Eisman JA, Bone HG, Hosking DJ, McClung MR, Reid IR, Rizzoli R, Resch H, Verbruggen N, Hustad CM, DaSilva C et al (2011) Odanacatib in the treatment of postmenopausal women with low bone mineral density: three-year continued therapy and resolution of effect. J Bone Miner Res 26(2):242–251
Press release announcing outcome of LOFT trial with odanacatib
Acknowledgement
This work was supported by a grant from the Dutch Arthritis Association (Reumafonds, no. 11-1-306).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alves, C.H., Farrell, E., Vis, M. et al. Animal Models of Bone Loss in Inflammatory Arthritis: from Cytokines in the Bench to Novel Treatments for Bone Loss in the Bedside—a Comprehensive Review. Clinic Rev Allerg Immunol 51, 27–47 (2016). https://doi.org/10.1007/s12016-015-8522-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-015-8522-7