
Abstract
MicroRNAs (miRNAs) are key players in multiple biological processes; therefore, analysis and characterization of these small regulatory RNAs is a critical step toward a better understanding of animal and plant biology. In apple (Malus domestica), 200 microRNAs are known, which most probably represent only a fraction of miRNAome diversity. As a result, more effort is required to better annotate miRNAs and their functions in this economically important species. We performed deep sequencing of 12 small RNA libraries obtained for fire blight-resistant and fire blight-sensitive trees. In the sequencing results, we identified 116 novel microRNAs and confirmed a majority of previously reported apple miRNAs. We then experimentally verified selected candidates with RT-PCR and stem-loop quantitative PCR (qPCR) and performed differential expression analysis. Finally, we identified and characterized putative targets of all known apple miRNAs. The gene ontology (GO) enrichment analysis suggests prominent roles of miRNAs in response to stresses, including pathogen infection. In this study, we identified 116 new and confirmed the expression of 143 already known miRNAs. Moreover, our data suggests that apple microRNAs might be considered as regulators and markers of fire blight resistance. The analyses we performed allowed us to define four apple miRNAs potentially involved in fire blight resistance in apple trees: mdm-miR169a, mdm-miR160e, mdm-miR167b-g, and mdm-miR168a,b. These miRNAs are known to be involved in response to stresses across other plant species, usually by targeting stress response proteins. The relatively low number of candidates may result from the high variance of biological replicates and the fact that stress response miRNAs are usually induced by the stress factors and frequently expressed at a low level, or not expressed at all, in normal conditions. The results of our studies are freely available in an online database at http://lemur.amu.edu.pl/share/apple_miRNAs/.
Similar content being viewed by others

Introduction
Fire blight (FB) is a contagious bacterial disease caused by Gram-negative bacteria Erwinia amylovora that affects apples and pears and leads to considerable reduction in fruit production worldwide (Vogt et al. 2013). Its incidence has increased in commercial apple orchards over the last 25 years (Norelli et al. 2003). There are several protection techniques used in agriculture, like antibiotic treatment, copper sprays, or pruning, but they have many drawbacks and are often ineffective (Norelli et al. 2003). Since apple trees do not reproduce true-to-type from seeds, they are propagated using grafting techniques (Mascall 1569). In addition, grafting can be used to alter the characteristics of the scion as the rootstock affects the tree phenotype. The best-known effect of rootstock is on tree stature (Wertheim 1998), but rootstock effect on scion disease susceptibility was also reported (Fallahi et al. 2002). Therefore, using fire blight-resistant rootstocks might be the most promising strategy for protection of apple trees (Kellerhals et al. 2009; Norelli et al. 2003; Vogt et al. 2013). The mechanism by which rootstock affects tree phenotype is not known, but it clearly influences gene expression pattern of their scion. In our previous studies, we identified genes demonstrating different expression patterns in scions from FB-susceptible and FB-resistant rootstocks (Jensen et al. 2012, 2010). Keeping in mind that microRNA (miRNA) molecules, being essential gene expression regulators, could be involved in this rootstock-scion regulation in apple, we assumed that miRNA expression profiles in scion should reflect rootstock characteristics, as is the case for protein coding genes.
The first apple (Malus domestica) miRNAs were found in expressed sequence tags (ESTs) using computational methods. Gleave et al. (2008) identified ten primary miRNA transcripts and Yu et al. (2011) found 16 conserved miRNA. Using a similar method, based on apple EST resource, Szczesniak and Makalowska (2014) identified 40 miRNAs in apple tree. Varkonyi-Gasic et al. (2010) provided the first experimental data. Using Northern blot and quantitative PCR (qPCR), they confirmed 18 conserved miRNAs in apple. To date, however, the most comprehensive list of apple miRNAs was delivered by Xia et al. (2012). Taking advantage of small RNA deep sequencing data, they found 33 conserved and 42 apple-specific miRNAs. Ma et al. (2013) further extended the list of apple miRNAs by 11 novel sequences. Altogether, there are 206 apple miRNAs deposited in miRBase release 20.
Although a number of miRNAs were shown to play a role in response to various stress conditions, miRNA involvement in bacterial resistance in plants is not well known. miR393 is suggested to be responsive to bacterial inoculation (Navarro et al. 2006; Fahlgren et al. 2007; Zhang et al. 2011), by suppressing auxin signaling. Additionally, miR160, miR167, miR159, miR398, and miR393* seem to be involved in antibacterial mechanisms, by showing up- or downregulation upon bacterial inoculation in different studies on Arabidopsis thaliana (Fahlgren et al. 2007; Zhang et al. 2011; Jagadeeswaran et al. 2009; Eldem et al. 2013).
Here, we present studies of 12 small RNA deep sequencing libraries from uninfected Gala apple trees grafted on four different rootstocks—fire blight susceptible (M.27 and B.9) and fire blight resistant (G.30 and M.111). Our analyses allowed us to extend the apple miRNA repertoire by 116 miRNAs as well as verify 143 miRNAs from previous studies. We confirmed five of new miRNAs using qPCR or RT-PCR. We also identified miRNAs with significantly changed expression among the analyzed rootstocks. In addition, we searched for potential miRNA targets using psRNATarget (Dai and Zhao 2011) focusing on transcripts with significantly higher expression in fire blight-resistant trees. Finally, we analyzed the apple degradome (Xia et al. 2012) to obtain experimentally supported miRNA/messenger RNA (mRNA) associations.
