
Abstract
Physicochemical properties like density, surface tension, and viscosity of liquid binary Al-Li and Li-Zn alloys have been measured using draining crucible method. The experimentally measured surface-tension values have been compared to theoretical results based either on the Butler model or the compound formation model assuming the existence of the most favored A 1 B 2 and A 2 B 3 clusters. Several models for viscosity calculation have been also applied and discussed in confrontation with measured data. Finally, the clustering effects in the liquid Al-Li and Li-Zn alloys have been examined using two microscopic functions, i.e., the concentration fluctuation function in the long-wavelength limit and the Warren-Cowley short-range order parameter.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Several different types of Li-based rechargeable batteries have attracted substantial interest. This results from an expectation of high specific energies and energy densities for such batteries system.[1] The attention has been paid for an even longer time to the use of lithium alloys as an alternative to elemental lithium. Especially interesting are the batteries working with molten salt electrolytes that operate above the melting point of lithium, the lithium-aluminum alloys,[2] for example. Apart from these, metallic systems: Li-Zn, Li-Sn, Li-Pb, etc. with organic solvent-based electrolytes at ambient temperatures were also investigated.[3,4]
Thermodynamic properties of both investigated liquid alloys Al-Li[5] and Li-Zn[6,7]: indicate a negative deviation from Raoult’s law due to large number of intermetallic compounds being present in the solid state.[8, 9] An associative tendency in the liquid state can be observed.[10] This energetic effect can be explained by studying the concentration dependence of mixing functions, for which a well-pronounced peak is positioned in the vicinity of the stoichiometric compositions of the intermetallic compounds. [11–14] Thermodynamic modeling in the framework of CFM formalism[15,16] showed that Al2Li3 and LiZn2 clusters can exist and stabilize the liquid Al-Li and Li-Zn alloys. The nature of ordering in liquid investigated binaries was described and quantified using Bhatia-Thornton theory.[17,18] The degree of ordering in the melts has been studied applying Warren-Cowley short-range order parameter (α 1). [19,20]
However, physicochemical properties such as density, viscosity, and surface tension of liquid Al-Li and Li-Zn alloys were not measured and they are unknown. These properties were only determined for pure liquid constituents: Al, Li, and Zn. They were reported for Al and Zn metals by many authors, e.g., Keene[21] and Gancarz et al.[22,23] etc. Data for liquid lithium are more scarce than for Al and Zn elements and were taken from.[24]
Thus, the aim of this work was to (a) measure the three physicochemical properties: density, surface tension, and viscosity of liquid Al-Li and Li-Zn alloys using Draining Crucible method[25–27] over a broad temperature range (from 773 K (500 °C) to 973 K (700 °C)) (b) calculate them by applying CFM formalism[15,16] (c) explain and understand the influence of short-range ordering on measured properties. Surface tension modeling was also performed using the Butler model[28] but viscosity by applying common known empirical models e.g., Kucharski,[29] Kaptay,[30] Kozlov et al. [31]
As final result we plan to obtain a complete database of physicochemical properties for liquid Al–Li and Li-Zn alloys.
2 Methods: Experimental and Modeling
2.1 Draining Crucible Method
The experimental method for simultaneous measurements of surface tension, viscosity, and density of fluids was first proposed by Roach and Henein.[25–27] This method is based on the measurement of fluid mass flowing out of an orifice under gravity. Schematic draining vessel system depicting flow rate of a fluid through an orifice placed at the bottom is presented in Figure 1. A proposition given by Roach and Henein is similar to the traditional Bernoulli formulation (\( Q = \pi r_{0}^{2} \sqrt {2gh} \)) with one exception including an additional term. It corresponds to the pressure effect resulting from the surface tension, which is quantified in the applied model. The complete mathematical description of DC method is given elsewhere.[25–27,32]

Schematic presentation of apparatus for draining crucible method used for measurement of physical properties of alloy
The volumetric flow Q in dimensionless form is written:
where Fr is Froude number representing the ratio of the inertial force of the stream to the potential force of the liquid head above the discharge orifice. Bo represents the ratio of the potential force to the surface force of the stream flowing out through the orifice.
Using liquids of known physical properties and an orifice of a given diameter, a calibration curve of C d vs Re is developed and expressed by a polynomial as follows:
C d is discharge coefficient characterizing friction losses in the orifice and Re is Reynolds number defining the inertial forces relative to viscous losses in the orifice expressed in terms of experimental volumetric flow rate; a 1 and a 2 are constants in the polynomial describing the relation C d vs Re. They are crucial for determination of the unknown properties of melts such as[25]:
2.1.1 Surface tension
2.1.2 Density
2.1.3 Viscosity
where h is height of the melt in a crucible and Q exp is a mass flux (experimental volumetric flow rate).
Combining the Eqs. [3–5], the relationship between measured height of the melt and three measured physical properties is expressed as follows:
In practice, the Eq. [6] is numerically solved basing on the Hooke–Jeeves method[33] and finally, density, surface tension, and viscosity of the melts are determined.
2.2 Compound Formation Model
The compound formation model (CFM) assumes that a binary system consists of A- and B- atoms and forming an appropriate privileged chemical compound of A m B n type (m and n are integer numbers). The complete mathematical formalism of the CFM approach is reported in References 13 through 16, 34, 35.
The excess Gibbs energy, \( G_{M}^{xc} \), can be expressed in the following form when standard thermodynamic relations are applied and some algebraic rearrangements are made[34]:
where Φ ij (i, j = A, B) are concentration functions depending on m and n values, R is the gas constant, T absolute temperature and W, ΔW AB , ΔW AA , ΔW BB are energy parameters.
Different expressions can be distinguished for computation of the concentration functions in relevance to the assumed type of clusters existing in liquid state. Performed study of concentration dependence of mixing function for both investigated alloys shows that the Al2Li3 and LiZn2 clusters dominate in liquid Al-Li and Li-Zn alloys, respectively.
Thus, the concentration functions for two assumed clusters are as follows[12–14,34]:
-
(a)
m = 1 and n = 2 for LiZn2 compound:
$$ \begin{aligned} \Phi_{AB} & = \frac{1}{6}(1 - C) + (1 - C)^{2} - \frac{5}{3}(1 - C)^{3} + \frac{1}{2}(1 - C)^{4} ; \\ \Phi_{AA} & = 0; \\ \Phi_{BB} & = - \frac{1}{4}(1 - C) + \frac{1}{2}(1 - C)^{2} - \frac{1}{4}(1 - C)^{4} \\ \end{aligned} $$(8) -
(b)
m = 2 and n = 3 for Al2Li3 clusters:
$$ \begin{aligned} \Phi_{AB} & = \frac{13}{420}(1 - C) + \frac{2}{3}(1 - C)^{3} - \frac{3}{2}(1 - C)^{4} + \frac{2}{3}(1 - C)^{6} - \frac{5}{7}(1 - C)^{7} + \frac{1}{4}(1 - C)^{8} ; \\ \Phi_{AA} & = - \frac{53}{840}(1 - C) + \frac{2}{3}(1 - C)^{3} - \frac{5}{4}(1 - C)^{4} - (1 - C)^{6} + \frac{4}{7}(1 - C)^{7} - \frac{1}{8}(1 - C)^{8} ; \\ \Phi_{BB} & = \frac{23}{280}(1 - C) - \frac{1}{2}(1 - C)^{4} + \frac{2}{5}(1 - C)^{6} + \frac{1}{7}(1 - C)^{7} - \frac{1}{8}(1 - C)^{8} \\ \end{aligned} $$(9)
2.2.1 Surface tension
In the framework of CFM formalism, the grand partition functions provide the connection between bulk and surface properties which allows to compute surface tension as[34,35]:
where σ A , σ B are surface tension of the component A and B, respectively, p and q parameters represent the surface coordination fractions. For closed packed structure, these values equal to 0.5 and 0.25 as a consequence p + 2q = 1.[36]
S is a mean surface area of the binary system computed by
and S i represents surface area of each atomic species given as
while V i is the molar volume of i-th component.
CFM formalism for computing the surface tension also includes concentration functions which depend on m and n small integer numbers. They are given by the equations as follows[35]:
-
(a)
m = 1 and n = 2 for LiZn2 compounds:
$$ \begin{aligned} \phi & = C^{2} ; \\ \phi_{AB} & = \frac{1}{6} + 2(1 - C) - 6(1 - C)^{2} + \frac{16}{3}(1 - C)^{3} - \frac{3}{2}(1 - C)^{4} ; \\ \phi_{AA} & = 0; \\ \phi_{BB} & = - \frac{1}{4} + (1 - C) - \frac{1}{2}(1 - C)^{2} + (1 - C)^{3} - \frac{3}{4}(1 - C)^{4} \\ f & = (1 - C)^{2} ; \\ f_{AB} & = - (1 - C)^{2} + \frac{10}{3}(1 - C)^{3} - \frac{3}{2}(1 - C)^{4} ; \\ f_{AA} & = 0; \\ f_{BB} & = - \frac{1}{2}(1 - C)^{2} + \frac{3}{4}(1 - C)^{4} \\ \end{aligned} $$(13) -
(b)
m = 2 and n = 3 for Al2Li3 clusters
$$ \begin{aligned} \phi & = C^{2} ; \\ \phi_{AB} & = \frac{13}{420} + 2(1 - C)^{2} - \frac{22}{3}(1 - C)^{3} - \frac{15}{2}(1 - C)^{4} + \frac{8}{5}(1 - C)^{5} - \frac{25}{3}(1 - C)^{6} + \frac{44}{4}(1 - C)^{7} - \frac{7}{4}(1 - C)^{8} ; \\ \phi_{AA} & = - \frac{53}{840} + 2(1 - C)^{2} - \frac{19}{3}(1 - C)^{3} - \frac{39}{4}(1 - C)^{4} - \frac{54}{5}(1 - C)^{5} + 9(1 - C)^{6} - \frac{31}{4}(1 - C)^{7} + \frac{7}{8}(1 - C)^{8} ; \\ \phi_{BB} & = \frac{23}{280} - 2(1 - C)^{3} + \frac{7}{2}(1 - C)^{4} - \frac{8}{5}(1 - C)^{5} + (1 - C)^{6} - \frac{13}{4}(1 - C)^{7} + \frac{7}{8}(1 - C)^{8} \\ f & = (1 - C)^{2} \\ f_{AB} & = - \frac{4}{3}(1 - C)^{3} + \frac{9}{2}(1 - C)^{4} - \frac{12}{5}(1 - C)^{5} - \frac{10}{3}(1 - C)^{6} + \frac{30}{4}(1 - C)^{7} - \frac{7}{4}(1 - C)^{8} ; \\ f_{AA} & = - \frac{4}{3}(1 - C)^{3} + \frac{15}{4}(1 - C)^{4} - \frac{24}{5}(1 - C)^{5} + 5(1 - C)^{6} - \frac{24}{7}(1 - C)^{7} + \frac{7}{8}(1 - C)^{8} ; \\ f_{BB} & = - \frac{3}{2}(1 - C)^{4} - \frac{8}{5}(1 - C)^{5} - \frac{6}{7}(1 - C)^{7} + \frac{7}{8}(1 - C)^{8} \\ \end{aligned} $$(14)
where ϕ ij and f ij are concentration functions describing the bulk phase while ϕ s ij and f s ij functions represent the surface phase.
2.3 Microscopic Functions: Concentration Fluctuation in the Long-Wavelength Limit and Chemical Short-Range Order Parameter (CSRO)
The concentration fluctuation function, S CC (0), is an important function which allows to understand the structure and the binding of atoms at the microscopic level. It is associated to binary liquid alloys having a tendency to compound formation or phase segregation. It is related to dynamic and physical properties: viscosity, diffusion[37], and surface tension.[38–40] The concentration fluctuation function S CC (0) is thermodynamically associated to Gibbs energy of mixing, \( G_{M} = G_{M}^{xs} + RT \sum _{i} C_{i} { \ln } C_{i} , \) and activity of pure components (a A and a B ). It can be expressed in the following form[17,18]:
Formula for determination of S CC (0) within CFM formalism is reported in the paper.[35]
For ideal mixing the energy parameters, W and W ij equal to zero, and the concentration–concentration fluctuation function (S CC (0, id)) is written as
The presence of a chemical ordering is found for such inequality S CC (0) < S CC (0, id), while the phase segregation occurs for S CC (0) > S CC (0, id).[41,42] A quantification of ordering or segregation effects in the melt is studied in terms of the Warren-Cowley short-range parameter (α 1).[19,20] It gives an insight into the local arrangement of atoms in the molten systems. This parameter is related to \( S_{CC} (0) \) by the relation[43]:
Z is the coordination number and was taken as 10.[36] For equiatomic composition, α 1 is found to be within the range of \( \left\langle { - 1,1} \right\rangle \), the value −1 means a complete ordering in the AB alloy and suggests a tendency for compound formation. Whereas, α 1 = 1 indicates tendency for segregation of AB melt elements.
2.3.1 Diffusivity
We can use this formalism to discuss the impact of ordering effects on diffusivity. Based on Darken’s thermodynamic equation,[44] the relation between diffusion and S CC (0) can be written as[43]
where D M is the mutual diffusion coefficient and D id is the intrinsic diffusion coefficient for ideal mixture. It is given as follows:
D A , D B define the self-diffusion coefficients of A and B constituent of the alloy. For a system having a tendency to compound formation, both inequalities, i.e., (S CC (0) < S CC (0, id)) and D M > D id are satisfied. The pronounced peak in the concentration dependence of D M /D id is related to the formation of the most probable associates in the liquid phase.[45,46]
2.4 Viscosity
Various expressions for viscosity (η) calculation are reported in the literature: Moelwyn-Hughes[47], Kucharski[29], Kaptay[30], Kozlov[31], Gasior[48] etc. In this work, the expressions of Kucharski (K), Kozlov–Romanov–Petrov (KRP), and Kaptay (Kap) models were applied to calculate the viscosity of liquid binary alloys. Viscosity formulae of K (η K), KRP (η KRP), and Kapt (η Kap) are as follows:
Parameters in Eqs. [20–22] i.e. η i , γ i , V i , (i = A, B) are viscosity, activity coefficient, and atomic volume of pure components of alloy, respectively. V is the molar volume, l is a fitted parameter in Kucharski model and α coefficient equal to 0.155 ± 0.055.G i *, H M are activation Gibbs energy of i-th component and mixing enthalpy of the binary system, correspondingly.
2.5 Butler Formulation: Surface Tension[28]
Surface tension of a binary liquid metallic system, σ, in Butler approach is expressed as
where G xc,b i is the partial excess Gibbs energy of A and B component in the bulk and it is expressed by Redlich–Kister polynomials[49]:
where L k ij , (i, j = A, B; k = 0, 1, 2…) are temperature dependent parameters.
The partial excess Gibbs energy of an i-th component in the surface layer, \( G_{i}^{xc,s} \), is assumed to be smaller than that in the bulk phase \( G_{i}^{xc} \), due to a smaller value of the coordination number in the surface layer with respect to the bulk phase.
The adjustable parameter β = 0.83 in Eq [25] was applied in an assessment of excess Gibbs energy of a monolayer as Tanaka et al.[50] suggest for surface tension calculation.
3 Discussion: Experimental- and Modeling-Based Results
3.1 Draining Crucible Method (DC): Density, Surface Tension, and Viscosity Measurement
The draining crucible method was used to determine three physicochemical properties: density, surface tension, and viscosity of liquid Al-Li and Li-Zn alloys. The studied liquid binary alloys were prepared directly in a molybdenum crucible in testing equipment of DC method presented in Figure 1. The equipment for the DC method was constructed and placed in a glove box with high purity argon, to provide the best possible protective atmosphere during the experiment. The level of O2 and H2O was hold below 1 ppm and measured by solid-state analyzers for absorption of oxygen and moisture. To held very low concentration of the above mentioned impurities in Ar, the continuous circulation of Ar between the glove-box and purification system was kept.
Pure metals of Al 99,999 pct, Zn 99,999 pct, and Li 99,9 pct (Alfa Aesar) were used for the preparation of Al-Li and Li-Zn alloys. These components of Al-Li and Li-Zn alloys were kept in a crucible and heated up to desired temperature which was controlled with a thermocouple placed inside the crucible and connected to a temperature controller. After melting and temperature stabilization, the mass of molten alloy flowing out of the crucible as a function of time was measured. The cumulative mass C m vs time curve for pure Al, Al-Li, and Li-Zn alloys was obtained and fitted by polynomial. Knowing the C m , the experimental volumetric flow rate V exp was determined and applied in formula (3–6). This information was also used for the determination of the discharge coefficient C d and Reynolds number (Re) which are necessary for calibration of a crucible. It means that the calibration C d vs Re is developed by expressing this relation by polynomial Eq. [2]. It was done for pure aluminum because of known three determined physical properties. Determined parameters of this equation for pure Al are applied for determination of unknown physicochemical properties for binary Al-Li and Li-Zn alloys using Eq. [6]. Calibration procedure was in detail described elsewhere.[22,25] The crucible used in these experiments had the same dimensions as the one used in our previous work.[22,32] The results presented in this work were measured in the temperature range from 773 K to 973 K (500 °C to 700 °C) with 50 K temperature step, and the mass of the measured alloys was between 0.5 and 0.7 kg.
In this work, we present the measured physicochemical properties in the liquid range for 10, 20, and 25 at. pct of lithium content in Al-Li alloys and from 9 to 41 at. pct of Li content in Li-Zn alloys. The choice of lithium concentrations was limited by technical difficulties to perform an experiment at high temperatures as well as high lithium reactivity.
The least-squares fitting procedure for three determined physicochemical properties: ρ, σ, η vs temperature (T) was applied. Eq. [26] was used for the description of linear temperature dependence of density and surface tension, whereas viscosity was expressed in Arrhenius form Eq. [27].
where Y represents ρ or σ, A is the ordinate intercept and B is the slope, A′ is pre-exponential parameter, and E act is an activation energy.
Experimental data described with linear and exponential temperature dependencies of the form (26) and (27) are collected for Al-Li and Li-Zn alloys in Tables I and II, correspondingly.
3.1.1 Surface tension
The surface tension of liquid binary Al-Li and Li-Zn alloys has not yet been investigated; only the pure constituents of both alloys were measured. The determined surface tension as a function of temperature is presented in Figure 2 for Al-Li system and for Li-Zn alloy in Figure 3. Both investigated liquid systems show that surface tension decreases with increasing temperature and with increasing lithium content.

Temperature dependence of the measured surface tension of liquid Al-Li alloys, i.e., triangle—pure Al,[23] diamond—10 at. pct Li, circles—20 at. pct Li and stars—25 at. pct Li

Temperature dependence of the measured surface tension of liquid Li-Zn alloys, i.e., triangle—pure Zn,[22] diamond—9 at. pct Li, circles—22 at. pct Li, stars—33 at. pct Li and squares—41 at. pct Li
3.1.2 Density
To our knowledge, density of liquid Al-Li and Li-Zn alloys has not yet been investigated. The temperature dependencies of the measured density in liquid Al-Li and Li-Zn systems are shown in Figures 4 and 5. Density of both alloys decreases with the Li addition, which is more pronounced in the case of Al-Li alloys.

Temperature dependence of the measured density of liquid Al-Li alloys, i.e., triangle—pure Al,[23] diamond—10 at. pct Li, circles—20 at. pct Li and stars—25 at. pct Li

Temperature dependence of the measured density of liquid Li-Zn alloys, i.e., triangle—pure Zn,[22] diamond—9 at. pct Li, circles—22 at. pct Li, stars—33 at. pct Li and squares—41 at. pct Li
3.1.3 Viscosity
Viscosity of pure liquid metals was examined by many authors. However, the Li-Zn alloys have not been investigated, while liquid Al-Li alloys have been studied only by Kononenko et al.[51] The temperature dependencies of viscosity are presented in Figures 6 and 7. It is observed a different trend in temperature dependence of viscosity for aluminum- and to zinc-based alloys. Namely, viscosity increases with increasing Li content for the liquid Al-Li alloys in opposition to the liquid Li-Zn system where it decreases with increasing Li content.

Temperature dependence of the measured viscosity of liquid Al–Li alloys, i.e., triangle—pure Al,[23] diamond—10 at. pct Li, circles—20 at. pct Li and stars—25 at. pct Li

Temperature dependence of the measured viscosity of liquid Li-Zn alloys, i.e., triangle—pure Zn,[22] diamond—9 at. pct Li, circles—22 at. pct Li, stars—33 at. pct Li and squares—41 at. pct Li
3.2 Compound Formation Model
An optimization procedure of energy parameters: W, ΔW AB , ΔW AA , ΔW BB in Eq. [7] was performed using a program developed by one of co-authors, utilizing the least-squares method. These parameters were obtained by fitting the mixing Gibbs energy Eq. [7] to the experimental data taken for Li-Zn and Al-Li systems from,[7, 52] respectively. They were used to compute surface tension (Eq. [10a], [10b]), two microscopic functions (Eqs. [15] and [17]), and diffusivity Eq. [18]. Each physical property was computed for two assumed cluster types.
3.2.1 Thermodynamic properties of liquid Al-Li and Li-Zn alloys
The CFM formalism has been applied to assess the interaction energy parameters required for evaluation of two microscopic functions, surface tension and diffusion in liquid Al-Li and Li-Zn alloys at temperature of 973 K and 873 K (700 °C and 600 °C), respectively. Thus, available experimental thermodynamic data, phase equilibria information, and concentration dependence of mixing Gibbs energy were used to determine m and n integer numbers in Eq. [7]. They are used for determination of thermodynamic and surface properties as well as microscopic functions of liquid systems. The examined binary liquid alloys, i.e., Al-Li and Li-Zn, characterize by the presence of many intermetallic phases in the solid state. The mixing functions of investigated liquid alloys display a negative deviation from Raoult’s law in the entire concentrations range.
Based on available thermodynamic data, G m /RT as a function of lithium concentration was computed for liquid Al-Li and Li-Zn alloys. It exhibits a visible minimum positioned in the vicinity of equiatomic composition (Figures 8 and 9). Values of G m /RT at minimum position for both liquid alloys are −1.345RT and −1.462RT, correspondingly. This suggests the tendency for a weak compound formation in the liquid state of both metallic systems.


The calculated interaction energy parameters in RT units [J/mol] are W = −0.88, W AA = −1.2, W AB = −13.6, and W BB = −1.8 of the liquid Al-Li system for m = 2 and n = 3. Their choice is due to the position of G m /RT minimum in the liquid Al-Li alloy locating in the range of 0.50 to 0.60 lithium concentration for data optimized by Hallstedt.[51] It can be assumed that the formation of Al2Li3 compounds in the liquid state of Al-Li alloys is energetically favored in comparison to the formation of other possible associates.
The concentration dependence of mixing Gibbs energy in the liquid Li-Zn alloys, obtained using Gasior data[7], is asymmetric, and its minimum position at zinc content of 0.60 to 0.65 is observed (Figure 9). It can suggest that LiZn2 chemical compound enriches the liquid phase of the Li-Zn system. Then, the calculated interaction energy parameters in RT units are W = 0.78, W AA = 0, W AB = −6.45, and W BB = −2.4 of the Li-Zn system for m = 1 and n = 2.
They were kept constant for further computations. It can be seen that the predicted mixing Gibbs energies using CFM formalism are in a good agreement with experimental data for both investigated systems, including asymmetry behavior.
3.2.2 Surface properties: surface concentration and surface tension
In this work, the experimental data of surface tension of pure elements, i.e., Al from,[23] Li from[21] and Zn from[22] are used to compute this property for studied alloys. They have been taken as
Based on formulae (Eq. [10a], [10b] and (21(a,b)), related to the CFM formalism and Butler model[28], respectively, surface properties of liquid Al-Li and Li-Zn alloys were computed. After rearranging mathematical formula, the obtained equation has been numerically solved with respect to the surface concentration of the i-th component. Computations of surface composition of liquid Al-Li and Li-Zn alloys were carried out at 973 K and 873 K(700 °C and 600 °C), respectively. Results are presented in Figures 10 and 11, respectively.

Surface concentration (C s Li ) with vs Li (C Li ) concentration in the bulk of liquid Al-Li alloys at 973 K (700 °C), i.e., solid line—linear, dashed line—Butler model,[28] dash-dotted line—for CFM formalism

Surface concentration (C s Li ) vs Li (C Li ) concentration in the bulk of liquid Li-Zn alloys at 873 K (600 °C), i.e., solid line—linear, dashed line—Butler model,[28] dash-dotted line—for CFM formalism
Tendency to accumulation of Li-atoms at the monoatomic surface is observed in the whole concentration range for both Al-Li and Li-Zn alloys. It is less pronounced for liquid Al-Li than for Li-Zn systems.
The measured surface tensions of the liquid Al-Li alloys have higher values than those computed using of both applied models in this work. Moreover, they are closer to data calculated applying Butler model (Eqs. [23a] and [23b]) than CFM formalism (Eqs. [10a] and [10b]). A wave curve describes the concentration dependency of surface concentration of Li, C s Li , (Figure 12) with two inflection points and one minimum positioned in the vicinity of assumed clusters. Moreover, the measured and modeled surface tension isotherms show negative deviation with respect to the linear concentration dependence. Thus, the tendency to compound formation of these alloys is confirmed.

Concentration dependence of surface tension of the Al-Li alloys at 973 K (700 °C), i.e., solid line—linear, dashed line—Butler model,[28] dash-dotted line—for CFM formalism, squares—the data measured in this work
For the Li-Zn system, the measured surface properties correlate better with the results obtained using Butler model than those within CFM formalism. The deviation of surface tension, both measured and modeled (only Butler case), is negative with respect to the linear dependence contrary to CFM results displaying changeable behavior (Figure 13). The varying tendency in surface tension, observed for CFM model, indicates that the monoatomic surface layer of zinc-rich alloys is enriched in Zn. The changeable behavior appears at about 0.35 to 0.4 lithium content when first intermetallic phase starts to grow.[8]

Concentration dependence of surface tension of the Li-Zn alloys at 873 K (600 °C), i.e., solid line—linear, dashed line—Butler model,[28] dash-dotted line—CFM formalism, squares—the data measured in this work
3.2.3 Microscopic functions: the concentration–concentration function and short-range order parameter
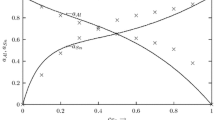
The ordering tendency in liquid Al-Li and Li-Zn alloys has been analyzed through the concentration fluctuation function in the long wavelength limit, S CC (0)[12] and the Warren-Cowley short-range order parameter, α 1,[19, 20] as a function of bulk concentration. The computed values of S CC (0) and α 1 of the liquid Al-Li and Li-Zn alloys are shown in Figures 14 and 15, respectively. Experimental determination of S CC (0) is very difficult due to measurement complexity. However, Bathia-Thornton theory[17, 18] allows to determine this function using Eq. [15].

Concentration dependence of S CC (0), S CC (0, id) functions and the Warren-Cowley short-range order parameter of the Al-Li alloys at 973 K (700 °C), i.e., solid line—S CC (0, id), dashed line—S CC (0) with CFM, triangle—S CC (0) with literature data[52], diamonds—α 1literature data[52] and dash-dotted line—α 1within CFM

Concentration dependence of S CC (0), S CC (0, id) function and Warren-Cowley short-range order parameter of the Li-Zn alloys at 873 K (600 °C), i.e., solid line—S CC (0, id), dotted line—S CC (0) with CFM, triangle—S CC (0) with literature data,[7] diamonds—α 1literature data[7] and dashed line—α 1within CFM
The values of S CC (0), obtained applying Eq. [15] for liquid Al–Li alloy (Figure 14), show a shallow minimum in the range where intermetallic phases occur. The computed S CC (0) has lower values than S CC (0, id) for ideal mixing, indicating the tendency of dissimilar atoms to bond. The computed Warren-Cowley short-range parameter using Eq. [17] shows the changeable ordering phenomena at Al- and Li-rich sides, and the result is shown in Figure 14. Namely, on Al- and Li-rich sides, flat maxima appear signifying random mixing contrary to a well-pronounced peak at 0.6 and as hallow minimum at 0.5 to 0.7 lithium concentration. The position of this peak signifies the possibility of Al2Li3 occurrence in the Al-Li melt. Generally, the most pronounced difference between functions of concentration fluctuations in the long-wavelength limit (S CC (0) and S CC (0, id)) indicates that the compound formation may take place.
The concentration dependencies of concentration fluctuation function, S CC (0), and Warren-Cowley short-range parameter of liquid Li-Zn alloys are analogous to those observed for liquid Al-Li alloys and presented in Figure 15. The computed S CC (0) shows that liquid Li-Zn alloys have tendency to compound formation which is weaker than this observed for liquid Al-Li alloys. The curve is smooth without characteristic points which are present for the liquid Al-Li. The short-range order parameter (α 1) of the liquid Li-Zn system exhibits one maximum in the Zn-rich side and a visible shallow minimum at the zinc content of 0.6 to 0.7. It also signifies the occurrence of clustering effects of unlike atoms.
Thus, it can be said that LiZn2 clusters in the Li-Zn melt are favored in comparison to the other complexes that may exist.
Although, the minimum position of mixing Gibbs energy obtained using CFM formalism is shifted to the Zn-rich side due to a non-perfect matching between experimental and calculated data (Figure 9).
3.2.4 Diffusivity
Based on the CFM formalism, the diffusivity of liquid Al-Li and Li-Zn alloys has been investigated. Figures 16 and 17 present concentration dependencies of D m /D id . The observed changes of analyzed diffusivity are related to changes observed for the short-range order parameter, α 1. To describe the relationship between S CC (0) and diffusivity, the Eq. [18] was used, which expresses the ratio of the mutual diffusion to intrinsic diffusion coefficients, D m /D id . It describes the mixing behavior of the liquid alloy, i.e., tendency to phase segregation or compound formation, since D m /D id < 1 orD m /D id > 1, respectively.[37, 53] The modeled data in the framework of CFM formalism are in better agreement with experimental results for the liquid Al-Li alloys[52] than for the liquid Li-Zn alloys.[7] Both examined liquid systems display changeable mixing behavior, in the Al-rich part of the Al-Li system and the Zn-poor part of the Li-Zn alloys. In these cases, the phase segregation tendency occurs. However, the tendency to compound formation corresponds to the maintained maxima. Positions of these maxima are in the concentration range of intermetallic phases occurrence (in the solid state). These are Al2Li3 in the Al-Li melt for C Li ≈ 0.55 and LiZn2 in the Li-Zn melt for C Zn ≈ 0.66. The values of D m /D id at maximum position, calculated in the framework of CFM, for both investigated alloys, are equal to 3.63 and 3.16 for Al-Li and Li-Zn, respectively.

Concentration dependence of D m /D id of the liquid Al–Li alloys at 973 K (700 °C), i.e., solid line—obtained using the assessed energy parameters within CFM formalism, stars—obtained using literature data[52]

Concentration dependence of D m /D id of the liquid Li-Zn alloys at 873 K (600 °C), i.e., solid line—obtained using the assessed energy parameters within CFM formalism, stars—obtained using literature data[7]
3.2.5 Viscosity
Viscosity isotherms of the Al-Li alloys at 973 K (700 °C) are presented in Figure 18. The results of Kononeko et al.[51] were also considered for the comparison to the measured kinematic viscosity. However, they had to be converted from dynamic to kinematic viscosity. It was done using density data obtained in this work; the present results at 973 K (700 °C) are plotted in Figure 18. The measured viscosity data and those obtained by Kononenko et al.,[51] increase with increasing Li content. The Kononenko viscosity values for pure aluminum and Al-based alloys are lower than the corresponding ones determined by authors of this work. Also, the data modeled using Kucharski[29] and KRP[31] models are plotted in Figure 18. These results are in good agreement with those measured. The presence of a maximum or an inflection point at a concentration close to the stoichiometric composition of an intermetallic compound is often observed for systems having a tendency for compound formation.[10] In the case of the Al-Li alloys, the maximum is located at a position corresponding to the Al2Li3 compounds.

The viscosity isotherm of the Li-Zn alloys at 873 K (600 °C) is plotted in Figure 19. Positive deviation from linearity in the viscosity isotherm has been observed for Li-Zn system. The computed viscosities using Kucharski model give values closest to those measured in this work in comparison to Kozlov et al.[31] and Kaptay[30] models. Moreover, a smooth inflection point on η − C Li curve is noticed and its location correlates with the chemical compounds existing in the melt, i.e., LiZn2.

The application of Kucharski model[29] confirms the ability to form compounds and the existence of LiZn2. This ability is also confirmed by investigation of other properties presented in this work, i.e., diffusion, S CC (0) function or short-range order parameter (α 1).
4 Conclusions
The present work provides an experimental of the surface, dynamic, and volume properties of liquid Al-Li and Li-Zn alloys. In addition, we performed calculations of the same systems, as well.
Performed experiments show the lowering of density, surface tension, and viscosity with increasing Li content for both investigated liquid alloys. For viscosity, our experimental results of liquid Al-Li alloys are comparable to the Kononenko et al.[51] data.
The applied CFM formalism with an assumption of the existence of Al2Li3 and LiZn2 compounds in the corresponding alloys (Al-Li and Li-Zn) predicts surface properties in correct agreement with experimental ones. However, data obtained using Butler model[28] yield to a better agreement with the measured data.
Tendency for the formation of abovementioned transient compounds corresponds to the maintained maxima of S CC (0) and diffusion coefficient ratio positioned at the concentration range of the existence Al2Li3 and LiZn2 clusters. The computed viscosity isotherm for Al-Li at 973 K (700 °C) has stronger positive deviation from ideal mixing than for Li-Zn alloys.
References
R.A. Huggins, J. Power Sources, 1999, vol. 81, pp. 13.
C.J. Wen, J. Electrochem. Soc., 1979, vol. 126, pp. 2258.
J. Wang, P. King, R.A. Huggins, Solid State Ionics, 1986, vol. 20, pp. 185.
J. Wang, J. Electrochem. Soc., 1986, vol. 133, pp. 457.
Z. Moser, F. Sommer, B. Predel, Z. Metallkd., 1988, vol. 79, pp. 705.
Z. Moser, F. Sommer, J. Lee, B. Predel, Thermochim. Acta, 1989, vol. 142, pp. 117.
W. Gasior, Z. Moser, J. Chim. Phys., 1993, vol. 90, pp. 387.
A. Pelton, J. Phase Equilibria, 1991, vol. 12, pp.42.
H. Okamoto, J. Phase Equilibria Diffus., 2012, vol. 33, pp 500.
J.H. Westbrook, R.L. Fleischer, Intermetallic Compounds: Principles and Practice - Vol. 1: Principles, Wiley, Chichester, 1995.
A.B. Bhatia, W.H. Hargrove, Lett. Al Nuovo Cim. Ser., 1973, vol. 2, pp. 1025.
A. Bhatia, W. Hargrove, Phys. Rev. B, 1974, vol. 10, pp. 3186.
A.B. Bhatia, R.N. Singh, Phys. Chem. Liq., 1982, vol. 11, pp. 285.
A.B. Bhatia, R.N. Singh, Phys. Chem. Liq., 1982, vol. 11, pp. 343.
A.B. Bhatia, R.N. Singh, Phys. Chem. Liq., 1984, vol. 13, pp. 177.
L.C. Prasad, R.N. Singh, Phys. Chem. Liq., 1990, vol. 22, pp. 1.
A.B. Bhatia, W.H. Hargrove, N.H. March, J. Phys. C Solid State Phys., 1973, vol. 6, pp. 621.
A. Bhatia, W. Hargrove, D. Thornton, Phys. Rev. B, 1974, vol. 9, pp. 435.
J.M. Cowley: Diffraction Physics, North-Holland, Amsterdam and Elsevier, New York, 1975.
B.E. Warren, X-Ray Diffraction, Addison-Wesley, Reading MA, 1969.
B.J. Keene, Int. Mater. Rev., 1993, vol. 38(4), pp. 157.
T. Gancarz, W. Gąsior, H. Henein: Int. J. Thermophys., 2013, vol. 34, pp. 250.
H. Henein, T. Gancarz, J. Jordan, and W. Gasior: Metall. Mater. Trans. B (in press).
W.F.Gale, T.C.Totemeier, Smithells Metals Reference Book (Elsevier, Butterworth-Heinemann, Oxford), 2003.
S.J. Roach, H. Henein, Can. Metall. Quat., 2003, vol. 42, pp. 175.
H. Henein, Can. Metall. Quat., 2004, vol. 44, pp. 261.
S.J. Roach, H. Henein, Metall. Mater. Trans. B, 2005, vol. 36, pp. 667.
J. A. V. Butler, Proc. R. Soc. A Math. Phys. Eng. Sci., 1932, vol. 135, pp. 348.
M. Kucharski, Z. Metallkd., 1986, vol. 77, pp. 393.
I. Budai, M.Z. Benkő, G. Kaptay, Mater. Sci. Forum, 2007, vol. 537-538, pp. 489.
L. Kozlov, L. Romanov, N. Petrov, Chernaya Metall., 3:7, 1983
T. Gancarz, Z. Moser, W. Gąsior, J. Pstruś, H. Henein, Int. J. Thermophys., 2011, vol. 32, pp. 1210.
M. Johnson and C. Hooke: http://www.netlib.org/opt/hooke.c, 1994.
R. Novakovic, T. Tanaka, M. L. Muolo, J. Lee, A. Passerone, Surf. Sci., 2005, vol. 591, pp. 56-69.
R. Novakovic, E. Ricci, M.L. Muolo, D. Giuranno, A. Passerone, Intermetallics, 2003, vol. 11, pp. 1301.
[36] E. A. Guggenheim, Trans. Faraday Soc., 1945, vol. 41, pp. 348.
R.N. Singh, F. Sommer, Phys. Chem. Liq., 1998, vol. 36, pp. 17.
L. Prasad, R. Singh, Phys. Rev. B, 1991, vol. 44, pp. 13768.
L.C. Prasad, R.K. Jha, Phys. Status Solidi, 2005, vol. 202, pp. 2709.
L.C. Prasad, R.N. Singh, G.P. Singh, Phys. Chem. Liq., 1994, vol. 27, pp. 179.
R.N. Singh, Can. J. Phys., 1987, vol. 65, pp. 309.
R.N. Singh, Phys. Chem. Liq., 1993, vol. 25, pp. 251.
R.N. Singh, F. Sommer, Reports Prog. Phys., 1997, vol. 60, pp. 57.
L. Darken, Trans. Met. Soc. Aime, 1948, vol. 175, pp. 184.
R. Novakovic, M.. Muolo, A. Passerone, Surf. Sci., 2004, vol. 549, pp. 281.
R. Novakovic, E. Ricci, D. Giuranno, A. Passerone, Surf. Sci., 2005, vol. 576, pp. 175.
E. Moelwyn-Hughes: Phys. Chem., 1970, vol. 434.
W. Gąsior, CALPHAD, 2014, vol. 44, pp. 119.
O. Redlich, A.T. Kister, Ind. Eng. Chem., 1948, vol. 40, pp. 345.
T. Tanaka, K. Hack, T. Iida, S. Hara, Z. Metallkd., 1996, vol. 87, pp. 380.
V.I. Kononenko, A.A. Razhabov, A. V. Ryabina, Russ. Metall., 2011, vol. 8, pp. 774.
B. Hallstedt, O. Kim, Int. J. Mater. Res., 2007, vol. 98, pp. 961.
L. Prasad, R.. Singh, V. Singh, S.. Chatterjee, Phys. B Condens. Matter. 215 (1995) 225.
Acknowledgments
Present work was financed by National Science Centre Poland, PRELUDIUM Grant (Project No 2011/03/N/ST8/05308).
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted March 10, 2014.
Appendix
Appendix
1.1 Symbols
- a 1, a 2 :
-
Constants in the polynomial C d vs Re
- A′:
-
Pre-exponential parameter in the Arrhenius equation
- A, B :
-
Components of a binary AB system
- Bo :
-
Bond number
- C, (1 − C):
-
Concentration of A and B atoms in molar fraction, respectively
- C s, (1 − C s):
-
Concentration of A and B atoms at surface phase in molar fraction, respectively
- C d :
-
Discharge coefficient
- D M :
-
Inter-diffusion coefficient of an binary alloy
- D id :
-
Intrinsic diffusion coefficient for an ideal solution
- D i (i = A, B):
-
Self-diffusion coefficient of component i
- E act :
-
Activation energy in Arrhenius equation
- Fr :
-
Froude number
- g :
-
Gravitational acceleration constant
- G i *:
-
Activation energy of i-th component in Eq. [22]
- \( G_{M}^{xc} \) :
-
Excess Gibbs energy of system
- \( G_{i}^{xc,b} \)(i = A,B):
-
Partial excess Gibbs energy of component i of the bulk phase
- \( G_{i}^{xc,s} \)(i = A,B):
-
Partial excess Gibbs energy of component i of the surface phase
- h :
-
High of melt in a crucible
- H M :
-
Mixing enthalpy of an alloy
- l :
-
Fitted parameter in viscosity model of Kucharski (Eq. [20])
- L k ij , (i, j = A, B; k = 0, 1, 2…):
-
Parameters in Redlich–Kister equation
- M :
-
Molar mass of an alloy
- N :
-
Avogadro’s number
- n, m :
-
Stoichiometric coefficient
- p, q :
-
Surface coordination
- Q :
-
Flow rate
- r 0 :
-
Orifice radius at the bottom of a crucible
- R :
-
Gas constant
- Re:
-
Reynolds number
- S :
-
Surface area, mean surface area of an alloy
- S i :
-
Mean surface area of component i
- S CC (0, id):
-
Concentration fluctuation function for ideal mixing
- S CC (0):
-
Concentration fluctuation function
- T :
-
Absolute temperature
- V exp :
-
Mass flux
- V i :
-
Partial molar volume of i-th component
- V :
-
Molar volume of a liquid system
- W, ΔW AB , ΔW AA , ΔW BB :
-
Energy parameters in Eq. [7]
- Z :
-
Coordination number
- α :
-
Adjustable parameter in Eq. [22]
- α 1 :
-
Warren-Cowley short-range order parameter
- β :
-
Adjustable parameter in Eq. [25]
- ϕ ij and f ij :
-
Concentration functions describing bulk phase
- ϕ s ij and f s ij :
-
Concentration functions describing surface phase
- η :
-
Viscosity
- ρ :
-
Density
- σ :
-
Surface tension
- γ i (i = A, B):
-
Activity coefficient of component i
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Trybula, M., Gancarz, T., Gasior, W. et al. Bulk and Surface Properties of Liquid Al-Li and Li-Zn Alloys. Metall Mater Trans A 45, 5517–5530 (2014). https://doi.org/10.1007/s11661-014-2524-6
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-014-2524-6