
Abstract
Colorectal cancer is the third most common cancer worldwide. A significant proportion of patients presents with unresectable metastatic disease or develops metachronous metastases following surgical resection of the primary tumor. The prognosis of the disease has improved over the past two decades, with extended multimodality treatment options and the development of increasingly intensified chemotherapy regimens that now typically include targeted biologics. A recent advance in therapy is a treatment regimen composed of three chemotherapeutic agents (i.e., triplet chemotherapy: 5-fluorouracil [5-FU]/leucovorin [LV], oxaliplatin, and irinotecan; FOLFOXIRI) in combination with the vascular endothelial growth factor inhibitor bevacizumab. This regimen has been shown to elicit significantly improved objective response rates and median progression-free survival compared with 5-FU/LV and irinotecan in combination with bevacizumab. However, triplet chemotherapy has been associated with increased rates of chemotherapy-related adverse events, and treatment-emergent adverse events should be properly managed to minimize treatment interruption or discontinuation. We present herein a review of clinical studies evaluating the safety and efficacy of FOLFOXIRI with bevacizumab in metastatic colorectal cancer, and propose a practical guide for the management of adverse events associated with the regimen.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
A large proportion of patients diagnosed with colorectal cancer (CRC) will develop metastatic disease (mCRC). The majority (20–90 %) of metastases are found in the liver (up to 25 % of which are synchronous), and 10–20 % are found in the lung [1–3]. Studies have shown that, in suitable patients, curative resection of colorectal metastases is a reasonable goal [4, 5] and dramatically improves survival prospects. Chemotherapy treatment may facilitate tumor resection, even in previously nonresectable tumors from hepatic metastases of CRC [6, 7].
Chemotherapy for CRC has evolved over time to improve patient outcomes compared with earlier regimens. Standard two-drug chemotherapeutic regimens include 5-fluorouracil (5-FU)/leucovorin (LV) plus either irinotecan (FOLFIRI) or oxaliplatin (FOLFOX) [8, 9]. The addition of a third chemotherapeutic agent (i.e., FOLFIRI combined with oxaliplatin or FOLFOX combined with irinotecan), resulting in a FOLFIRINOX or a FOLFOXIRI regimen, respectively, has recently been evaluated in clinical trials [10–17]. Phase II studies of triplet chemotherapy on a biweekly schedule showed promising efficacy with regard to both progression-free survival (PFS) (e.g., 10.4 months [11], 10.8 months [12]) and overall survival (OS) (e.g., 26.5 months [11], 28.4 months [12]).
Moreover, a more recent study comparing FOLFIRINOX to standard 2-agent chemotherapy (FOLFIRI or FOLFOX4) or other intensified doublet regimens with either high-dose irinotecan (FOLFIRI-HD) or oxaliplatin (FOLFOX7) revealed that patients in the FOLFIRINOX arm had the longest numerical PFS (standard: 9.2 months [95 % confidence interval (CI): 6.8–13.4]; FOLFIRI-HD: 12.1 months [95 % CI: 10.3–16.6]; FOLFOX7: 8.5 months [95 % CI: 6.4–10.9]; FOLFIRINOX: 14.1 months [95 % CI: 11.2–21.7]) and OS (standard: 17.7 months [95 % CI: 13.7–43.0]; FOLFIRI-HD: 29.4 months [95 % CI: 26.1–42.4]; FOLFOX7: 26.9 months [95 % CI: 18.7–45.0]; FOLFIRNIOX: 48.8 months [95 % CI: 21.9–not reached]) compared with patients in the other arms [17]. FOLFIRINOX, furthermore, elicited the highest 3-year OS rate (58 % [95 % CI 38–74] vs. 38 % [95 % CI 21–55], 43 % [95 % CI 26–59], 44 % [95 % CI 25–61] in the control, FOLFIRI-HD, and FOLFOX7 arms, respectively) [17]. Additionally, one phase III study showed that FOLFOXIRI led to a numerically greater time to disease progression (8.4 vs. 6.9 months, p = 0.17), OS (21.5 vs. 19.5 months; p = 0.337), and response rate (43 vs. 33.6 %; p = 0.168) compared with FOLFIRI [14], while another study showed that FOLFOXIRI conferred a significant improvement in PFS (9.8 vs. 6.9 months; hazard ratio [HR] = 0.63; p = 0.006) and OS (22.6 vs. 16.7 months; HR = 0.70; p = 0.032) [13].
Common grade 3/4 adverse events associated with triplet chemotherapy include neutropenia (33–59 %) [12–14, 17], diarrhea (16–30 %) [12–14, 17], mucositis/stomatitis (5–10 %) [12–14, 17], thrombocytopenia (2-13 %) [12–14, 17], neurotoxicity (2–17 %) [12–14, 17], nausea/vomiting (0–7 %) [12–14, 17], asthenia (6–13 %) [13–17], and alopecia (0–32 %) [12–14]. Adverse events for which the FOLFOXIRI arm had a significantly higher frequency than the FOLFIRI arm were grade 3/4 alopecia (32 vs. 12 %, p = 0.0001) [14], diarrhea (27.7 vs. 10.9 %, p = 0.0001) [14], neurosensory toxicity (5.8 vs. 0 %, p = 0.001) [14], and neutropenia [50 vs. 28, p < 0.001) [13]. The dosing schedule for standard triplet chemotherapy is illustrated in Table 1.
The addition of a targeted biologic agent (i.e., the vascular endothelial growth factor inhibitor bevacizumab or the epidermal growth factor receptor [EGFR] inhibitors cetuximab or panitumumab in patients with mCRC that is wild-type for RAS [KRAS/NRAS exon 2–4]) has been another advance to standard chemotherapy doublets [18–23]. Improved rates of response, PFS and OS have been reported for doublet + targeted biologic treatments compared with doublets alone in clinical trials [18–23]. FOLFOX or FOLFIRI in combination with bevacizumab (first-line or second-line) [4, 24, 25] or in combination with cetuximab or panitumumab are indicated for first-line, RAS-wild-type mCRC [4, 26–31], and have become widely adopted for the first-line treatment of mCRC.
The next step in the evolution of chemotherapy for mCRC was the combination of a biologic agent with triplet chemotherapy. This combination has recently been explored to evaluate clinical outcomes and to potentially increase opportunities for surgical resection. Clinical studies have demonstrated the feasibility of combining cetuximab or panitumumab and FOLFOXIRI with promising response and resection rates; however, significant toxicity (grade 3/4) was observed [32–36]. Commonly reported grade ≥ 3 adverse events included diarrhea (25–94 %) [32–36], neutropenia (23–48 %) [32, 35, 36], nausea/vomiting (5–17 %) [32–36], and stomatitis (10–14 %) [34, 35]. Dose reductions to mitigate adverse events were common [33, 34]. Alternatively, given the well-described and tolerable safety profile of bevacizumab, the combination of triplet chemotherapy with bevacizumab has also been studied as a treatment option.
FOLFOXIRI with bevacizumab has recently been evaluated in clinical settings, and has been associated with improved survival and/or activity compared with two-drug regimens combined with bevacizumab [37–41]. However, the observed incidence rates of certain adverse events that are associated with the regimen (i.e., diarrhea, nausea/vomiting, neutropenia, stomatitis, and neutropenia) have resulted in treatment delays and dose reductions, and have led to debate regarding whether the regimen should be considered a standard of care [42].
Herein, we review recently published efficacy and safety outcomes from phase II and phase III clinical trials of bevacizumab in combination with triplet chemotherapeutic regimens in patients with mCRC. Additionally, we propose a practical guide for the management of commonly occurring adverse events to improve the tolerability of the regimen.
2 Clinical Studies
2.1 First-line FOLFOXIRI in Combination with Bevacizumab for mCRC (FOIB)
Treatment composed of bevacizumab in combination with triplet (FOLFOXIRI) chemotherapy for induction (6-month maximum) therapy, followed by bevacizumab maintenance therapy, was assessed in 57 patients [43]. The primary endpoint was PFS rate at 10 months from study entry; secondary endpoints included response rate and safety profile.
Approximately half (53 %) of the patients had liver-only metastases, and KRAS and BRAF mutations were identified in 38 % and 18 % of patients, respectively. The study met its primary endpoint, with a PFS rate at 10 months of 74 % (95 % CI: 62–85 %). Overall response rate was 77 % (95 % CI: 66–88 %); median PFS and OS were 13.1 months and 30.9 months, respectively. Among patients with liver-only metastases at baseline (n = 30), median PFS was 16.0 months (95 % CI: 12.5–21.5), the response rate was 80 % (n = 24), and the radical resection rate was 40 % (n = 12). No associations between KRAS and BRAF mutations and treatment response were found.
Neutropenia, diarrhea, and hypertension were the most common grade 3/4 adverse events reported, which is similar to other clinical trials (Table 2). There were no treatment-related deaths, and grade 4 toxicities were few (with the exception of uncomplicated neutropenia). Of the 14 patients who discontinued induction treatment, two discontinued because of adverse events. All 37 patients who continued to maintenance therapy eventually discontinued treatment; of these, two were due to adverse events.
2.2 Bevacizumab in Combination with mFOLFOX6 or FOLFOXIRI in Patients with Unresectable Liver-limited mCRC (OLIVIA)
OLIVIA assessed the efficacy of bevacizumab in combination with either FOLFOXIRI or modified FOLFOX6 (mFOLFOX6) in 80 previously untreated patients with CRC and initially unresectable liver metastases [37]. The primary endpoint was the overall resection rate for first resections. Secondary endpoints included PFS, overall response rate, and safety.
The majority of enrolled patients (94 %; n = 75) had ≥ 3 metastatic lesions at baseline. Following treatment, a 12 % difference in overall resection rate for first resection between the FOLFOXIRI + bevacizumab and the mFOLFOX-6 + bevacizumab groups was observed (61 % vs. 49 %, respectively; difference 12 %, 95 % CI: 11–36 %). Overall, tumor response rate was also higher (81 % [95 % CI: 65–91 %] vs. 62 % [95 % CI: 45–77 %]) and median PFS was numerically longer (18.6 [95 % CI: 12.9–22.3] vs. 11.5 [95 % CI: 9.6–13.6] months) in the FOLFOXIRI + bevacizumab group compared with the mFOLFOX-6 + bevacizumab group, respectively.
Adverse events grade ≥ 3 occurred in 95 % of patients in the triplet group and 84 % of patients in the doublet + bevacizumab group, with neutropenia, diarrhea, and febrile neutropenia as the most frequently reported (Table 2). Eight patients in the FOLFOXIRI + bevacizumab group and 13 patients in the mFOLFOX6 + bevacizumab group discontinued the study due to adverse events.
2.3 OPAL: A Study of Bevacizumab in Combination with FOLFOXIRI in Patients with Previously Untreated mCRC
The OPAL study evaluated the feasibility of administering bevacizumab plus FOLFOXIRI followed by maintenance with bevacizumab plus 5-FU/LV until disease progression to treat 90 treatment-naive patients with unresectable mCRC [44]. The primary endpoint was PFS; secondary endpoints included OS, resectability rate, and safety.
At baseline, the majority of patients (57 %) had multiple sites of metastases, and 39 % had metastases confined to the liver. Overall, induction plus maintenance treatment led to a median PFS of 11.1 months and OS of 32.2 months. These results are similar to those of the TRIBE study (FOLFOXIRI arm) [41]. Of the 24 patients (27 %) who underwent secondary tumor resections, 67 % were considered curative (R0). The R0 resection rate (18 %) was lower than that reported in the FOIB study (26 %) [43], which may be attributed to the lower proportion of patients with liver-limited metastases at baseline in the OPAL study compared with the FOIB study (39 vs. 53 %).
The most frequently reported grade 3/4 adverse events (≥ 10 %) during induction were leukopenia/neutropenia and diarrhea (see Table 2). Due to adverse events, 16 % of patients discontinued the study and 32 % of the patients required dose reductions.
2.4 TRIBE: Combination Chemotherapy and Bevacizumab as First-line Therapy in Treating Patients with mCRC
Based on the favorable results of the FOIB phase II trial, the phase III TRIBE study was initiated to compare FOLFOXIRI + bevacizumab with FOLFIRI + bevacizumab as a first-line treatment in 508 patients with unresectable mCRC [39]. The primary endpoint was PFS; secondary endpoints included response rate, OS, resection rate for metastases, and safety.
At baseline, metastases were confined to the liver in 18.0 % of the triplet and 23.4 % of the doublet chemotherapy treatment groups, and KRAS and BRAF mutations were found in 39.4 % and 5.5 % of patients analyzed, respectively. In the triplet arm, significantly more cycles were delayed (16.4 vs. 6.1 %, p < 0.001) and more chemotherapy doses were reduced (21.4 vs. 8.2 %, p < 0.001) compared with the doublet chemotherapy arm. However, disease progression led to significantly more discontinuations in the doublet than in the triplet chemotherapy treatment group (20.1 vs. 12.8 %, p = 0.03).
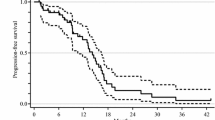
An improvement in the objective response rate was observed in the triplet + bevacizumab arm compared with the doublet + bevacizumab arm (65 vs. 53 %, p = 0.006). Likewise, the median PFS increased to 12.1 months from 9.7 months (hazard ratio [HR] for progression = 0.75; 95 % CI: 0.62–0.90; p = 0.003) (Fig. 1). Subgroup analysis of the treatment effect on PFS indicated that having received only a previous adjuvant treatment significantly affected PFS (p = 0.04). The risk of progression analyzed according to Eastern Cooperative Oncology Group (ECOG) performance status, primary tumor site, time to metastases, liver-only disease, surgery on the primary tumor, Köhne index score, or KRAS or BRAF mutation status was otherwise consistent, although a trend was observed for improved outcomes for patients with BRAF mutations treated with FOLFOXIRI + bevacizumab. Furthermore, triplet + bevacizumab conferred an OS benefit compared with doublet + bevacizumab (29.8 vs. 25.8 months, respectively; HR for death, 0.80; 95 % CI: 0.65–0.98; p = 0.03) [41]. The treatment groups, however, did not have a significantly different rate of R0 resection of metastases (FOLFOXIRI + bevacizumab: 12 % vs. FOLFIRI + bevacizumab: 15 %; p = 0.33) [39].

TRIBE phase III study: Kaplan–Meier estimates of (a) PFS [39] and (b) OS [41], according to treatment group. FOLFIRI, 5-fluorouracil, leucovorin, irinotecan; FOLFOXIRI, 5-fluorouracil, leucovorin, oxaliplatin, irinotecan; OS, overall survival; PFS, progression-free survival. a. Reprinted with permission [39]. b. Reprinted from The Lancet Oncology, Vol 16, C Cremolini, F Loupakis, C Antoniotti, Cristiana Lupi, Elisa Sensi, Sara Lonardi, Silvia Mezi, Gianluca Tomasello, Monica Ronzoni, Alberto Zaniboni, Giuseppe Tonini, C Carlomagno, G Allegrini, S Chiara, M D'Amico, C Granetto, M Cazzaniga, L Boni, G Fontanini, A Falcone. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study, 1306-1315, Copyright (2015), with permission from Elsevier [41]
No statistically significant differences were observed between treatment groups in the rates of serious adverse events (20.4 vs. 19.7 %), death due to disease progression (48.4 vs. 55.9 %), or treatment-related toxicity (2.4 vs. 1.6 %); however, as expected, treatment intensification was associated with a significant increase in grade 3/4 rates of neurotoxicity (p < 0.001), stomatitis (p = 0.048), diarrhea (p = 0.01), and neutropenia (p < 0.001) (see Table 2).
3 Overall Feasibility of Triplet Chemotherapy + Bevacizumab
3.1 Feasibility for All Patients
Overall, data from phase II and phase III clinical studies indicate that treatment with bevacizumab in combination with triplet chemotherapy is feasible in patients with mCRC, and no new safety signals were observed [37–39, 43]. While grade ≥ 3 neutropenia, diarrhea, stomatitis, and peripheral neuropathy were significantly higher with triplet compared with doublet chemotherapy regimens in the phase III TRIBE study, adverse events were considered manageable [39]. Incidences of bevacizumab-related adverse events were similar to those seen in previous studies of bevacizumab combined with chemotherapy (e.g., irinotecan, 5-FU, and LV, or oxaliplatin-based doublets) in the first-line setting [30, 31, 45].
Due to the known toxicity associated with FOLFOXIRI + bevacizumab, an early evaluation of response may be important, so that patients who do not benefit from this regimen may avoid unnecessary toxicity and delay in initiating another line of treatment. An analysis of the TRIBE results revealed that a significantly higher proportion of patients in the FOLFOXIRI + bevacizumab arm compared with the FOLFIRI + bevacizumab arm experienced early tumor shrinkage of ≥ 20 % (62.7 vs. 51.9 %, p = 0.025) and depth of response (43.4 vs. 37.8 %, p = 0.003), and demonstrated an association between these parameters and PFS, post-progression survival, and OS [40].
3.2 Feasibility in the Conversion Setting (i.e., to Render Patients with Unresectable mCRC Liver Metastases Potentially Resectable)
Because of the potential impact of bevacizumab on wound healing and liver regeneration, the addition of bevacizumab to triplet regimens comes with an increased risk of perioperative toxicity, although recent studies have shown safe use of bevacizumab if treatment breaks, beginning 4–6 weeks prior to surgery and lasting through complete wound healing are observed [46]. Results of recent studies have shown that this regimen has activity in eliciting a pathological response of liver metastases without increasing liver toxicity, and that the risk of perioperative complications may be reduced by discontinuing bevacizumab prior to surgery [37, 47].
A study that evaluated hepatic tissue from patients showed that FOLFOXIRI + bevacizumab elicited a greater histopathologic response than chemotherapy alone (either FOLFOXIRI or XELOXIRI) with regard to tumor regression grades of 1, 2, and 3 (FOLFOXIRI + bevacizumab: 63 %; chemotherapy: 28 %; p = 0.033) [47]. Furthermore, a tumor necrosis exploratory cutoff of ≥ 50 % was achieved in a greater proportion of patients who received bevacizumab compared with those who did not (FOLFOXIRI + bevacizumab: 52 %; chemotherapy: 12.5 %; p = 0.017), while there was no significant difference in liver toxicity as measured by sinusoidal dilation, parenchymal steatosis, steatohepatitis, and parenchymal necrosis. Notably, the replacement of liver metastases by a necrosis rate of ≥ 50 % conferred a PFS benefit compared with patients with a necrosis rate of < 50 % (HR = 0.38; 95 % CI: 0.19–0.97; p = 0.041).
In the OLIVIA study of patients with unresectable liver-only metastasis, data suggested that secondary tumor resection was occasionally feasible in patients treated with FOLFOXIRI + bevacizumab [37]. Response rates, primary resection rates, and PFS all showed improvement compared with treatment with doublet chemotherapy (i.e., mFOLFOX6 + bevacizumab). Notably, OLIVIA achieved these results in an unselected patient population with regard to molecular factors and extent of disease. While treatment-related toxicity was increased with triplet chemotherapy (i.e., FOLFOXIRI + bevacizumab), no new or unexpected safety signals were observed. Additionally, data from the surgical safety population (n = 25, FOLFOXIRI + bevacizumab group; n = 19, mFOLFOX6 + bevacizumab group) showed comparable incidences of all-grade surgery-related adverse events with FOLFOXIRI + bevacizumab compared with the mFOLFOX6 + bevacizumab group (60 vs. 79 %), with no anastomotic leaks following primary tumor resection. This is similar to previous observations [46, 48] that showed that by discontinuing bevacizumab 4–5 weeks prior to surgery, a marked increase in postoperative bleeding and wound-healing complications (which are potential bevacizumab-related adverse events that could interfere with perioperative treatment continuation) may be avoided, and resection of liver metastases may thus be safely performed.
3.3 Perspective on the Importance of Bevacizumab + Triplet Chemotherapy for Patients Eligible for Aggressive Chemotherapy
3.3.1 RAS Wild-type Disease
Data from a meta-analysis indicated that there was a benefit to using EGFR inhibitors with doublet chemotherapy as first-line therapy in patients with RAS wild-type mCRC [21, 49]. Furthermore, results from the FIRE-3 study, in which patients with KRAS exon2 wild-type mCRC were treated with FOLFIRI in combination with either bevacizumab or cetuximab, showed that while response rate and PFS were similar between treatment groups, median OS was longer in the cetuximab group than in the bevacizumab group (28.7 vs. 25.0 months; HR 0.77, 95 % CI: 0.62–0.96; p = 0·017) [45]. However, the first-line treatment of RAS wild-type mCRC with doublet chemotherapy in combination with a targeted agent is still a matter of debate, as data from relevant prospective trials are still pending. Thus far, in the phase III TRIBE study, a similar proportion of patients in both FOLFIRI + bevacizumab (39 %) and FOLFOXIRI + bevacizumab (37 %) arms had KRAS wild-type disease, and subgroup analyses showed that the treatment effect (i.e., reduced the risk of disease progression for patients receiving FOLFIXIRI + bevacizumab compared with those receiving FOLFIRI + bevacizumab) was independent of KRAS mutation status.
3.3.2 BRAF-mutated Disease
The BRAF V600E mutation is considered a strong prognostic marker in mCRC, and a putative predictive role of this BRAF mutation for resistance to anti-EGFR treatment has been hypothesized based on retrospective analyses. However, the hypothesis has not been universally accepted despite two recent meta-analyses showing that there is no clinically relevant benefit for the addition of an EGFR antibody to chemotherapy for patients with BRAF-mutated disease [50, 51]. The use of the BRAF V600E mutation as a prognostic marker has been demonstrated in survival analyses of mCRC first-line trials, in which patients with BRAF-mutated mCRC had a worse prognosis than those without BRAF mutations [52–54]. Further, results in the TRIBE study indicated that BRAF-mutated disease (5.5 % of the study population) is an adverse prognostic factor for both PFS and OS (univariate analysis) [39].
Data suggest that triplet chemotherapy + bevacizumab may be a reasonable option in patients with BRAF-mutated disease. The TRIBE study, for example, showed that treatment effect was not significantly different between BRAF-wild-type and BRAF-mutant patient subgroups [41]. Moreover, in a phase II trial of 15 patients with BRAF-mutated mCRC who were treated with first-line FOLFOXIRI + bevacizumab, the 6-month progression-free rate (primary endpoint), median PFS, and OS were 73 %, 9.2 months, and 24.1 months, respectively [55]. These results, however, should be interpreted with caution due to small sample sizes.
4 Ongoing Trials of Triplet Chemotherapy in Combination with Targeted Biologics
4.1 Anti-EGFR Agents
Anti-EGFR agents have shown efficacy in patients with mCRC, and cetuximab and panitumumab are thus indicated, in combination with doublet chemotherapy, for the treatment of patients with mCRC [26, 27]. These agents are currently being investigated in several phase II clinical trials of anti-EGFR agents in combination with triplet chemotherapy in patients with previously untreated mCRC. These include MACBETH (FOLFOXIRI induction + cetuximab and maintenance cetuximab or bevacizumab; NCT02295930; primary completion date: April 2015), FOCULM (FOLFOXIRI ± cetuximab in patients with liver metastases only; NCT02063529; primary completion date: February 2016), VOLFI (FOLFOXIRI ± panitumumab; NCT01328171; primary completion date: December 2016), and CELIM2 (FOLFOXIRI or FOLFIRI + cetuximab in patients with RAS/BRAF wild-type tumors and FOLFOXIRI ± bevacizumab in patients with RAS/BRAF mutant tumors; NCT01802645; primary completion date: July 2017).
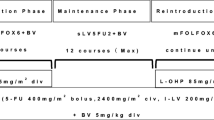
4.2 Bevacizumab (Fig. 2)

Study schemas for key ongoing trials investigating triplet chemotherapy + biologics as first-line therapy in patients with mCRC. (a) CHARTA phase II study (NCT01321957): FOLFOX and bevacizumab with or without irinotecan [38]. (b) TRIBE-2 phase III study: first-line FOLFOXIRI + bevacizumab, followed by reintroduction of FOLFOXIRI + bevacizumab at progression vs. FOLFOX + bevacizumab, followed by FOLFIRI + bevacizumab at progression, in first- and second-line treatment of unresectable mCRC (TRIBE-2 clinical protocol). (c) METHEP-2 phase 2 study: FOLFIRI or FOLFOX vs. FOLFIRINOX + bevacizumab or cetuximab (NCT01442935). (d) STEAM phase 2 study: FOLFOXIRI + bevacizumab (STEAM study protocol; NCT01765582). aStrata: clinical groups (unresectable liver and/or lung metastasis potentially resectable after treatment-induced downsizing, comorbidities allowing surgery; or multiple metastasis, rapid progression, risk of rapid deterioration, unlikely to become resectable; or never resectable and no symptoms or risk of deterioration). bThe third and subsequent lines of treatment will be at investigators’ choice. cBiologic agent is chosen according to tumor KRAS status: bevacizumab for mutated KRAS, cetuximab for wild-type KRAS. dPatients stratified for extent of metastatic disease (liver-limited disease vs. non-liver-limited disease), primary tumor location (right vs. left), and study center. esFOLFOXIRI (sequential FOLFOXIRI) consists of alternating 4-week administrations (2 × 2-week cycles) of FOLFOX and FOLFIRI. fIf the patient exhibits a good response (complete response, partial response, or stable disease) and still tolerates the regimen after 4 months of induction, the therapy can be continued at the investigator's discretion for up to an additional 2 months (discussion with the Medical Monitor). gTreatment will continue until progression, death, withdrawal of consent, or unacceptable toxicities occur, according to dose modification guidance. 5-FU, 5-fluorouracil; ECOG, Eastern Cooperative Oncology Group; FOLFIRI, 5-fluorouracil, leucovorin, irinotecan; FOLFOX, 5-fluorouracil, leucovorin, oxaliplatin; FOLFOXIRI, 5-fluorouracil, leucovorin, oxaliplatin, irinotecan; LV, leucovorin; mCRC, metastatic colorectal cancer; PD, progressive disease; sFOLFOXIRI, sequential 5-fluorouracil, leucovorin, oxaliplatin, irinotecan
4.2.1 Efficacy of FOLFOX + Bevacizumab in Combination with Irinotecan in the Treatment of Patients with mCRC (CHARTA, NCT01321957)
CHARTA is a randomized phase II trial that seeks to evaluate the activity of an intensified first-line therapy for mCRC (FOLFOXIRI + bevacizumab) compared with standard treatment (FOLFOX + bevacizumab) in 250 patients with mCRC or recurrent CRC (Fig. 2a) [38]. Treatment will be administered until disease progression, intolerable toxicity, secondary resection, or for a maximum of 12 cycles (6 months). After 6 months of treatment and/or no disease progression, patients will continue with a maintenance regimen with bevacizumab and a fluoropyrimidine (choice of 5-FU or capecitabine at the investigator’s discretion) for up to 12 months in the absence of progression or intolerable toxicity. The maximum treatment duration, including maintenance therapy, is 18 months. The primary endpoint is PFS at 9 months. Notably, the CHARTA study includes a secondary endpoint of health-related quality of life (as assessed via the European Organisation for Research and Treatment of Cancer QLQ-C30, QLQ-CR29, and QLQ-CIPN20 scales), which may add valuable information regarding patient-reported outcomes of triplet chemotherapy + bevacizumab. Recruitment was completed in December 2014.
4.2.2 First-line FOLFOXIRI + Bevacizumab, Followed by Reintroduction of FOLFOXIRI + Bevacizumab at Progression vs. FOLFOX + Bevacizumab Followed by FOLFIRI + Bevacizumab in mCRC (TRIBE-2, NCT02339116)
The primary endpoint of the TRIBE-2 study is Progression-free survival 2 (PFS2); the planned study length is approximately 4.5 years (Fig. 2b). The first patient was enrolled in February 2015, and the last patient visit is expected in May 2019. With an expected enrollment rate of 200 patients per year, an enrollment period of 3 years, and a minimum follow-up period of 18 months, the first data release is projected to occur in early 2020. The end of the study is defined as the time when all randomized patients will show evidence of second disease progression or will be discontinued from treatment, per protocol, for toxicity or medical decision.
4.2.3 Chemotherapies Associated with Targeted Therapies on the Resection Rate of Hepatic Metastases (METHEP-2, NCT01442935)
METHEP-2 (ACCORD 21–PRODIGE 14) trial is a phase II randomized trial comparing doublet chemotherapy (FOLFIRI/FOLFOX) with FOLFIRINOX combined with a targeted therapy selected according to RAS mutational status (i.e., bevacizumab for tumors with mutated KRAS; cetuximab for wild-type KRAS) in patients with colorectal cancer and initially unresectable liver metastases (Fig. 2c). The primary endpoint is the comparison of secondary resection rates of liver metastases between the control arm (doublet chemotherapy + targeted therapy) vs. the experimental arm (triplet chemotherapy + targeted therapy). The total number of patients is 256, and accrual was closed in May 2015. Translational studies are planned.
4.2.4 A Study of FOLFOXIRI + Bevacizumab Regimens (Concurrent and Sequential) vs. FOLFOX + Bevacizumab in First-line mCRC (Sequential Triplet and Bevacizumab Maintenance; STEAM, NCT01765582)
It has been suggested that alternating treatment every two cycles with FOLFOX + bevacizumab and FOLFIRI + bevacizumab (i.e., sequential FOLFOXIRI + bevacizumab) may improve the tolerability of the regimen without affecting efficacy [56], and may help guide the choice of second-line treatment. The phase II STEAM trial is investigating concurrent or sequential FOLFOXIRI + bevacizumab vs. FOLFOX + bevacizumab in patients with previously untreated mCRC (Fig. 2d). The coprimary efficacy endpoints are overall response rate and PFS.
At the time of a preplanned interim safety analysis, 94 patients were randomized (intention-to-treat population), and 93 patients received at least one dose of study treatment (safety population) [57]. The overall adverse event profile of FOLFOXIRI + bevacizumab was similar to that observed in previous trials of triplet chemotherapy + bevacizumab, including the TRIBE study [58], although the incidence of treatment-emergent adverse events in the sequential FOLFOXIRI + bevacizumab group were similar to those in the FOLFOX + bevacizumab arm. This suggests that alternating treatment with FOLFOX + bevacizumab and FOLFIRI + bevacizumab may improve the tolerability of the regimen, although more mature data are needed to confirm these results [57].
Data from these ongoing studies and others may shed light on how different combinations of chemotherapeutic agents administered in combination with bevacizumab, and how the use of these combinations across multiple lines of therapy impacts efficacy and safety in patients with mCRC.
5 A Practical Guide for the Administration of Triplet Chemotherapy in Combination with Bevacizumab
5.1 Frequently Occurring Adverse Events
Adverse events are common in patients treated with chemotherapeutic agents. Particular associations include diarrhea with 5-FU and irinotecan [59–61], neutropenia with irinotecan [60], and neurotoxicity with oxaliplatin [62]. With treatment intensification to triplet chemotherapy combined with bevacizumab, the most common adverse events were neutropenia, febrile neutropenia, stomatitis, and diarrhea [37–39, 43].
Given the known safety profile of triplet chemotherapy ± bevacizumab, the FOLFOXIRI + bevacizumab regimen should only be considered for patients < 75 years old with an ECOG performance status of ≤ 1, as these patients have a lower risk of requiring dose reductions or treatment discontinuations due to chemotherapy-related toxicities, compared with older patients and those that have an ECOG performance status ≥ 2. Patients aged 71–75 years should have an ECOG performance status < 2 if FOLFOXIRI + bevacizumab is considered, while those > 75 years should not be treated with the regimen. Alternatively, physicians may initiate treatment using a dose step-up approach with an upfront dose of 5-FU and/or irinotecan at 75 % of the approved dose, followed by escalation to the approved dose if no significant toxicity occurs.
5.2 Management of Treatment-related Adverse Events
Early recognition and the active management of treatment-related adverse events are critical for the overall care of patients [39]. Rapid intervention, particularly in the case of gastrointestinal adverse events, may improve tolerability [61], with the potential to decrease the need to reduce or interrupt treatment. Management options include symptomatic drug treatment (e.g., loperamide, granulocyte colony stimulating factor, and prophylactic antibiotics for diarrhea or antihypertensive agents for blood pressure control), immediate supportive care as needed, or dose reduction or interruption [63, 64].
Notably, polymorphisms in drug metabolism genes may leave some patients susceptible to severe adverse events [65]. Patients experiencing unexpected toxicity during treatment may be genotyped for polymorphisms known to impact tolerability to various agents [66]. For example, it is recommended that patients with loss-of-function mutations in dihydropyrimidine dehydrogenase or uridine diphosphoglucuronic genes receive reduced doses of 5-FU or irinotecan, respectively [65, 67].
Table 3 provides protocol-based recommendations for the management of grade 3 or 4 adverse events that were commonly observed in clinical trials of patients with mCRC who were treated with bevacizumab in combination with triplet chemotherapy; guidance for dose reduction/interruption or discontinuation of agents, when prudent, is also provided.
5.2.1 Diarrhea
It is estimated that more than half of all patients receiving chemotherapy for CRC experience diarrhea of any grade [59, 68, 69]. Severe (grade 3 and 4) diarrhea, observed in approximately one-fifth of the patients in the TRIBE study, imparts a significant health-related burden on the quality of life of patients.
Uncomplicated diarrhea may be managed through supportive care, including dietary modifications, and commonly used drugs [70, 71]. Several pharmacologic agents have been investigated for the prevention of treatment-induced diarrhea, but, to date, none have proven efficacy [71]. Loperamide (or codeine as an alternative), which works by binding to receptors in the gastrointestinal tract to slow transit time [61], is appropriate for the initial treatment of diarrhea [71]. For persistent diarrhea, despite loperamide treatment (or as initial treatment for grade 3 or 4 diarrhea), octreotide or tincture of opium may be effective [63, 71]. Racecadotril, which does not slow transit time, may be recommended in acute cases of diarrhea for which infection is suspected [72].
Probiotics, including lactobacillus and bifidobacterium, are thought to restore the balance of intestinal microbiota disrupted by chemotherapy [73, 74] and have been shown to reduce the incidence of grade ≥ 2 diarrhea [75, 76]. The Multinational Association of Supportive Care in Cancer/International Society of Oral Oncology has therefore recently recommended probiotic use for the prevention of diarrhea [77]. However, caution should be exercised in immunocompromised patients with cancer, but rare or no cases of sepsis have been observed in this patient population [75, 76].
5.2.2 Nausea/Vomiting
Nausea and vomiting (emesis) occur in approximately 29 % and 11 % of overall patients, respectively, receiving chemotherapy [78]. In the OLIVIA and TRIBE trials, patients were treated prophylactically (starting day 1 of chemotherapy) with 5-hydroxytryptamine-3 antagonists (i.e., palonosetron), dexamethasone, or with other antiemetics. Of note, the incidence of grade 3/4 nausea and vomiting was not significantly increased with the use of triplet + bevacizumab compared with doublet + bevacizumab in the two available randomized studies [37, 39]. In general, palonosetron and dexamethasone are recommended for patients receiving moderately emetogenic chemotherapy [79, 80].
5.2.3 Stomatitis/Mucositis
Guidelines for management of oral mucositis include maintaining oral hygiene by brushing with a soft toothbrush that is frequently replaced, flossing, and rinsing with saline and applying topical anesthetics or transdermal fentanyl; painful mucositis may be treated with doxepin [81, 82]. While mucositis prophylaxis may include cryotherapy [81], it should be avoided in patients receiving oxaliplatin, as it may exacerbate acute neurologic symptoms [62] and cause laryngospasm for up to 48 h.
Patients with gastrointestinal mucositis may be treated with ranitidine or omeprazole to prevent epigastric pain following 5-FU treatment [81]. Adequate hydration and the potential for transient lactose intolerance and presence of bacterial pathogens should also be considered for at-risk patients [81].
5.2.4 Neutropenia
Current guidelines recommend that primary granulocyte colony-stimulating factor (G-CSF) prophylaxis be used when the overall risk (i.e., due to regimen and patient factors) for febrile neutropenia is ≥ 20 % [83]. However, the use of G-CSF to treat febrile neutropenia should be carefully considered, as studies have shown that G-CSF has been overutilized in patients with low risk of febrile neutropenia, but underutilized in those with high risk [84, 85]. Individual patient factors are to be considered in cases with a 10–20 % risk, while G-CSF is not indicated for patients with a risk < 10 % [83]. The rate of febrile neutropenia with FOLFOXIRI and bevacizumab is reported to be 8.8–13 %; therefore, a general primary GCSF prophylaxis is not necessary [37, 39]. However, G-CSF has also been shown to be effective as a secondary prophylaxis in patients who experienced febrile neutropenia in a previous treatment cycle [86]. If the goal is to induce rapid regression of liver metastases to achieve secondary resection, G-CSF may be considered for primary prophylaxis to avoid delays or dose reductions in chemotherapy administration [17].
5.2.5 Hypertension
Because patients receiving bevacizumab have an increased incidence of severe hypertension, blood pressure monitoring every 2–3 weeks is recommended [24], and antihypertensive treatment should be provided [24] on the basis of recommended guidelines [87]. Patients with grade 2 hypertension may be treated with antihypertensive monotherapy (e.g., an angiotensin-converting enzyme inhibitor), and may resume bevacizumab treatment once blood pressure is controlled to < 150/100 mm Hg. Patients experiencing grade 3 hypertension may require more than one antihypertensive agent or more intensive therapy (i.e., a diuretic or calcium channel blocker). Bevacizumab treatment should be suspended in patients with grade 3 hypertension that is not controlled by medical management, and it should be discontinued in patients with hypertensive crisis or hypertensive encephalopathy.
5.3 Dosage Reductions, Interruptions, or Discontinuations
5.3.1 Bevacizumab
There are no recommended dose reductions for bevacizumab [24], and no reductions were permitted in the phase II and phase III clinical trials, which assessed bevacizumab in combination with triplet chemotherapy. However, management of adverse events, dose interruptions, or discontinuations of therapy were recommended in the study protocols in certain, well-defined situations (see Table 3). The prescribing information recommends that bevacizumab be temporarily suspended in patients with uncontrolled, severe hypertension and severe proteinuria [24]. In addition to these recommendations, the protocols also specified guidelines for the management of hypertension (i.e., angiotensin-converting enzyme inhibitor for grade 2 and a three-drug medication for grade 3) and monitoring of proteinuria (see Table 3). Furthermore, because bevacizumab impairs wound healing, suspension of bevacizumab dosing is recommended prior to elective surgery (4 weeks according to the guidelines of the US Food and Drug Administration and 6–8 weeks according to the guidelines of the European Medicines Agency) until the patient is fully healed [24, 25]. In the event of severe infusion reactions, gastrointestinal perforation, surgery and wound-healing complications, severe arterial thromboembolic events, life-threatening venous thromboembolism, hypertensive crisis/encephalopathy, posterior reversible encephalopathy syndrome, or nephritic syndrome, bevacizumab should be permanently discontinued [24]. The protocols, moreover, recommended a delay in bevacizumab dosing in the event of grade 4 febrile neutropenia, a delay or discontinuation for grade ≥ 2 pulmonary hemorrhage and grade 2 nonthromboembolic fistulae, and a discontinuation for any-grade central nervous system (CNS) hemorrhage, grade ≥ 3 nonpulmonary and non-CNS hemorrhage, and any-grade thromboembolic fistulae (see Table 3).
5.3.2 Triplet Chemotherapy
According to the TRIBE study protocol, dose adjustments were recommended for all three components or a subset of the triplet regimen for specific adverse events (see Table 3). Generally, dose delays may be implemented until an adverse event resolves to a lower grade or baseline. For example, a dose should be delayed in patients with grade ≥ 3 neutropenia occurring for < 5 days, grade ≥ 2 thrombocytopenia, or grade ≥ 2 diarrhea and/or stomatitis. Dose reductions to 75 % are recommended at the recurrence of a prior adverse event that led to a dose reduction (e.g., third occurrence of neutropenia), or at the first occurrence of a higher-grade adverse event (e.g., grade 4 neutropenia lasting ≥ 5 days, febrile neutropenia, grade ≥ 3 thrombocytopenia, or grade ≥ 3 related significant organ toxicities). Triplet chemotherapy should be discontinued for grade 4 thrombocytopenia. For other adverse events, dose modifications of individual components of the triplet regimen are recommended. For example, 5-FU/LV dose may be reduced for grade ≥ 3 hand–foot syndrome or mucositis, or after a second occurrence for these events of grade ≥ 2. Furthermore, for persistent, unresolved grade ≥ 2 neurosensory events, oxaliplatin is recommended to be temporarily discontinued [62]. For the second occurrence of diarrhea that requires a dose delay or the first occurrence of a grade ≥ 3 event, both irinotecan and 5-FU/LV may be reduced to 75 % of the starting dose.
6 Conclusions
Data from phase II and phase III clinical trials of triplet chemotherapy in combination with bevacizumab suggest that efficacy and/or resection rates are improved compared with doublet chemotherapy + bevacizumab [37, 39]. Adverse events were manageable given the proper selection of patients and the careful monitoring for severe adverse events [37, 39]. We have herein described the clinical applicability of this regimen and provided a guide for its usage in first-line mCRC patients. Additional trials to evaluate efficacy, tolerability, and patient-reported outcomes of FOLFOXIRI + targeted biologic agents across multiple lines of treatment and in different settings (i.e., potentially resectable patients) are underway.
References
Penna C, Nordlinger B (2002) Colorectal metastasis (liver and lung). Surg Clin N Am 82:1075–90, x–xi
Castellanos JA, Merchant NB (2014) Strategies for management of synchronous colorectal metastases. Curr Surg Rep 2:62
Howlader N, Noone AM, Krapcho M, et al (eds) (2014) SEER cancer statistics review, 1975-2011, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2011/, based on November 2013 SEER data submission, posted to the SEER web site, April 2014. SEER Stat Fact Sheets: Colon and rectum cancer. Available from: http://seer.cancer.gov/statfacts/html/colorect.html.
National Comprehensive Cancer Network (2015) Clinical practice guidelines in oncology (NCCN guidelines): colon cancer, version 2.2015. Available from: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp.
Van Cutsem E, Cervantes A, Nordlinger B et al (2014) Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 25(Suppl 3):iii1–9
Smith JJ, D'Angelica MI (2015) Surgical management of hepatic metastases of colorectal cancer. Hematol Oncol Clin N Am 29:61–84
Jones RP, Malik HZ, Fenwick SW et al (2013) Perioperative chemotherapy for resectable colorectal liver metastases: where now? Eur J Surg Oncol 39:807–11
de Gramont A, Figer A, Seymour M et al (2000) Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 18:2938–47
Douillard JY, Cunningham D, Roth AD et al (2000) Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 355:1041–7
Falcone A, Masi G, Vasile E et al (2009) FOLFOXIRI (irinotecan, oxaliplatin, infusional 5FU/LV) vs. FOLFIRI as first-line treatment of metastatic colorectal cancer (mCRC): updated results after 5 years follow up and risk-stratified analysis. Ann Oncol 20(suppl 8):348, Abstract P-6085
Falcone A, Masi G, Allegrini G et al (2002) Biweekly chemotherapy with oxaliplatin, irinotecan, infusional fluorouracil, and leucovorin: a pilot study in patients with metastatic colorectal cancer. J Clin Oncol 20:4006–14
Masi G, Allegrini G, Cupini S et al (2004) First-line treatment of metastatic colorectal cancer with irinotecan, oxaliplatin and 5-fluorouracil/leucovorin (FOLFOXIRI): results of a phase II study with a simplified biweekly schedule. Ann Oncol 15:1766–72
Falcone A, Ricci S, Brunetti I et al (2007) Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ouest. J Clin Oncol 25:1670–6
Souglakos J, Androulakis N, Syrigos K et al (2006) FOLFOXIRI (folinic acid, 5-fluorouracil, oxaliplatin and irinotecan) vs. FOLFIRI (folinic acid, 5-fluorouracil and irinotecan) as first-line treatment in metastatic colorectal cancer (MCC): a multicentre randomised phase III trial from the Hellenic Oncology Research Group (HORG). Br J Cancer 94:798–805
Ychou M, Conroy T, Seitz JF et al (2003) An open phase I study assessing the feasibility of the triple combination: oxaliplatin plus irinotecan plus leucovorin/ 5-fluorouracil every 2 weeks in patients with advanced solid tumors. Ann Oncol 14:481–9
Ychou M, Viret F, Kramar A et al (2008) Tritherapy with fluorouracil/leucovorin, irinotecan and oxaliplatin (FOLFIRINOX): a phase II study in colorectal cancer patients with non-resectable liver metastases. Cancer Chemother Pharmacol 62:195–201
Ychou M, Rivoire M, Thezenas S et al (2013) A randomized phase II trial of three intensified chemotherapy regimens in first-line treatment of colorectal cancer patients with initially unresectable or not optimally resectable liver metastases. The METHEP trial. Ann Surg Oncol 20:4289–97
Folprecht G, Gruenberger T, Bechstein WO et al (2010) Tumour response and secondary resectability of colorectal liver metastases following neoadjuvant chemotherapy with cetuximab: the CELIM randomised phase 2 trial. Lancet Oncol 11:38–47
Tan BR, Zubal B, Hawkins W, et al (2009) Preoperative FOLFOX plus cetuximab or panitumumab therapy for patients with potentially resectable hepatic colorectal metastases. Presented at: 2009 gastrointestinal cancers symposium; January 15–17, 2009; San Francisco, CA. Abstract 497.
Douillard JY, Siena S, Cassidy J et al (2014) Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol 25:1346–55
Sorich MJ, Wiese MD, Rowland A et al (2015) Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 26:13–21
Ye LC, Liu TS, Ren L et al (2013) Randomized controlled trial of cetuximab plus chemotherapy for patients with KRAS wild-type unresectable colorectal liver-limited metastases. J Clin Oncol 31:1931–8
Van Cutsem E, Lenz HJ, Köhne CH et al (2015) Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 33:692–700
Avastin [prescribing information]. South San Francisco, CA: Genentech, Inc; 2014.
Avastin EPAR summary for the public. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000582/WC500029260.pdf.
Cetuximab [prescribing information]. Bridgewater, NJ: Imclone LLC; 2015.
Panitumumab [prescribing information] Thousand Oaks, CA: Amgen, Inc; 2015.
Cetuximab EPAR summary for the public. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000558/WC500029119.pdf.
Panitumumab EPAR summary for the public. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000741/WC500047710.pdf.
Hurwitz H, Fehrenbacher L, Novotny W et al (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–42
Saltz LB, Clarke S, Díaz-Rubio E et al (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26:2013–9
Folprecht G, Hamann S, Schütte K et al (2014) Dose escalating study of cetuximab and 5-FU/folinic acid (FA)/oxaliplatin/irinotecan (FOLFOXIRI) in first line therapy of patients with metastatic colorectal cancer. BMC Cancer 14:521
Garufi C, Torsello A, Tumolo S et al (2010) Cetuximab plus chronomodulated irinotecan, 5-fluorouracil, leucovorin and oxaliplatin as neoadjuvant chemotherapy in colorectal liver metastases: POCHER trial. Br J Cancer 103:1542–7
Fornaro L, Lonardi S, Masi G et al (2013) FOLFOXIRI in combination with panitumumab as first-line treatment in quadruple wild-type (KRAS, NRAS, HRAS, BRAF) metastatic colorectal cancer patients: a phase II trial by the Gruppo Oncologico Nord Ouest (GONO). Ann Oncol 24:2062–7
Saridaki Z, Androulakis N, Vardakis N et al (2012) A triplet combination with irinotecan (CPT-11), oxaliplatin (LOHP), continuous infusion 5-fluorouracil and leucovorin (FOLFOXIRI) plus cetuximab as first-line treatment in KRAS wt, metastatic colorectal cancer: a pilot phase II trial. Br J Cancer 107:1932–7
Assenat E, Desseigne F, Thezenas S et al (2011) Cetuximab plus FOLFIRINOX (ERBIRINOX) as first-line treatment for unresectable metastatic colorectal cancer: a phase II trial. Oncologist 16:1557–64
Gruenberger T, Bridgewater J, Chau I, et al (2014) Bevacizumab plus mFOLFOX-6 or FOLFOXIRI in patients with initially unresectable liver metastases from colorectal cancer: the OLIVIA multinational randomised phase II trial. Ann Oncol 2014 Dec 23.
Stein A, Glockzin G, Wienke A, Arnold D et al (2012) Treatment with bevacizumab and FOLFOXIRI in patients with advanced colorectal cancer: presentation of two novel trials (CHARTA and PERIMAX) and review of the literature. BMC Cancer 12:356
Loupakis F, Cremolini C, Masi G et al (2014) Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med 371:1609–18
Cremolini C, Loupakis F, Antoniotti C, et al (2015) Early tumor shrinkage and depth of response predict long-term outcome in metastatic colorectal cancer patients treated with first-line chemotherapy plus bevacizumab: results from phase III TRIBE trial by the Gruppo Oncologico del Nord Ouest. Ann Oncol 2015 Feb 23.
Cremolini C, Loupakis F, Antoniotti C et al (2015) FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. doi:10.1016/S1470-2045(15)00122-9
Nipp RD, Ryan DP (2015) Should FOLFOXIRI plus bevacizumab be the standard first-line therapy in metastatic colorectal cancer? Oncologist 20:236–8
Masi G, Loupakis F, Salvatore L et al (2010) Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a phase 2 trial. Lancet Oncol 11:845–52
Stein A, Atanackovic D, Hildebrandt B, Stübs P et al (2015) Upfront FOLFOXIRI+bevacizumab followed by fluoropyrimidin and bevacizumab maintenance in patients with molecularly unselected metastatic colorectal cancer. Br J Cancer 113:872–7
Heinemann V, von Weikersthal LF, Decker T et al (2014) FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol 15:1065–75
Gruenberger B, Tamandl D, Schueller J et al (2008) Bevacizumab, capecitabine, and oxaliplatin as neoadjuvant therapy for patients with potentially curable metastatic colorectal cancer. J Clin Oncol 26:1830–5
Loupakis F, Schirripa M, Caparello C et al (2013) Histopathologic evaluation of liver metastases from colorectal cancer in patients treated with FOLFOXIRI plus bevacizumab. Br J Cancer 108:2549–56
Reddy SK, Morse MA, Hurwitz HI et al (2008) Addition of bevacizumab to irinotecan- and oxaliplatin-based preoperative chemotherapy regimens does not increase morbidity after resection of colorectal liver metastases. J Am Coll Surg 206:96–106
Vale CL, Tierney JF, Fisher D et al (2011) Does anti-EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta-analysis. Cancer Treat Rev 38:618–25
Pietrantonio F, Petrelli F, Coinu A et al (2015) Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer 51:587–94
Rowland A, Dias MM, Wiese MD et al (2015) Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer 112:1888–94
Tol J, Koopman M, Cats A et al (2009) Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 360:563–72
Bokemeyer C, Van Cutsem E, Rougier P et al (2012) Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer 48:1466–75
Douillard JY, Oliner KS, Siena S et al (2013) Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369:1023–34
Loupakis F, Cremolini C, Salvatore L et al (2014) FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur J Cancer 50:57–63
Hebbar M, Chibaudel B, André T et al (2013) Randomized trial of simplified LV5FU2 versus FOLFOX7 followed by FOLFIRI (MIROX) in patients with initially resectable metastatic colorectal cancer: a GERCOR study. J Chemother 25:104–11
Hurwitz H, Tan B, Reeves J, et al (2014) Interim safety results from STEAM: a randomized phase 2 trial of sequential and concurrent FOLFOXIRI-bevacizumab (BEV) vs. FOLFOX-BEV for the first-line (1L) treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC). Ann Oncol 25(suppl 4): poster 521-P.
Falcone A, Cremolini C, Masi G, et al (2013) FOLFOXIRI/bevacizumab (bev) versus FOLFIRI/bev as first-line treatment in unresectable metastatic colorectal cancer (mCRC) patients (pts): results of the phase III TRIBE trial by GONO group. J Clin Oncol 31(Suppl): abstract 3505.
National Cancer Institute (2015) Gastrointestinal complications (PDQ): diarrhea. Available from: http://www.cancer.gov/cancertopics/pdq/supportivecare/gastrointestinalcomplications/HealthProfessional/page5.
Camptosar [prescribing information]. New York, NY: Pflzer, Inc; 2012.
Maroun JA, Anthony LB, Blais N et al (2007) Prevention and management of chemotherapy-induced diarrhea in patients with colorectal cancer: a consensus statement by the Canadian Working Group on Chemotherapy-Induced Diarrhea. Curr Oncol 14:13–20
Eloxatin [prescribing information]. Bridgewater, NJ: Sanofi-Aventis; 2014.
Benson AB III, Ajani JA, Catalano RB et al (2004) Recommended guidelines for the treatment of cancer treatment–induced diarrhea. J Clin Oncol 22:2918–26
BCCA guidelines for the management of chemotherapy-induced diarrhea. Available from: http://www.bccancer.bc.ca/nr/rdonlyres/4e7ef86a-eaa5-4f3c-b147-b2512799f6b3/7371/guidelinesformanagementofcid.pdf.
Mini E, Nobili S (2009) Pharmacogenetics: implementing personalized medicine. Clin Cases Miner Bone Metab 6:17–24
Eng C (2009) Toxic effects and their management: daily clinical challenges in the treatment of colorectal cancer. Nat Rev Clin Oncol 6:207–18
Caudle KE, Thorn CF, Klein TE et al (2013) Clinical pharmacogenetics implementation consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin Pharmacol Ther 94:640–5
Arbuckle RB, Huber SL, Zacker C (2000) The consequences of diarrhea occurring during chemotherapy for colorectal cancer: a retrospective study. Oncologist 5:250–9
National Institutes of Health (2010) Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. Available from: http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf.
McQuade RM, Bornstein JC, Nurgali K (2014) Anti-colorectal cancer chemotherapy-induced diarrhoea: current treatments and side-effects. Int J Clin Med 5:393–406
Andreyev J, Ross P, Donnellan C et al (2014) Guidance on the management of diarrhoea during cancer chemotherapy. Lancet Oncol 15:e447–60
Eberlin M, Mück T, Michel MC (2012) A comprehensive review of the pharmacodynamics, pharmacokinetics, and clinical effects of the neutral endopeptidase inhibitor racecadotril. Front Pharmacol 3:93
Wang H, Geier MS, Howarth GS (2014) Prebiotics: a potential treatment strategy for the chemotherapy-damaged gut? Crit Rev Food Sci Nutr 2014 Aug 27.
Prisciandaro LD, Geier MS, Butler RN, Cummins AG et al (2011) Evidence supporting the use of probiotics for the prevention and treatment of chemotherapy-induced intestinal mucositis. Crit Rev Food Sci Nutr 51:239–47
Redman MG, Ward EJ, Phillips RS (2014) The efficacy and safety of probiotics in people with cancer: a systematic review. Ann Oncol 25:1919–29
Österlund P, Ruotsalainen T, Korpela R et al (2007) Lactobacillus supplementation for diarrhoea related to chemotherapy of colorectal cancer: a randomised study. Br J Cancer 97:1028–34
Gibson RJ, Keefe DM, Lalla RV et al (2013) Systematic review of agents for the management of gastrointestinal mucositis in cancer patients. Support Care Cancer 21:313–26
National Cancer Institute. Nausea and vomiting (PDQ) (2015) Available from: http://www.cancer.gov/cancertopics/pdq/supportivecare/nausea/HealthProfessional.
Roila F, Herrstedt J, Aapro M et al (2010) Guideline update for MASCC and ESMO in the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting: results of the Perugia consensus conference. Ann Oncol 21(Suppl 5):v232–43
Basch E, Prestrud AA, Hesketh PJ et al (2011) Antiemetics: American society of clinical oncology clinical practice guideline update. J Clin Oncol 29:4189–98
Peterson DE, Bensadoun RJ, Roila F, ESMO Guidelines Working Group (2011) Management of oral and gastrointestinal mucositis: ESMO clinical practice guidelines. Ann Oncol 22(Suppl 6):vi78–84
Lalla RV, Bowen J, Barasch A et al (2014) MASCC/ISOO clinical practice guidelines for the management of mucositis secondary to cancer therapy. Cancer 120:1453–61
Aapro M, Crawford J, Kamioner D (2010) Prophylaxis of chemotherapy-induced febrile neutropenia with granulocyte colony-stimulating factors: where are we now? Support Care Cancer 18:529–41
Barnes G, Pathak A, Schwartzberg L (2014) G-CSF utilization rate and prescribing patterns in United States: associations between physician and patient factors and GCSF use. Cancer Med 3:1477–84
Ramsey SD, McCune JS, Blough DK et al (2010) Colony-stimulating factor prescribing patterns in patients receiving chemotherapy for cancer. Am J Manag Care 16:678–86
Gupta S, Singh PK, Bhatt ML et al (2010) Efficacy of granulocyte colony stimulating factor as a secondary prophylaxis along with full-dose chemotherapy following a prior cycle of febrile neutropenia. Biosci Trends 4:273–8
Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (2004) The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure – complete report. Available from: http://www.nhlbi.nih.gov/guidelines/hypertension/jnc7full.htm.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Support for third-party medical writing assistance was provided by Genentech, Inc.
Conflicts of Interest
F. Loupakis has served as an Advisory Board member for Roche, Amgen, Sanofi, Merck and Bayer; as a consultant for Amgen, Roche, and Lilly; and as a speaker for Roche, Amgen and Bayer. A. Stein has served as an Advisory Board member for Merck; as a speaker for Roche, Merck, Amgen, Bayer and Sanofi, and has received research funding from Roche and Sanofi. M. Ychou has served as an Advisory Board member for Roche, Amgen, Bayer, Merck, Celgene, Lilly and Sanofi. P Österlund has served as an Advisory Board member/consultant for Amgen, Bayer, Celgene, Eli Lilly, Merck, Roche and Sanofi and has received research funding from Amgen, Merck, Roche and Sanofi. F Hermann is an employee of Roche. A Salud declares no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Loupakis, F., Stein, A., Ychou, M. et al. A Review of Clinical Studies and Practical Guide for the Administration of Triplet Chemotherapy Regimens with Bevacizumab in First-line Metastatic Colorectal Cancer. Targ Oncol 11, 293–308 (2016). https://doi.org/10.1007/s11523-015-0400-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-015-0400-y