Materials and methods
Plant material
Plant material was purchased from Cameron Nursery (Eltopia, WA, USA). ‘Crimson Gala’ scions were bench grafted onto four distinct rootstocks: Malling 27 EMLA (M.27), Budagovsky 9 (B.9), Geneva 30 (G.30), and Malling Merton 111 EMLA (MM.111). The trees were bench grafted by the nursery, by splicing a two-bud dormant shoot and planted at the research orchards in Rock Springs, PA, USA in March 2005. Actively growing shoot tip tissue samples were collected in the summer of 2011. Three biological replicates of each of the following scion-rootstock combinations—B.9, G.30, M.111, and M.27—were used in further analysis (12 apple trees). The samples (approximately 0.5 g each) included the meristem, stem, and the leaves but not including the first fully expanded leaf. Shoot tips were flash frozen in liquid nitrogen and stored at −80 °C for later RNA isolation.
RNA extraction
Samples were ground with a mortar and pestle chilled with liquid nitrogen. The frozen powder was mixed with extraction buffer (2 % cetyl trimethylammonium bromide, 2 % polyvinylpyrrolidone, 100 mM Tris pH 8.0, 25 mM ethylenediaminetetraacetic acid, 2 M NaCl, 0.5 g L−1 (w/v) spermidine, 2 % ß-mercaptoethanol) and total RNA was isolated using 0.8 M LiCl and isopropanol (Sigma). Total RNA concentration and purity were determined using NanoDrop (Thermo Fischer Scientific). Total RNA quality was determined on the BioAnalyzer (Agilent). The RNA samples had high and consistent RNA integrity values.
SOLiD sequencing
Total RNAs were sent to the Genomics Core Facility of the Pennsylvania State University at University Park, PA. Small RNA libraries were prepared using the Applied Biosystems (Life Technologies) Total RNA-Seq Kit according to the Small RNA Library Preparation protocol. Barcoded libraries were sequenced on a SOLiD 5500 xl machine using 5 nt barcode sequencing and 35 nt fragment sequencing according to the manufacturer’s protocols.
Processing of small RNA libraries
Following SOLiD sequencing of small RNAs, 12 small RNA (sRNA) libraries were obtained. Adapter sequences were removed and the reads were filtered by quality using fastx-trimmer (http://hannonlab.cshl.edu/fastx_toolkit/) to make sure all the bases have the score value of at least 20. Redundant reads in each library were counted, and reads per million (RPM) values were calculated.
De novo miRNA search
Using Bowtie, we mapped sRNA reads to the apple genome (http://www.rosaceae.org/species/malus/malus_x_domestica/genome_v1.0) with no mismatches allowed. Based on genomic coordinates of mapped reads, the genomic sequences containing putative miRNA as well as 250 bases upstream and downstream were extracted. The putative mature miRNA was required to have RPM (Reads Per Million) >2 and length between 19 and 22 bases. The latter parameter is justified by the fact that in plants 23–25 nt long reads contain only a small fraction of miRNAs, which are hugely outnumbered by other classes of small regulatory RNAs, like ca-siRNAs. For extracted genomic fragments, a secondary structure was predicted using hybrid-ss-min from UNAFold package (Markham and Zuker 2008) with default settings. We kept only those structures, where (1) there were no more than five mismatches and two bulges between mature miRNA candidate and the opposite hairpin arm, and (2) mature miRNA candidate was entirely found in one hairpin arm. At this step, the hairpin was cut out from a longer sequence if needed and it was required that the hairpin was at least 40 bases long. Next, we made sure that there is a miRNA-like profile of reads mapped to the hairpin. To this point, we kept only the hairpins where (i) mature miRNA was represented by the most abundant read in at least one library, (ii) mature miRNA abundance constituted at least 20 % of total read counts in at least one library, and (iii) the total count of reads starting at mature miRNA 5′ end was the maximal one in at least one library. The sequences that bore 60 % or more low-complexity regions were identified using Dustmasker from BLAST package (Camacho et al. 2009) and discarded. The settings were default except for “-level” set from 20 to 15. Finally, we performed BLAST search against proteins from UniProtKB/Swiss-Prot and noncoding RNAs other than miRNAs from RFAM, and we removed candidates that produced the E-value of 1e-20 (proteins) or 1e-10 (RNAs).
Target search
mRNAs being potentially under control of miRNAs were identified using psRNATarget (Dai and Zhao 2011) with default settings except for length for complementarity scoring adjusted to mature miRNA lengths. An mRNA set consisted of predicted CDS sequences from the Genome Database for Rosaceae (GDR) (http://www.rosaceae.org/) complemented with transcripts from UniGene. Additionally, PAREsnip (Folkes et al. 2012) was used to identify miRNA targets with deep sequencing support. The mRNA dataset was the same as above, and the degradome library, GSM880656, was downloaded from Gene Expression Omnibus (Edgar et al. 2002). The settings were modified so that the program returned only category 0, 1, and 2 targets, which correspond to highest quality hits. Minimal and maximal degradome read lengths were set to 20 and 21, respectively, maximal number of mismatches in miRNA/mRNA duplex was set to 4.0, while p value remained default (0.05). Then, we used custom Python scripts to remove redundant miRNA target pairs. Finally, we used Bowtie (Langmead et al. 2009) to map degradome reads to targeted mRNAs and prepared a graphical representation of mapping results.
Functional categories for miRNA targets
We downloaded A. thaliana complementary DNAs (cDNAs) from Ensembl Plants (Kersey et al. 2010). Using BLASTN, we identified A. thaliana counterparts for apple targets generated with psRNATarget. The E-value threshold was set to 0.001 and the best hits per target were selected. The A. thaliana gene names were used as an input in the analysis performed with GOrilla (Eden et al. 2009). We applied the following settings: (i) all ontologies: process, function, component; and (ii) two unranked lists of genes: target and background, the latter being all gene names downloaded from Ensembl Plants via BioMart.
miRNA splice site prediction
In the first step, EST sequences that correspond to apple pre-miRNAs from miRBase (Kozomara and Griffiths-Jones 2014) were identified. To this point, ESTs from dbEST (Boguski et al. 1993) were searched using Megablast (Camacho et al. 2009), and it was required that the identity was 97 % or higher and that the EST sequence overlapped with at least 90 % of the pre-miRNA sequence. The selected ESTs were subsequently mapped to the M. domestica genome using Splign (Kapustin et al. 2008) with default settings, and finally, splice site data was inferred from the obtained alignments using custom Python scripts.
Stem-loop qPCR
Selected mature miRNA molecules were detected using stem-loop RT-PCR method according to the published protocol (Chen et al. 2005; Varkonyi-Gasic et al. 2007). The amplification product was obtained in standard PCR reaction as well as in real-time PCR reaction. Primers used for each miRNA are provided in online resources (Table OR1). U6 gene was used as an internal control for ΔCt calculation. In detail, the procedure was discussed in the next two sections that follow.
Pulsed reverse transcription
Total RNA extracted from 12 apple trees was treated with DNAse I (Thermo Scientific) according to the manufacturer’s instructions; 125 ng of DNase-treated RNA was applied during miRNA and U6-specific cDNA synthesis using Superscript III RT kit (Invitrogen) in 20 μL reactions. Pulsed reverse transcription reaction was performed as follows: 1 cycle of 16 °C for 30 min; 60 cycles of 30 °C for 30 s, 42 °C for 30 s, and 50 °C for 1 s; and 1 cycle of 70 °C for 15 min.
Real-time PCR
Real-time PCR (Kubista et al. 2006) was performed in Applied Biosystems 7900HT System with Power SYBR® Green PCR Master Mix (Applied Biosystems) in 35 cycles and with T m = 53 °C. After RT reaction, the cDNA template was diluted ten times. The reactions were performed in 10 μL tubes on 384-well plates. The results were interpreted using SDS Software 2.3. Specificity of amplification was evaluated with the melt curve analysis; no primer dimers were detected and only specific products were obtained. A series of dilutions of cDNA was analyzed to determine the amplification efficiencies. The relative amount of each miRNA against U6 RNA was calculated using the equation 2−ΔCt × 106, where Ct = (CtmiRNA − CtU6RNA).
RT-PCR of novel miRNA precursors
Total RNA samples were DNase treated (Thermo Scientific) and RT reaction was performed; 125 ng of RNA was used along with 250 ng of random primers (Promega) and reverse transcription kit (Invitrogen or Lucigen) in 20 μL reaction volume according to the manufacturer’s protocol. The PCR reaction was performed using primers designed to span the pre-miRNA molecule in 20 μL reaction volume consisting of 0.1 μL (10 μM) of forward and reverse primers, 2 μL of 10× reaction buffer, 1.6 μL of dNTP (4 × 2.5 mM), and 0.25 U of Taq DNA Polymerase per reaction (Thermo Scientific). Primers used for RT-PCR are provided in online resources (Table OR2).
Results
Identification of apple miRNAs
To identify miRNAs that might play a role in rootstock-regulated apple resistance to fire blight, sRNA libraries were generated from the shoots of ‘Gala’ apple grafted on four different rootstocks: two fire blight resistant: G.30 and M.111 and two susceptible: M.27 and B.9. Three biological samples were collected from each scion-rootstock combination, and 12 separate libraries were sequenced. All reads were filtered based on the quality value (see “Materials and methods”) and a total of 105,646,008 high quality reads were obtained. They represent 63,365,291 unique sRNA sequences. A summary of reads for each library is presented in Table 1. The lengths of small RNA reads ranged from 17 to 29 nt. The majority of sRNAs were, in all libraries, in the range of 21 to 24 nt; however, the 24 nt sRNAs were by far the most abundant species. They constituted around 60 % of all redundant and nonredundant reads in each library (Online Resource—Fig. OR3). The next most represented classes were 23 nt sRNA and 21 nt sRNAs. The 23- and 24-mers most frequently possess 5′-adenosine at the first position, a feature often observed in endogenous siRNAs (Axtell 2013), representing in plants the most abundant class of small RNAs (Online Resource—Fig. OR4).
sRNA sequences were mapped to the apple genome, and those, which mapped without any mismatches, were subjected to further rigorous analyses for miRNA identification (see “Materials and methods”). As a result, we obtained 252 miRNA candidates, 114 of which are conserved across plant species. Based on similarity to miRNA precursors from apple, we divided them into two categories: known (136 miRNAs) and novel (116 miRNAs). Names, mature sequences, genomic coordinates, structures, and expression of the complete set of miRNAs identified in the study are stored in the locally developed database (http://lemur.amu.edu.pl/share/apple_miRNAs).
Using RT-PCR, we confirmed the expression of five novel miRNAs: mdm-MIR227N, mdm-MIR260N, mdm-MIR285N, mdm-MIR200N, and mdm-MIR132N (Fig. 1). Experimentally confirmed novel miRNAs were selected from 13 most abundant ones. In addition, for four novel and 15 known miRNAs, we found ESTs that provide evidence for the existence of their precursors (Online Resource—Table OR5). Further analysis of EST sequences led us to identify splice sites in four miRNA genes: mdm-MIR85N, mdm-MIR160c, mdm-MIR403b, and mdm-MIR167j (Fig. 2). There were between one and three introns per gene: one intron in mdm-MIR85N and mdm-MIR160c genes and three introns in mdm-MIR403b and mdm-MIR167j. However, in reality these values might be higher as our method does not guarantee full gene structure discovery.We found that mdm-MIR167j is subject to alternative 3′ splice site selection. The pre-miRNA sequences of intron containing MIR genes are not disrupted by intronic sequences. The gene structures can be viewed at http://lemur.amu.edu.pl/share/apple_miRNAs.

Agarose gel showing five novel miRNA precursors (mdm-MIR227N, mdm-MIR260N, mdm-MIR285N, mdm-MIR200N, and mdm-MIR132N) confirmed with RT-PCR

Gene structures of four MIRNA genes: mdm-miR85N, mdm-MIR160c, mdm-MIR403b, and mdm-MIR167j; precursor sequences are shown in red
Analysis of miRNA expression
Results obtained from the high-throughput sequencing were next used in the analysis of miRNA expression in different rootstocks. To normalize the data for each miRNA, the number of copies per million filtered reads was calculated. First, we compared sets of miRNAs expressed in each rootstock. No rootstock-specific miRNAs were identified; however, mdm-miR535b was absent in all three G.30 biological replicates and two libraries from M.111 rootstock. Both of these rootstocks are fire blight resistant.
Based on the average RPMs for each rootstock, we identified the most abundant miRNAs. The top 10 miRNAs for respective rootstocks did not differ significantly and nearly all of them belong to miRNAs conserved across plant species. This is not surprising since conserved miRNAs tend to be expressed at higher levels than lineage-specific ones (Montes et al. 2014; Axtell 2013; Fahlgren et al. 2010). The only exception is novel apple-specific mdm-miR227N, which is one of the most abundant miRNAs in rootstock G.30. However, the expression level of this miRNA did not differ significantly between rootstocks. Considering the average expression level in all 12 libraries, the most abundant mature miRNAs were as follows: mdm-miR482a, mdm-miR399a,b,c, mdm-miR172e,h, mdm-miR1511, mdm-miR167b-g, mdm-miR482c, three miRNAs from the miR159 family (mdm-miR34N, mdm-miR35N, mdm-miR36N), mdm-miR24N, mdm-miR395a-d,g-i, and mdm-miR396c-e (Table 2).
To assess the biological variance among three replicates of each rootstock/scion combination as well as differential expression of apple miRNAs, one-way ANOVA test was performed. Significant differences in expression were discovered in case of 15 mature miRNAs—eight novel and seven known, with p value < 0.05 and RPM >6. However, the abundance of novel miRNAs was relatively low (below 30 RPM) and the differences may be biologically insignificant. Figure 3 shows the average expression levels, together with ANOVA p values, in all four scion/rootstock combinations for selected conserved and apple-specific miRNAs. A relatively small number of differentially expressed miRNAs can be explained by high variance within biological replicates as the coefficient of variation exceeded 35 % in most groups. In addition, for differentially expressed miRNAs, we performed follow-up Tukey’s range test to better understand which pairs of libraries contribute most to the observed differences. In four out of nine miRNAs, there is at least one pair of libraries with adjusted p value below 0.05, and they include M.27 vs G.30, M.111 vs G.30, G.30 vs B.9, and M.111 vs B.9. Out of them, M.111 and G.30 represent fire blight-resistant trees, while the three remaining pairs contain both fire blight-resistant and fire blight-susceptible trees (Online Resources—Table OR6, Fig. OR7).

Average expression levels in all four scion/rootstock combinations for differentially expressed miRNAs (according to ANOVA statistics)
Nine miRNAs, mdm-miR169a, mdm-miR125N to mdm-miR129N (all from the MIR169 family), mdm-miR160e, mdm-miR160d, and mdm-miR1511 had significantly higher expression level in scions growing on G.30 rootstock. The expression of mdm-miR403a,b was more abundant in M.27 and B.9 scion/rootstock combinations, and the level of mdm-miR403a,b was significantly higher in M.27 and B.9, both fire blight susceptible. mdm-miR7121a-c was overexpressed in G.30 and B.9, and the abundance of mdm-miR7121f-h was significantly lower in M.111.
Target prediction
To assign possible functions to the analyzed miRNAs, we performed target prediction using psRNATarget (Dai and Zhao 2011) against apple coding sequences from GDR (http://www.rosaceae.org/) and UniGene transcripts from NCBI (http://www.ncbi.nlm.nih.gov/unigene). This allowed us to find targets for 118 out of 143 distinct mature known miRNAs and over 200 targets for novel miRNA. Additionally, we analyzed apple degradome (Xia et al. 2012) with PAREsnip (Folkes et al. 2012) and found 72 targets for 27 apple miRNAs. The detailed targets data are available at our database (http://lemur.amu.edu.pl/share/apple_miRNAs). Target interpretation was mainly focused on their putative roles in fire blight resistance in apple and is described in the next section.
Putative miRNA-gene associations in fire blight resistance
Deep sequencing data showed that mdm-miR169a and novel miRNAs from the same family, mdm-miR125N–mdm-miR129N, are preferentially expressed in FB-resistant trees. We confirmed this differential expression by qPCR experiment. It was found that in other plants, including A. thaliana, this miRNA targets NF-Y, a heterotrimeric transcription factor composed of subunits A, B, and C (Leyva-Gonzalez et al. 2012). Consistent with these reports, we found three putative targets, all highly similar to Fragaria vesca nuclear transcription factor Y subunit A (NF-YA) transcript. It has been suggested that miR169-dependent NF-YA regulation can be involved in response to stresses in A. thaliana and maize (Leyva-Gonzalez et al. 2012; Li et al. 2008; Nelson et al. 2007; Pant et al. 2009). Its implications in fungal response in wheat (Inal et al. 2014), as well as in drought tolerance and stem sugar content, were also described (Calvino and Messing 2013). Additionally, miR169 is involved in nodule development and nutritional balance in legume plants (Simon et al. 2009).
Another miRNA showing significantly increased expression in resistant trees is mdm-miR160e. However, identified targets cannot be easily associated with fire blight resistance. Nevertheless, results from other works (Chen et al. 2007; Xin et al. 2010; Zhang et al. 2011) indicate that miR160 targets auxin response factors (ARFs), transcription factors, which are usually upregulated during bacterial infection in plants (Zhang et al. 2011). Moreover, miR160 was reported to positively regulate PAMP callose deposition in response to bacterial infection in A. thaliana (Li et al. 2010).
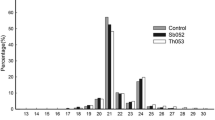
In addition to the abovementioned miRNAs, we also examined targets of mdm-miR167b-g and mdm-miR168a,b, which showed higher expression in fire blight-susceptible trees. Although ANOVA test did not show significant differences in expression, we included these miRNAs since both of them were previously reported to be stress responsive in plants (Liu et al. 2008; Navarro et al. 2006; Sunkar 2010). Overexpression of mdm-miR167b-g in trees growing on fire blight-susceptible rootstocks (M.27 and B.9) was confirmed with qPCR (Fig. 4). For mdm-miR168a,b, we found one target similar to A. thaliana and F. vesca Argonaute protein. Argonaute is a core component of the RISC complex (Caudy et al. 2002; Hammond et al. 2001). The same target was found for miR168 in other plants (Jones-Rhoades and Bartel 2004; Vazquez et al. 2004). miR167 is known to target auxin response factors and participates in bacterial response in plants (Navarro et al. 2006; Sunkar 2010; Varkonyi-Gasic et al. 2010; Zhang et al. 2011). However, we did not find any miR167 targets that correlated with bacterial resistance in apple.

Mature mdm-miR167b-g expression levels detected by RT stem-loop PCR (qPCR) and deep sequencing in four scion/rootstock combinations. Asterisk indicates there are significant differences in mdm-miR167 expression as measured with qPCR (significance level = 0.05, AVOVA statistics)
Some of the abovementioned miRNAs, mdm-miR169a, mdm-miR160e, mdm-miR167b-g, and mdm-miR168a,b, are associated with response to stresses across plant species and are differentially expressed between fire blight-susceptible and fire blight-resistant trees. They represent putative regulators of FB resistance and might be considered as possible markers of FB resistance, but further research is needed.
In our earlier studies, we identified 690 genes up- or downregulated in FB-resistant trees (Jensen et al. 2012). This set of genes was obtained for the same scion/rootstock combinations as in this study. Therefore, to further investigate miRNA-gene associations in FB resistance, we performed reverse target search with psRNATarget, i.e., we looked for miRNAs that potentially target identified differentially expressed genes. We identified three target candidates that are overexpressed in FB-resistant trees: (i) NHX1 (Na+/H+ antiporter), (ii) plant nodulin, and (iii) dinelactone hydrolase. NHX1 might be targeted by four novel miRNAs: mdm-miR142N, mdm-miR143N, mdm-miR144N, and mdm-miR145N. This gene is involved in preventing leakage of electrolytes and nutrients through the plant cell membrane, which appears during the bacterial pathogen infection in plants. The electrolyte leakage from pear treated with E. amylovora was reported previously (Brisset et al. 1990). Apple nodulin is potentially targeted by apple-specific mdm-miR535b,c,d. Nodulins are expressed in response to bacterial infection and are involved in nodule formation in legumes. Nodulin-related proteins were also reported to play a role in pathogen and heat stress tolerance in A. thaliana (Fu et al. 2010) as well as they may be markers of Rhisobium infection in Vicia faba (Becker et al. 2001). The third gene, a transcript highly similar to dinelactone hydrolase protein family, was predicted to be targeted by conserved miR159c. Upregulation of this protein was observed in Glycine max after fungal inoculation (Tremblay et al. 2010) as well as in Japanese black pine after infection with pine wood nematode. It has been suggested that dinelactone hydrolase and other differentially expressed genes may reflect differences between resistant and nonresistant pines (Nose and Shiraishi 2011).
Gene set enrichment analysis
In order to investigate miRNA targets in the miRNAome scale, we performed gene ontology (GO) terms enrichment analysis. First, using BLAST, we found A. thaliana counterparts of apple genes putatively targeted by microRNAs. Then, using GOrilla, a tool for gene set enrichment analysis, we identified enriched GO terms. There are three enriched terms in the category of GO biological process—defense response, response to stress, and primary metabolic process—each with FDR q value < 0.05 (i.e., p value corrected with the Benjamini and Hochberg method). Nucleotide-binding site leucine-rich repeat (NBS-LRR) class disease resistance proteins constitute the prevailing group of genes there. Those proteins are encoded by hundreds of diverse genes per genome and are thought to monitor the status of plant proteins that are targeted by pathogen effectors (McHale et al. 2006). In the molecular function GO category, the six enriched GO terms are dominated by ABC transporters. ABC transporters are driven by ATP hydrolysis and can act as metabolite exporters or importers. Originally identified as transporters involved in detoxification processes, they have also been shown to participate in plant development, organ growth, plant nutrition, response to abiotic stresses, or pathogen resistance. The latter could be supported with the observation that expression levels of more than half of ABC transporters are elevated upon treatment with microorganisms (Kang et al. 2011). In the third GO category, cellular component, there are two terms that meet our q value threshold: proteasome complex and proteasome regulatory particle, base subcomplex. Occurrence of those two terms could be explained by the fact that proteasome-dependent protein degradation is involved in almost all aspects of plant growth and responses to environmental stresses including pathogen resistance. This observation as well as results from the two remaining GO categories (biological process and molecular function) is in compliance with the well-established notion that plant microRNAs are important players in response to abiotic and biotic stresses, including pathogen invasion. The latter is quite strongly supported by the occurrence of genes implicated in plant immune response to biotic stresses, including pathogens (ABC transporters, NBS-LRR proteins, and proteasome complex). We also performed KEGG gene set enrichment analysis using plantGSEA (Yi et al. 2013), but the tool returned no results for our data and applied settings.
Apple miRNA database
The findings described in this work are collected in our apple miRNA database. miRNAs section grants access to all mature miRNA records stored in the database, along with their annotation and expression levels in 12 rootstock-scion combinations. Evidence provides deep sequencing support for miRNAs in the form of alignment of short reads to the precursor. The psRNAtarget and PAREsnip sections provide miRNA targets predicted with psRNAtarget and PAREsnip accordingly. In Introns, miRNA splice sites information is available. GOrilla provides results of GO terms enrichment analysis. Finally, the data can be downloaded in the Downloads section. The database is freely available at http://lemur.amu.edu.pl/share/apple_miRNAs.
Discussion
In this study, we applied small RNA deep sequencing technology to discover apple miRNAs. We found 116 new and confirmed the expression of 143 already known miRNAs. Out of the 13 most abundant novel miRNA precursors, we confirmed five with RT-PCR. We identified one miRNA, mdm-miR535b, which was absent in all three G.30 biological replicates and two replicates from M.111 rootstock (both fire blight-resistant). It was observed that this miRNA is differentially expressed in the leaves and roots (Xia et al. 2012) and does not seem to be related to the bacterial pathogen resistance in apple or other plants. It is rather involved in other processes like fruit ripening in grapes (Wang et al. 2011).
Using small RNA deep sequencing data, we assessed the expression profiles of newly identified and known miRNAs. The most abundant miRNAs corresponded to those, which are highly conserved. The expression level of novel miRNAs was relatively low, which is often seen in species-specific miRNAs (Axtell 2013; Fahlgren et al. 2010). The only exception was mdm-miR227N, which belongs to the ten most abundant miRNAs in rootstock G.30.
Using stem-loop qPCR, we checked the expression of four mature miRNAs, but the results were not always consistent with NGS-based estimations. This can be explained by biases potentially introduced at different steps of sequencing process, e.g., adapter ligation, reverse transcription, or PCR amplification. It is also noteworthy that gene expression is a stochastic process; hence, one can always expect biological variability among measurements (Hansen et al. 2011; McCormick et al. 2011; Zhang et al. 2010). Applying multiple biological replicates may mitigate biological variability. In this study, we used three biological replicates for each scion/rootstock combinations although, due to quite high cost of the sequencing procedure, only one biological replicate was used in most of the published experiments (McCormick et al. 2011). High variance between these replicates was most likely responsible for the fact that only a few differentially expressed miRNAs were identified in the ANOVA analysis. Nevertheless, rootstock-dependent fold change in miR expression levels, together with qPCR confirmations, allow us to define four apple miRNAs potentially involved in fire blight resistance in apple trees: mdm-miR169a, mdm-miR160e, mdm-miR167b-g, and mdm-miR168a,b.
In silico target analysis showed that mdm-miR169a is targeting three apple transcripts, all highly similar to NF-YA, which is known to be involved in response to stresses in other plants (Leyva-Gonzalez et al. 2012; Li et al. 2008; Nelson et al. 2007; Pant et al. 2009). For the remaining three miRNAs, we did not identify targets, which could be easily associated with fire blight; however, all of them are known to be involved in response to stresses across plant species, usually by targeting stress response proteins (Zhang et al. 2011; Liu et al. 2008; Navarro et al. 2006; Sunkar 2010), and therefore, they might be considered as putative regulators of FB resistance and possible markers of FB resistance.
In addition to the abovementioned miRNAs, we also examined miRNAs targeting transcripts previously identified by us as up-or downregulated in FB-resistant trees (Jensen et al. 2012). We identified three candidates, which might be targeted by miRNAs expressed in analyzed trees: NHX1, nodulin, and dinelactone hydrolase. NHX1, involved in preventing leakage of electrolytes and nutrients through the plant cell membrane, which appears during the bacterial pathogen infection in plants (Brisset et al. 1990), is putatively targeted by four novel miRNAs: mdm-miR142N, mdm-miR143N, mdm-miR144N, and mdm-miR145N. Nodulin, which is known to be expressed in response to bacterial infection (Fu et al. 2010; Becker et al. 2001), is potentially targeted by apple-specific mdm-miR535b,c,d. The third gene, a transcript highly similar to dinelactone hydrolase protein family, was predicted to be targeted by conserved miR159c. Upregulation of this protein was observed in other plants after fungal or other infections (Tremblay et al. 2010).
In summary, our studies revealed four miRNAs differentially expressed in fire blight-resistant and fire blight-susceptible trees. In addition, we identified several miRNAs targeting transcripts overexpressed in fire blight-resistant trees. The relatively low number of singled out candidates may result, besides the high variance of biological replicates, from the fact that stress response miRNAs are usually induced by the stress factors and frequently expressed at a low level, or not expressed at all, in normal conditions (i.e., without particular stress) (Kawashima et al. 2009; Zhao et al. 2009). Therefore, further studies, based on healthy and inoculated plants, are currently conducted to confirm identified miRNA candidates and pinpoint additional ones.
References
Axtell MJ (2013) Classification and comparison of small RNAs from plants. Annu Rev Plant Biol 64:137–159
Becker JD, Moreira LM, Kapp D, Frosch SC, Puhler A, Perlic AM (2001) The nodulin vfENOD18 is an ATP-binding protein in infected cells of Vicia faba L. nodules. Plant Mol Biol 47:749–759
Boguski MS, Lowe TM, Tolstoshev CM (1993) dbEST—database for “expressed sequence tags”. Nat Genet 4:332–333
Brisset MN, Ochatt SJ, Paulin JP (1990) Evidence for quantitative responses during co-culture of Pyrus communis protoplasts and Erwinia amylovora. Plant Cell Rep 9:272–275
Calvino M, Messing J (2013) Discovery of microRNA169 gene copies in genomes of flowering plants through positional information. Genome Biol Evol 5:402–417
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinforma 10:421
Caudy AA, Myers M, Hannon GJ, Hammond SM (2002) Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev 16:2491–2496
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179
Chen Z, Agnew JL, Cohen JD, He P, Shan L, Sheen J, Kunkel BN (2007) Pseudomonas syringae type III effector AvrRpt2 alters Arabidopsis thaliana auxin physiology. Proc Natl Acad Sci U S A 104:20131–20136
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinforma 10:48
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210
Eldem V, Okay S, Ünver T (2013) Plant microRNAs: new players in functional genomics. Turk J Agric For 37:1–21
Fahlgren N, Howell MD, Kasschau KD et al (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219
Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, Smith LM, Dasenko M, Givan SA, Weigel D, Carrington JC (2010) MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22:1074–1089
Fallahi E, Colt WM, Fallahi B, Chun IJ (2002) The importance of apple rootstocks on tree growth, yield, fruit quality, leaf nutrition, and photosynthesis with an emphasis on ‘Fuji’. Hort Technol 12:38–44
Folkes L, Moxon S, Woolfenden HC, Stocks MB, Szittya G, Dalmay T, Moulton V (2012) PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res 40:e103
Fu Q, Li S, Yu D (2010) Identification of an Arabidopsis Nodulin-related protein in heat stress. Mol Cells 29:77–84
Gleave AP, Ampomah-Dwamena C, Berthold S, Dejnoprat S, Karunairetnam S, Nain B, Wang Y, Crowhurst RN, MacDiarmid RM (2008) Identification and characterisation of primary microRNAs from apple (Malus domestica cv. Royal Gala) expressed sequence tags. Tree Genet Genomes 4:343–358
Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ (2001) Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293:1146–1150
Hansen KD, Wu Z, Irizarry RA, Leek JT (2011) Sequencing technology does not eliminate biological variability. Nat Biotechnol 29:572–573
Inal B, Türktaş M, Eren H, Ilhan E, Okay S, Atak M, Erayman M, Unver T (2014) Genome-wide fungal stress responsive miRNA expression in wheat. Planta. doi:10.1007/s00425-014-2153-8
Jagadeeswaran G, Saini A, Sunkar R (2009) Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 229(4):1009–1014
Jensen PJ, Makalowska I, Altman N, Fazio G, Praul C, Maximova SN, Crassweller RM, Travis JW, McNellis TW (2010) Rootstock-regulated gene expression patterns in apple tree scions. Tree Genet Genomes 6:57–72
Jensen PJ, Halbrendt N, Fazio G, Makalowska I, Altman N, Praul C, Maximova SN, Ngugi HK, Crassweller RM, Travis JW, McNellis TW (2012) Rootstock-regulated gene expression patterns associated with fire blight resistance in apple. BMC Genomics 13:9
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787–799
Kang J, Park J, Choi H, Burla B, Kretzschmar T, Lee Y, Martinoia E (2011) Plant ABC transporters. Arabidopsis Book 9:e0153
Kapustin Y, Souvorov A, Tatusova T, Lipman D (2008) Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct 3:20
Kawashima CG, Yoshimoto N, Maruyama-Nakashita A, Tsuchiya YN, Saito K, Takahashi H, Dalmay T (2009) Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J 57:313–321
Kellerhals M, Székely T, Sauer C, Frey J, Patocchi A (2009) Pyramiding scab resistances in apple breeding. ISHS Acta Horticult 976:21–28
Kersey PJ, Lawson D, Birney E, Derwent PS, Haimel M, Herrero J, Keenan S, Kerhornou A, Koscielny G, Kähäri A, Kinsella RJ, Kulesha E, Maheswari U, Megy K, Nuhn M, Proctor G, Staines D, Valentin F, Vilella AJ, Yates A (2010) Ensembl genomes: extending Ensembl across the taxonomic space. Nucleic Acids Res 38:D563–D569
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, Sindelka R, Sjoback R, Sjogreen B, Strombom L, Stahlberg A, Zoric N (2006) The real-time polymerase chain reaction. Mol Asp Med 27:95–125
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Leyva-Gonzalez MA, Ibarra-Laclette E, Cruz-Ramirez A, Herrera-Estrella L (2012) Functional and transcriptome analysis reveals an acclimatization strategy for abiotic stress tolerance mediated by Arabidopsis NF-YA family members. PLoS One 7:e48138
Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20:2238–2251
Li Y, Zhang Q, Zhang J, Wu L, Qi Y, Zhou JM (2010) Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol 152:2222–2231
Liu HH, Tian X, Li YJ, Wu CA, Zheng CC (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14:836–843
Ma C, Lu Y, Bai S, Zhang W, Duan X, Meng D, Wang Z, Wang A, Zhou Z, Li T (2013) Cloning and characterization of miRNAs and their targets, including a novel miRNA-targeted NBS-LRR protein class gene in apple (Golden Delicious). Mol Plant 7:218–230
Markham NR, Zuker M (2008) UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 453:3–31
Mascall L (ed) (1569) A booke of the arte and manner how to plant and graffe all sorts of trees: how to sette stones and sovv pipins, to make wild trees to graffe on, as also remedies and medicines: with divers other new practises. H. Bynnemann for J. Wight, London
McCormick KP, Willmann MR, Meyers BC (2011) Experimental design, preprocessing, normalization and differential expression analysis of small RNA sequencing experiments. Silence 2:2
McHale L, Tan X, Koehl P, Michelmore RW (2006) Plant NBS-LRR proteins: adaptable guards. Genome Biol 7:212
Montes RA, de Fátima Rosas-Cárdenas F, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, Marsch-Martínez N, Meyers BC, Green PJ, de Folter S (2014) Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun. doi:10.1038/ncomms4722
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JD (2006) A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 312:436–439
Nelson DE, Repetti PP, Adams TR, Creelman RA, Wu J, Warner DC, Anstrom DC, Bensen RJ, Castiglioni PP, Donnarummo MG, Hinchey BS, Kumimoto RW, Maszle DR, Canales RD, Krolikowski KA, Dotson SB, Gutterson N, Ratcliffe OJ, Heard JE (2007) Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved corn yields on water-limited acres. Proc Natl Acad Sci U S A 104:16450–16455
Norelli JL, Jones AL, Aldwinckle HS (2003) Fire blight management in the twenty-first century: using new technologies that enhance host resistance in apple. Plant Dis 87:756–765
Nose M, Shiraishi S (2011) Comparison of the gene expression profiles of resistant and non-resistant Japanese black pine inoculated with pine wood nematode using a modified LongSAGE technique. For Pathol 41:143–155
Pant BD, Musialak-Lange M, Nuc P, May P, Buhtz A, Kehr J, Walther D, Scheible WR (2009) Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Plant Physiol 150:1541–1555
Simon SA, Meyers BC, Sherrier DJ (2009) MicroRNAs in the rhizobia legume symbiosis. Plant Physiol 151:1002–1008
Sunkar R (2010) MicroRNAs with macro-effects on plant stress responses. Semin Cell Dev Biol 21:805–811
Szczesniak MW, Makalowska I (2014) miRNEST 2.0: a database of plant and animal microRNAs. Nucleic Acids Res 42:D74–D77
Tremblay A, Hosseinib P, Alkharoufb NW, Lic S, Matthews BF (2010) Transcriptome analysis of a compatible response by Glycine max to Phakopsora pachyrhizi infection. Plant Sci 179:183–193
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12
Varkonyi-Gasic E, Gould N, Sandanayaka M, Sutherland P, MacDiarmid RM (2010) Characterisation of microRNAs from apple (Malus domestica ‘Royal Gala’) vascular tissue and phloem sap. BMC Plant Biol 10:159
Vazquez F, Gasciolli V, Crete P, Vaucheret H (2004) The nuclear dsRNA binding protein HYL1 is required for microRNA accumulation and plant development, but not posttranscriptional transgene silencing. Curr Biol 14:346–351
Vogt I, Wohner T, Richter K, Flachowsky H, Sundin GW, Wensing A, Savory EA, Geider K, Day B, Hanke MV, Peil A (2013) Gene-for-gene relationship in the host-pathogen system Malus x robusta 5-Erwinia amylovora. New Phytol 197:1262–1275
Wang C, Shangguan L, Kibet KN, Wang X, Han J, Song C, Fang J (2011) Characterization of microRNAs identified in a table grapevine cultivar with validation of computationally predicted grapevine miRNAs by miR-RACE. PLoS One 6:e21259
Wertheim SJ (ed) (1998) Rootstock guide: apple, pear, cherry, European plum. Fruit Research Station, Wilhelminadorp
Xia R, Zhu H, An YQ, Beers EP, Liu Z (2012) Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol 13:R47
Xin M, Wang Y, Yao Y, Xie C, Peng H, Ni Z, Sun Q (2010) Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol 10:123
Yi X, Du Z, Su Z (2013) PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res 41(Web Server issue):W98–W103
Yu H, Song C, Jia Q, Wang C, Li F, Nicholas KK, Zhang X, Fang J (2011) Computational identification of microRNAs in apple expressed sequence tags and validation of their precise sequences by miR-RACE. