
Abstract
Ganoderma boninense is a telluric lignicolous basidiomycete and the causal agent of basal stem rot, one of the most devastating diseases of the oil palm (Elaeis guineensis). While the fight against G. boninense is of major concern in Southeast Asia, little information is available regarding the genetic diversity and evolutionary history of the fungus. In this context, the development of an informative molecular marker set to characterize the diversity of G. boninense is a key step towards understanding the biology of this pathogen. A G. boninense draft genome sequence of 63 Mbp, assembled using 454 and Illumina sequencing technology, was used to identify and develop a set of microsatellite markers (simple sequence repeats, SSRs). A total of 2487 SSRs were identified, for which 145 SSR primer pairs were designed. These SSRs are characterized by di- to hexanucleotide motifs with 5 to 34 repetitions. Ninety-seven SSR loci were successfully amplified on an initial small set of G. boninense isolates from Indonesia. A collection of 107 isolates from several regions in Southeast Asia were screened to characterize each locus for allele number, polymorphism information criterion and the presence or absence of null alleles at each locus. These results allowed us to propose an effective set of 17 SSRs for studying genetic diversity within G. boninense.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The availability of new molecular biology tools, along with the exploration of genomic information, has made it possible to develop a large number of genetic markers, including microsatellites. Microsatellites, also known as simple sequence repeats (SSRs), are DNA regions comprising short tandem repeat units from one to six base pairs in length. Polymorphic microsatellites have become one of the most popular and commonly used molecular markers for studying genetic diversity, genealogy and molecular ecology (Jarne and Lagoda 1996). In particular, microsatellites remain the most powerful and widely used tool for studying population genetics and the geographical spread of plant pathogens (Schoebel et al. 2013a, b; Presti and Wasko 2014). This success results from their codominance, high polymorphism, high allele number and relatively high abundance in most genomes, and the ease of typing by polymerase chain reaction (PCR).
Until recently, developing microsatellite markers for a previously unsequenced species involved the construction of an enriched DNA library by cloning sequences in plasmids and sequencing them using the Sanger method (Zane et al. 2002), which was expensive and time-consuming. With the development of next-generation sequencing (NGS) technologies, large amounts of genomic sequence data can be made available at a low cost ($0.05 for 1 Mbp in April 2014 against $5292 for 1 Mbp in September 2001; NIH estimate: http://www.genome.gov/sequencingcosts), providing a faster and cheaper solution for finding microsatellites without a priori knowledge of the target species. In recent years, NGS platforms such as Illumina GAII and Roche 454 have enabled the availability of larger sets of microsatellite markers by exploring complete genome sequences, even for non-model species (Abdelkrim et al. 2009; Zalapa et al. 2012; Cai et al. 2013; Schoebel et al. 2013a, b). Until now, most studies have searched raw NGS data for microsatellite patterns for the design of primers (Gardner et al. 2011). The use of NGS raw data, i.e. raw reads, has a number of limitations. The length of raw reads from the 454 (500 bp) and Illumina (250 bp) systems is often not sufficient to design primers for both sides of the SSR, which is a critical step in developing microsatellite markers (Castoe et al. 2010). Proper primer design for SSR genotyping, therefore, should satisfy several conditions, including a minimum PCR product size of 100 bp, in order to avoid confounding artefacts caused by primer peak or the same melting temperature (Tm) for both forward and reverse primers. Castoe et al. (2010) were able to design primers for only 49.6 % of the SSRs they identified in their data set of 454 raw reads, and Cai et al. (2013) were able to design primers for only 32.7 % to 40.1 % of the SSRs they identified in their Illumina raw read data set. Moreover, sequence redundancy in raw reads poses a major challenge in the design of locus-specific primers, as determining whether reads with similar sequences correspond to a single locus or to duplicate regions is not a trivial task (Castoe et al. 2010). New approaches based on exploring genome assemblies seem to be a promising way of circumventing both problems caused by the use of raw reads (Cai et al. 2013). Read assembly consists in aligning and merging reads into much longer DNA/RNA fragments in order to reconstruct the original molecule. This method thus provides much longer sequences—about 1 to 10 kbp for a raw assembly against a maximum of 600 bp for 454 raw reads or 300 bp for Illumina raw reads (Zalapa et al. 2012; Wei et al. 2014)—and reduces data complexity considerably.
Despite the advent of NGS, developing fungal microsatellite markers is still challenging (Dutech et al. 2007; Schoebel et al. 2013a, b). While the small size of fungal genomes, most ranging from 10 to 60 Mbp (Gregory et al. 2007), makes it easier to explore the genome to find microsatellite loci, it appears that microsatellites are less abundant in fungi than in any other organism (Dutech et al. 2007). Fungal microsatellite motifs are also known to have low numbers of repetitions (most fewer than eight), with a high proportion of mononucleotide motifs, making it difficult to find highly polymorphic SSRs (Dutech et al. 2007). Finally, the cross-species transferability of SSRs in fungi seems to be lower than in other organisms (Barbara et al. 2007; Dutech et al. 2007).
In this study, we developed an SSR marker set from a draft genome assembly of Ganoderma boninense, a telluric white rot basidiomycete that is the causal agent of oil palm basal stem rot (BSR). G. boninense is a non-model plant pathogen fungus whose biology is poorly known (Ho and Nawawi 1986; Pilotti et al. 2002), but it belongs to the same genus as Ganoderma lucidum, a very popular medicinal fungus that has already been sequenced (Chen et al. 2012). G. boninense can affect up to 50 % of palm trees in a plantation, ranging from lower productivity to tree death (Corley and Tinker 2003), and is of major economic importance in Southeast Asia (Turner 1981). As palm oil represents the largest share of the world’s vegetable oil market, with 54 million tons produced in 2012 (United States Department of Agriculture: http://apps.fas.usda.gov/), 80 % of which is used by the food industry, BSR may have a serious effect on global food security. Most studies of G. boninense have been carried out to select resistant oil palm progenies (Idris et al. 2004; Durand-Gasselin et al. 2005; Breton et al. 2006) or to develop management practices to limit the spread of the disease (Susanto et al. 2005). To fight G. boninense more effectively, we must improve our understanding of its biology. Our knowledge of the physiology (Pilotti 2005; Rees et al. 2007) and genetics (Miller et al. 1999; Pilotti et al. 2003) of G. boninense is insufficient to explain its current range across an area as large as Southeast Asia. In recent years, population genetics has proved to be an important tool for understanding the biology and spread of plant pathogens (Jarne and Lagoda 1996; Travadon et al. 2012a, b). Genetic diversity patterns can provide information about the respective contributions of clonal and sexual reproduction (Travadon et al. 2012b) and the existence of gene flow barriers (Ali et al. 2014). Neutral molecular markers can be used to reconstruct the main dispersion paths of cosmopolitan or invasive plant pathogens (Montarry et al. 2010), and can even lead to the identification of the original hotspot of an emerging disease (Dutech et al. 2012).
The current work contributes to our knowledge of the genetic diversity of G. boninense. In this study, we used a “Seq-Assembly-SSR” approach (Cai et al. 2013) in our preliminary genome assembly, which was based on two sequencing methods (454 and Illumina), in order to develop a suitable set of SSR markers that was then used to produce an initial description of the genetic diversity of a collection of G. boninense isolates from southern Asia. As the microsatellite compatibility between closely related fungal species is often described as anecdotal (Dutech et al. 2007), we also tested the transferability of those markers to a closely related species, G. resinaceum. The development of this microsatellite marker set would seem to be essential for gaining a better understanding of the diversity of G. boninense, and may be a first step towards improving crop and disease management practices.
Materials and methods
Fungal material
As G. boninense is the main species found on oil palms (Ho and Nawawi 1985), we based our sampling on the morphological traits of basidiocarps as described by Ho and Nawawi (1986) and Flood et al. (2000). The basidiocarps may be stalked or sessile, flat or bracket-shaped. The dorsal surface is glossy, blackish-brown in color, with concentric markings. The edge is white when fresh, and the undersurface is also white. Five G. boninense specimens were collected from infected palms in industrial plantations owned by PT Socfin Indonesia in North Sumatra (Indonesia). Mycelium was purified and maintained in a standard potato dextrose agar (PDA) medium (39 g L−1 PDA from Sigma-Aldrich®, 0.5 g L−1 chloramphenicol, pH 7.0) in the dark at 34 °C. We assumed that these isolates are constituted of dikaryotic mycelium. One isolate, NJ3, which was also used for genome sequencing, and four others were used for the first SSR validation step. To assess the trans-specific amplification pattern for the SSRs, we obtained two isolates of G. resinaceum, a species closely related to G. boninense, from the CBS-KNAW Fungal Biodiversity Centre’s culture collection (http://www.cbs.knaw.nl/index.php/collection).
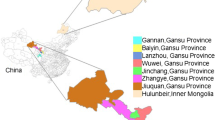
One hundred and seven fruiting bodies were collected from peninsular Malaysia (40 fruiting bodies), Borneo (20 fruiting bodies) and North Sumatra (47 fruiting bodies). Geographical locations and sample distribution are summarized in Table 1. Each sample was isolated from a single oil palm tree and identified as G. boninense using morphological criteria. Only dikaryotic tissues from the fruiting bodies were kept and freeze-dried before being packed with silica gel.
DNA extraction
Dikaryotic tissues from the fruiting bodies and 2-week-old pure cultures were crushed to powder, and the DNA was extracted using the MATAB [mixed alkyltrimethylammonium bromide] protocol, adapted from the CTAB [cetyltrimethylammonium bromide] protocol (Doyle 1987), with some modifications to improve the DNA extraction yield (Tris HCl 1 M pH 8, NaCl 5 M, EDTA 500 mM, MATAB, PEG 6000 and sodium sulfite). A first step with cell wall digestion by Glucanex (lysing enzymes from Trichoderma harzianum, Sigma-Aldrich®) buffer (0.5 g Glucanex®, 70 ml NaCl 1 M, 30 ml H2O, buffer at pH 6) produced a higher DNA yield and removed most of the polysaccharides. The MATAB protocol was also modified for 2-ml tubes to facilitate the extraction of a large set of samples. The DNA concentration (ng μl−1) of each sample was determined by fluorometry (Invitrogen Qubit® 2.0 Fluorometer; Thermo Fisher Scientific) to avoid measurement bias due to the high polysaccharide content.
Genome sequencing and assembly
The draft genome sequence was obtained using total genomic DNA extracted from the NJ3 isolate. A combination of 454 GS FLX reads (single reads with theoretical length of 400 bp) and Illumina HiSeq 2000 reads (paired reads with insert size of 3 Kb and theoretical length of 50 bp) were used for contig assembly. The reads were assembled using CLC Genomics Workbench 4.6.1 (CLC bio/QIAGEN), with standard parameters, and the N25, N50 and N75 genome assembly statistics were calculated. The N values are standard quality measurement values for genomic data (N25, N50, N75 are calculated by summing the lengths of the largest contigs until N % of the total contig length is reached, and the minimum contig length in this set is the number usually used to report the N value of a de novo assembly). This whole genome shotgun project has been deposited at DDBJ/EMBL/GenBank under accession number LFMK00000000. The version described in this paper is version LFMK01000000.
SSR development and primer design
The draft genome of G. boninense (NJ3 isolate) was screened with msatcommander 1.0.8 (Faircloth 2008) to identify SSRs. The parameters were set to search for di- to hexanucleotide perfect SSR motifs with a minimum of five repeats. In order to increase the success of the primer design and to allow for greater flexibility in the choice of fragment length, only candidate SSRs with flanking sequences of at least 400 bp were kept. Primers were designed using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), with the following requirements: optimum primer size of 20 bp, product size between 150 and 500 bp, primer melting temperatures (Tm) from 57 °C to 63 °C, with optimum at 60 °C, maximum 5bp complementarity between primer pairs, and primer location at least 2 bp away from targeted microsatellite locus. Each primer pair (forward and reverse) was tested with in silico PCR, using the NJ3 genome sequence and a local blastn procedure (Altschul et al. 1990) to select primer pairs that matched on a single genome location. Selected primer pairs were built with an M13 (5’CACGACGTTGTAAAACGA3’) (Boutin-Ganache et al. 2001) tail added on the 5’ side of the forward primer. This M13 sequence is complementary to the M13 fluoro-tagged primer.
The first in vitro validation of the designed primer pairs was performed on the five Indonesian G. boninense isolates maintained as pure cultures (the 5-Indonesian-isolate set) to identify primer pairs exhibiting a mono-locus amplification pattern and polymorphism. Only primer pairs producing mono-locus amplification and polymorphic products were used to genotype the set of 107 G. boninense fruiting bodies and two G. resinaceum isolates. All primer characteristics are given in Table 2.
PCR amplification and genotyping
Each microsatellite was amplified in simplex 10-μl PCR reactions containing 5 ng DNA, 100 nM of each of fluoro-tagged M13 (FAM, NED, VIC, PET), 100 nM of reverse primer, 80 nM of forward primer, 0.2 mM dNTPs, 0.6 mM MgCl2, 1X reaction buffer and 0.2 U QIAGEN Taq DNA polymerase, 5 % DMSO and BSA at 0.8 μg μl−1. After amplification, 2 μl of simplex PCR product for each dye (two markers per dye) were pooled in a plate, and 10 μl of a mixture of GeneScan 600 LIZ size standard and formamide (in a ratio of 1:125) was added to each well for genotyping.
For SSR validation using the 5-Indonesian-isolate set, genotyping was performed on a LI-COR 4300 automated DNA analyzer with IRDye sizing standards of 50–350 bp and 50–700 bp (LI-COR, Inc.). For SSR genotyping of the 107 G. boninense samples, PCR products were analyzed using a 3500xl genetic analyzer (Applied Biosystems; Thermo Fisher Scientific). All alleles were scored using GeneMapper software (version 4.1; Applied Biosystems).
SSR characteristics and diversity analysis
Allele number and genetic diversity are key marker characteristics, as they enable the quantification of marker information content. For each locus, the allele number, observed (H o ) and expected heterozygosity (H e ) were calculated using GENETIX 4.05.2 (Belkhir et al. 2004) for all samples together and for each geographical area: peninsular Malaysia, Borneo and Sumatra. Polymorphism information content (PIC) was calculated using the R package PopGenKit, based on Botstein et al. (1980). In addition, the number of observed genotypes was calculated for each locus for both the three geographical areas and the whole area.
Linkage disequilibrium (LD) is used to test for and quantify the statistical correlation between loci or alleles, and in particular, is able to detect whether markers convey redundant information due to their physical linkage. LD analysis between allele pairs was conducted at two levels, first on the whole set of samples and second for each geographical area, using FSTAT 2.9.3. Significance was assessed using a permutation test accounting for Bonferroni corrections with 40800 permutations [http://www2.unil.ch/popgen/softwares/fstat.htm, developed by Goudet (2002)].
A potential drawback to using SSRs is the presence of null alleles, which for various reasons (Dakin and Avise 2004) fail to amplify to the level of detection. Such null alleles generate false homozygote genotypes and may cause bias in population genetic estimates (Pemberton et al. 1995; Chapuis and Estoup 2007). For example, in genetic structure analysis, null alleles may reduce accuracy in assigning samples to populations and may overestimate Fst (Carlsson 2008). It is necessary, therefore, to identify loci with such null alleles and to exclude them from further analysis. Null alleles can be identified using indirect methods that account for excess homozygotes at null allele loci compared to other loci. As recommended by Dabrowski et al. (2014), we combined the results of two programs based on indirect methods, ML-NullFreq (Kalinowski and Taper 2006) and Micro-Checker (Van Oosterhout et al. 2004), to determine loci with null alleles. These two programs are complementary in the sense that Micro-Checker has a high detection rate associated with a high false-positive rate, while ML-NullFreq is less powerful but detects fewer false positives. Null allele detection was carried out independently for the three geographical areas, and we considered that loci with at least five negative results over six (three populations tested with two software programs) could be considered as having no null alleles and kept for further analysis.
A descriptive analysis of the genetic structure of the samples was carried out using the ade4 R package (Dray and Dufour 2007). To measure possible genetic differentiation between geographical areas, F st was calculated between samples from the three geographical areas and a principal component analysis (PCA) performed to study possible sample differentiation. The calculation of F st was based on Nei’s genetic distance (Nei 1973), with heterozygosities weighted by group size and significance assessed using a standard permutation test based on Markov chain (1000 permutations). Both PCA and Fst calculations were carried out twice, first with the SSR set with no null alleles and then with the full SSR set.
Results
Genome sequencing and assembly
The 454 pyrosequencing of the NJ3 isolate generated 2,599,370 reads, with an average read length of 378 bp. Illumina sequencing generated 276,922,790 reads, with an average read length of 51 bp. After assembly using the CLC Genomics Workbench 4.6.1 software, 21,425 contigs were obtained, with lengths varying from 501 to 89,905 bp and a mean length of 2864 bp. This assembly generated a genome with total length of 63,034,190 bp. The N25, N50 and N75 isolates were 10,965, 5913 and 2563 bp in length, respectively.
Characterization of microsatellite markers in G. boninense
Based on the draft genome assembly, msatcommander software identified a total of 2487 candidate SSR markers in the 21,425 contigs. The number of loci detected for each type of microsatellite loci and the number of repeats decreased from dinucleotide to hexanucleotide motifs, with the exception of the pentanucleotide motif, which had the lowest number of detected loci (Table 3).
The 1687 SSR markers with flanking sequences of at least 400 bp were selected to facilitate primer design. This selection was fairly equitable for most of the SSR types, with a mean of 32.5 % of loci discarded for di-, tri-, tetra- and hexanucleotide SSR markers and 61 % of the candidate pentanucleotide SSR markers. To save time, 145 SSR markers were randomly selected for the design of primer pairs to further assess marker properties. All primer pairs were designed to produce an expected PCR product size ranging from 150 to 400 bp. After in silico PCR was performed, only 110 primer pairs showed the expected mono-locus amplification pattern. Upon completion of in vitro PCR using these 110 primer pairs, 17 primer pairs (15.5 %) showed no amplification, while 93 (84.5 %) showed reproducible mono-locus amplification. Among these 93 primer pairs, 46 (49.5 %) exhibited polymorphic amplification products from the five Indonesian isolates. The SSR loci amplified by these 46 primer pairs were then used as markers for genotyping the collection of 107 G. boninense isolates and the two G. resinaceum isolates. After this step was completed, only 17 SSR markers showed proper amplification, i.e. migration patterns characterized by a main peak of over 200 RFU (relative fluorescent units), no more than two amplified peaks, and a maximum of 10 % missing data. As the primer pairs were designed with a wide range of expected PCR product lengths, for each dye, two markers (differing by at least 16 nucleotides) were able to be multiplexed for genotyping (i.e. 8 markers per plate), increasing genotyping speed for large-scale analysis. All properties of the 17 properly amplified polymorphic SSR markers are summarized in Table 2. The set comprises six dinucleotides, ten trinucleotides and one tetra-nucleotide, with the number of repeats ranging from 5 to 11 and with PCR products 171 to 356 bp in length. All primers have a theoretical melting temperature between 57.6 °C and 60.7 °C and a difference between forward and reverse primers of no more than 2.2 °C (Table 2).
SSR characteristics and diversity analysis
We identified 136 alleles for the 17-SSR set in genotyping the 107 G. boninense isolates. The allele number per locus varied from 3 (locus 17b) to 14 (locus 24d), with an average value of 7 (Table 4). The number of identified genotypes across all geographic areas varied from 5 (locus 17b) to 38 (locus 20d), with an average of 17 (Table 4). No PCR amplification failure was observed for five of the loci (locus 7a, 22a, 33a, 42d, 51d), whereas up to 8.4 % failure was found for locus 18a. There was no significant difference between mean observed (H o ) and expected heterozygosity (H e ) for any of the geographical areas. PIC values for the 17 SSR markers ranged from 0.298 (locus 17b) to 0.813 (locus 20d). LD was significant (p value < 0.01) between alleles for four pairs of loci in the full set of samples: 18a and 47d, 19a and 20d, 3d and 20d, and 11d and 47d (ranging from 0.001 to 0.009). All of these markers belonged to different contigs. Among the various geographical areas, LD was significant only for the pair 19a and 20d in the peninsular Malaysia samples (0.002) and the pair 3d and 20d in the Sumatra samples (0.001).
Based on our decision rules, nine loci (7a, 22a, 33a, 6d, 11d, 20d, 37d, 47d, 51d) showed no convincing indication of null alleles (see supplementary information Table 1), whereas eight loci (11a, 18a, 19a, 17b, 3d, 24d, 42d, 52d) showed evidence for the presence of null alleles.
Genetic structure
The main result shown by PCA (first axis, eigenvalue 4.36) was the clear separation between samples from Borneo and those from Sumatra and peninsular Malaysia. Inclusion or exclusion of loci with null alleles did not affect this result (first axis eigenvalue 4.8 %; see Fig. 1a and b). Samples collected from peninsular Malaysia and Sumatra were interspersed within a homogenous cluster, indicating that they belonged to the same genetic group. The PCA results are in agreement with pairwise F st values calculated between samples from the three geographical areas. For the nine markers without null alleles, F st was 0.0792 (p value 0.0009) between Sumatra and Borneo samples and 0.0773 (p value 0.0009) between peninsular Malaysia and Borneo samples, but only 0.0111 (p value 0.1778) between Sumatra and peninsular Malaysia samples (Table 5). The results were similar for the 17-marker set (Table 5), with slightly smaller but still significant F st values of 0.056 between the Borneo and Sumatra samples and 0.061 between the Borneo and peninsular Malaysia samples.

Global PCA results based on two sets of markers. a Global genetic diversity observed in PCA analyses based on total SSR marker set. b Global genetic diversity observed in PCA analyses based on the reduced 9-SSR marker set including only markers with no predicted null alleles
Cross-species transferability of SSR marker set
All 17 SSR markers in the G. resinaceum CBS19476 strain were successfully amplified, and 16 of the SSR markers in the G. resinaceum strain CBS22036 were able to be amplified, with locus 18a yielding no PCR products. For the complete 17-SSR marker set, only two new alleles were discovered on the G. resinaceum isolates. Locus 37d had a specific allele for strain CBS22036 only, with a length of 326 bp, which is within the range of detected allele sizes in G. boninense (311–350 bp). Locus 52d had a specific allele for both strains of G. resinaceum. This allele was 309 bp in length, outside the range of sizes found for G. boninense isolates (313–327 bp).
Discussion
In the present study, we first identified 2487 SSR loci in a G. boninense draft genome assembly. Then, focusing on the fastest way to obtain a workable minimum microsatellite set for genetic diversity analysis, we developed 17 SSR markers. These markers exhibited satisfactory amplification and polymorphism properties for large-scale population genotyping. To reduce the time and cost of genotyping, these SSR markers were designed to be used in multiplex. This is the first set of locus-specific SSR markers developed for G. boninense. Until now, only random amplified microsatellites (RAMS) (Zakaria et al. 2005), rapid amplification of polymorphic DNA (RAPD) (Abu-Seman et al. 1996; Zakaria et al. 2005) and internal transcribed spacer (ITS) (Moncalvo et al. 1995b; Smith and Sivasithamparam 2000; Utomo et al. 2005) markers have been developed for pathogenic strains of Ganoderma species found on oil palms.
Our study confirms the advantages of using assembled reads (Seq-Assembly-SSR) rather than raw reads for the detection and development of SSR markers for non-model species, as previously shown by Cai et al. (2013). We were able to design primers for 100 % of the selected SSRs, compared to 49.6 % for studies based on 454 raw reads, and 30 to 40 % for studies based on Illumina raw reads. Among the 145 primer pairs tested in vitro, 93 (84.5 %) showed mono-locus amplification, which is comparable to results reported by Cai et al. (2013), who obtained 90.7 % locus-specific amplification from primer pairs designed using a genome assembly. The effectiveness of the Seq-Assembly-SSR approach in targeting locus-specific markers and primer design is the result of two improvements. First, assembled contigs provide longer sequences, defining primers with an average contig length of 2864 bp after assembly, compared to raw reads of 51 bp for the Illumina and 378 bp for the 454 technique. Second, sequence redundancy is reduced by the assembly process: while the amount of raw reads was around 4 Gb, the assembly process reduced the total amount of data to around 61 Mb. This reduction in redundancy improves the efficiency of pre-screening for primers amplifying single-locus PCR products using in silico PCR (Cai et al. 2013). Furthermore, the selection of microsatellite loci with flanking sequences of at least 400 bp facilitates primer design and multiplexed genotyping, which reduces genotyping costs for large sets of samples.
The number of SSRs detected in the G. boninense genome draft assembly in this study falls within the range of numbers of SSRs detected in fungal genomes as reviewed by Karaoglu et al. (2005), which ranged from 124 (Encephalitozoon cuniculi) to 8376 (Neurospora crassa). However, the number detected here is almost twice that of SSRs identified in G. lucidum (1584) by Qian et al. (2013). As our G. boninense genome assembly is only a draft, no firm conclusions can be drawn. However, a comparison of the estimated genome size of these two fungi (43 Mb for G. lucidum and 65 Mb estimated for the G. boninense draft assembly), as well as the much larger number of SSRs, suggests that there may have been a large duplication or deletion event at genome scale in the Ganoderma genus.
While most studies recommend a minimum of 8 to 11 SSR markers for performing sound statistical analysis of population genetics data (van Asch et al. 2010), our study developed 17 SSR markers with good amplification and polymorphism properties, which should allow comprehensive large-scale population studies in G. boninense. Fungus population studies have used on average 10 SSR markers and generally no more than 16 SSR markers (Schoebel et al. 2013a, b; Wang et al. 2014). Of the 17 SSR markers developed in this study, the mean PIC value was approximately 0.604, and 14 of the 17 markers presented PIC values greater than 0.5, indicating that they are highly informative (PIC > 0.5), according to Botstein et al. (1980). Compared to a mean PIC value of 0.33 obtained for SSRs described on another Polyporales by Zhang et al. (2015), our SSRs show high polymorphic information content. Among our 17 SSRs, 9 had no null alleles according to criteria used in this study. While the LD detected at the whole sample scale may be the result of sample structure, LD was significant for only two pairs of alleles within one of three populations, indicating that the information conveyed by each SSR marker was non-redundant. Visualization of putative relationships among investigated samples by PCA showed clear differentiation of a group of samples from Borneo against a mixed group of samples from Sumatra and peninsular Malaysia. PCA results were confirmed by F st values: about 0.07 between Borneo and Sumatra or Borneo and peninsular Malaysia, while only about 0.01 between Sumatra and peninsular Malaysia. PCA and F st results were similar regardless of whether the full 17SSR set or the set of 9 SSR with no null alleles was used. This indicates a potential high gene flow between the Sumatra and peninsular Malaysia populations, even though they are separated by the Malacca Strait, and suggests that the Borneo population is partially isolated.
Amplification of 16 of the SSRs for two G. resinaceum isolates, a species closely related to G. boninense (Moncalvo et al. 1995a, b; Binder et al. 2013), raises the question of species discrimination using this SSR set and species delimitation within the Ganoderma genus. SSRs are often considered to be poorly transferable between fungal species (Dutech et al. 2007; Schoebel et al. 2013a, b), with the exception of certain particular cases involving species complexes (Perez et al. 2012). The non-transferability of SSR loci between G. lucidum and G. boninense, however, were able to be verified by comparing the flanking sequences of the SSRs we detected for G. boninense to sequences in the G lucidum genome (Chen et al. 2012), although it was impossible to identify motifs of at least 20 bp that could be used to design trans-specific SSR primers. Finally, the limited results for G. resinaceum prevent us from drawing any firm conclusions. A larger number of individuals from G. resinaceum and the sequencing of fungi barcode markers such as ITS or tubulin genes (Moncalvo et al. 1995a, b) would be necessary to determine the species status of these two taxa.
References
Abdelkrim J, Robertson B, Stanton JA, Gemmell N (2009) Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. Biotechniques 46(3):185–92
Abu-Seman, I., M. Thangavelu and T. Swinburne (1996). The use of RAPD for identification of species and detection of genetic variation in Ganoderma isolates from oil palm, rubber and other hardwood trees. Proceeding of Porim International palm oil congress, Kuala Lumpur, Malaysia
Ali S, Gladieux P, Rahman H, Saqib MS, Fiaz M, Ahmad H, Leconte M, Gautier A, Justesen AF, Hovmoller MS, Enjalbert J, de Vallavieille-Pope C (2014) Inferring the contribution of sexual reproduction, migration and off-season survival to the temporal maintenance of microbial populations: a case study on the wheat fungal pathogen Puccinia striiformis f.sp. tritici. Mol Ecol 23(3):603–17
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–10
Barbará T, Palma-Silva C, Paggi GM, Bered F, Fay MF, Lexer C (2007) Cross-species transfer of nuclear microsatellite markers: potential and limitations. Mol Ecol 16(18):3759–67
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004). GENETIX. Populations Genet Software Windows TM Univ Montp 4(05).
Binder M, Justo A, Riley R, Salamov A, Lopez-Giraldez F, Sjokvist E, Copeland A, Foster B, Sun H, Larsson E, Larsson KH, Townsend J, Grigoriev IV, Hibbett DS (2013) Phylogenetic and phylogenomic overview of the Polyporales. Mycologia 105(6):1350–1373
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32(3):314–31
Boutin-Ganache, I., M. Raposo, M. Raymond and C. F. Deschepper (2001). M13-tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques 31(1): 24–+
Breton F, Hasan Y, Hariadi S, Lubis Z, De Franqueville H (2006). Characterization of parameters for the development of an early screening test for basal stem rot tolerance in oil palm progenies. Journal of Oil Palm Research: 24–36
Cai G, Leadbetter CW, Muehlbauer MF, Molnar TJ, Hillman BI (2013) Genome-wide microsatellite identification in the fungus Anisogramma anomala using Illumina sequencing and genome assembly. PLoS One 8(11), e82408
Carlsson J (2008) Effects of microsatellite null alleles on assignment testing. J Hered 99(6):616–623
Castoe TA, Poole AW, Gu W, Jason de Koning AP, Daza JM, Smith EN, Pollock DD (2010) Rapid identification of thousands of copperhead snake (Agkistrodon contortrix) microsatellite loci from modest amounts of 454 shotgun genome sequence. Mol Ecol Resour 10(2):341–7
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24(3):621–31
Chen S, Xu J, Liu C, Zhu Y, Nelson DR, Zhou S, Li C, Wang L, Guo X, Sun Y, Luo H, Li Y, Song J, Henrissat B, Levasseur A, Qian J, Li J, Luo X, Shi L, He L, Xiang L, Xu X, Niu Y, Li Q, Han MV, Yan H, Zhang J, Chen H, Lv A, Wang Z, Liu M, Schwartz DC, Sun C (2012) Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat Commun 3:913
Corley, R. H. V. and P. B. Tinker (2003). The oil palm. Oxford ; Malden, MA, Blackwell Science
Dabrowski MJ, Bornelov S, Kruczyk M, Baltzer N, Komorowski J (2014) 'True' null allele detection in microsatellite loci: a comparison of methods, assessment of difficulties and survey of possible improvements. Mol Ecol Resour 15(3):477–88
Dakin EE, Avise JC (2004) Microsatellite null alleles in parentage analysis. Heredity (Edinb) 93(5):504–9
Doyle JJ (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Dray S, Dufour A-B (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22(4):1–20
Durand-Gasselin T, Asmady H, Flori A, Jacquemard JC, Hayun Z, Breton F, de Franqueville H (2005) Possible sources of genetic resistance in oil palm (Elaeis guineensis Jacq.) to basal stem rot caused by Ganoderma boninense--prospects for future breeding. Mycopathologia 159(1):93–100
Dutech C, Barres B, Bridier J, Robin C, Milgroom MG, Ravigne V (2012) The chestnut blight fungus world tour: successive introduction events from diverse origins in an invasive plant fungal pathogen. Mol Ecol 21(16):3931–46
Dutech C, Enjalbert J, Fournier E, Delmotte F, Barres B, Carlier J, Tharreau D, Giraud T (2007) Challenges of microsatellite isolation in fungi. Fungal Genet Biol 44(10):933–49
Faircloth BC (2008) msatcommander: detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol Ecol Resour 8(1):92–4
Flood, J., P. D. Bridge and M. Holderness (2000). Ganoderma diseases of perennial crops. Wallingford, Oxon England ; New York, CABI.
Gardner MG, Fitch AJ, Bertozzi T, Lowe AJ (2011) Rise of the machines--recommendations for ecologists when using next generation sequencing for microsatellite development. Mol Ecol Resour 11(6):1093–101
Goudet, J. (2002). Fstat. A program to estimate and test gene diversities and fixation indices, release 2(1).
Gregory TR, Nicol JA, Tamm H, Kullman B, Kullman K, Leitch IJ, Murray BG, Kapraun DF, Greilhuber J, Bennett MD (2007) Eukaryotic genome size databases. Nucleic Acids Res 35(Database issue):D332–8
Ho YW, Nawawi A (1986) Isolation, growth and sporophore development of Ganoderrna boninense from oil palm in Malaysia. Pertanika 9(1):69–73
Ho YW, Nawawi A (1985) Ganoderma boninense Pat. from basal stem rot of oil palm (Elaeis guineensis) in Peninsular Malaysia. Pertanika 8(3):425–428
Idris A, Kushairi A, Ismail S, Ariffin D (2004) Selection for partial resistance in oil palm progenies to Ganoderma basal stem rot. J Oil Palm Res 16(2):12–18
Jarne P, Lagoda PJL (1996) Microsatellites, from molecules to populations and back. Trends Ecol Evol 11(10):424–429
Kalinowski ST, Taper ML (2006) Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7(6):991–995
Karaoglu H, Lee CM, Meyer W (2005) Survey of simple sequence repeats in completed fungal genomes. Mol Biol Evol 22(3):639–49
Miller RNG, Holderness M, Bridge PD, Chung GF, Zakaria MH (1999) Genetic diversity of Ganoderma in oil palm plantings. Plant Pathol 48(5):595–603
Moncalvo JM, Wang HF, Hseu RS (1995a) Gene phylogeny of the Ganoderma lucidum complex based on ribosomal DNA sequences. Comparison with traditional taxonomic characters. Mycol Res 99:1489–1499
Moncalvo JM, Wang HH, Hseu RS (1995b) Phylogenetic relationships in Ganoderma inferred from the internal transcribed spacers and 25s ribosomal DNA sequences. Mycologia 87(2):223–238
Montarry J, Andrivon D, Glais I, Corbiere R, Mialdea G, Delmotte F (2010) Microsatellite markers reveal two admixed genetic groups and an ongoing displacement within the French population of the invasive plant pathogen Phytophthora infestans. Mol Ecol 19(9):1965–77
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70(12):3321–3
Pemberton JM, Slate J, Bancroft DR, Barrett JA (1995) Nonamplifying alleles at microsatellite loci: a caution for parentage and population studies. Mol Ecol 4(2):249–52
Perez G, Slippers B, Wingfield MJ, Wingfield BD, Carnegie AJ, Burgess TI (2012) Cryptic species, native populations and biological invasions by a eucalypt forest pathogen. Mol Ecol 21(18):4452–71
Pilotti CA (2005) Stem rots of oil palm caused by Ganoderma boninense: pathogen biology and epidemiology. Mycopathologia 159(1):129–37
Pilotti CA, Sanderson FR, Aitken EAB (2002) Sexuality and interactions of monokaryotic and dikaryotic mycelia of Ganoderma boninense. Mycol Res 106:1315–1322
Pilotti CA, Sanderson FR, Aitken EAB (2003) Genetic structure of a population of Ganoderma boninense on oil palm. Plant Pathol 52(4):455–463
Presti, FT, Wasko AP (2014). A Review of Microsatellite Markers and their Application on Genetic Diversity Studies in Parrots. Open Journal of Genetics 2014
Qian J, Xu H, Song J, Xu J, Zhu Y, Chen S (2013) Genome-wide analysis of simple sequence repeats in the model medicinal mushroom Ganoderma lucidum. Gene 512(2):331–6
Rees RW, Flood J, Hasan Y, Cooper RM (2007) Effects of inoculum potential, shading and soil temperature on root infection of oil palm seedlings by the basal stem rot pathogen Ganoderma boninense. Plant Pathol 56(5):862–870
Schoebel CN, Brodbeck S, Buehler D, Cornejo C, Gajurel J, Hartikainen H, Keller D, Leys M, Ricanova S, Segelbacher G, Werth S, Csencsics D (2013a) Lessons learned from microsatellite development for nonmodel organisms using 454 pyrosequencing. J Evol Biol 26(3):600–11
Schoebel CN, Jung E, Prospero S (2013b) Development of new polymorphic microsatellite markers for three closely related plant-pathogenic Phytophthora species using 454-pyrosequencing and their potential applications. Phytopathology 103(10):1020–7
Smith BJ, Sivasithamparam K (2000) Internal transcribed spacer ribosomal DNA sequence of five species of Ganoderma from Australia. Mycol Res 104:943–951
Susanto A, Sudharto PS, Purba RY (2005) Enhancing biological control of basal stem rot disease (Ganoderma boninense) in oil palm plantations. Mycopathologia 159(1):153–7
Travadon R, Baumgartner K, Rolshausen PE, Gubler WD, Sosnowski MR, Lecomte P, Halleen F, Peros JP (2012a) Genetic structure of the fungal grapevine pathogen Eutypa lata from four continents. Plant Pathol 61(1):85–95
Travadon R, Smith ME, Fujiyoshi P, Douhan GW, Rizzo DM, Baumgartner K (2012b) Inferring dispersal patterns of the generalist root fungus Armillaria mellea. New Phytol 193(4):959–969
Turner, P. D. (1981). Oil palm diseases and disorders. Oil palm diseases and disorders
Utomo C, Werner S, Niepold F, Deising HB (2005) Identification of Ganoderma, the causal agent of basal stem rot disease in oil palm using a molecular method. Mycopathologia 159(1):159–70
van Asch B, Pinheiro R, Pereira R, Alves C, Pereira V, Pereira F, Gusmão L, Amorim A (2010) A framework for the development of STR genotyping in domestic animal species: characterization and population study of 12 canine X–chromosome loci. Electrophoresis 31(2):303–308
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538
Wang M, Xue F, Yang P, Duan XY, Zhou YL, Shen CY, Zhang GZ, Wang BT (2014) Development of SSR markers for a phytopathogenic fungus, Blumeria graminis f.sp tritici, Using a FIASCO Protocol. J Integr Agric 13(1):100–104
Wei N, Bemmels JB, Dick CW (2014) The effects of read length, quality and quantity on microsatellite discovery and primer development: from Illumina to PacBio. Mol Ecol Resour 14(5):953–65
Zakaria L, Kulaveraasingham H, Guan TS, Abdullah F, Wan HY (2005) Random amplified polymorphic DNA (RAPD) and random amplified microsatellite (RAMS) of Ganoderma from infected oil palm and coconut stumps in Malaysia. Asia Pac J Mol Biol Biotechnol 13:23–34
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P (2012) Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot 99(2):193–208
Zane L, Bargelloni L, Patarnello T (2002) Strategies for microsatellite isolation: a review. Mol Ecol 11(1):1–16
Zhang Y, Chen Y, Wang R, Zeng A, Deyholos MK, Shu J (2015). Development of microsatellite markers derived from expressed sequence tags of Polyporales for genetic diversity analysis of endangered Polyporus umbellatus.
Acknowledgments
We should like to thank all the PT Socfin Indonesia team for the sampling in Indonesian plantations and especially Mr. Indra, Mr. Sri, Mr. Turnbull and Mr. Williams. We should also like to thank the entire FELDA team for the sampling in the Malaysian plantations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Marco Thines
Electronic supplementary material
Below is the link to the electronic supplementary material.
supplementary information Table 1
(XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mercière, M., Laybats, A., Carasco-Lacombe, C. et al. Identification and development of new polymorphic microsatellite markers using genome assembly for Ganoderma boninense, causal agent of oil palm basal stem rot disease. Mycol Progress 14, 103 (2015). https://doi.org/10.1007/s11557-015-1123-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11557-015-1123-2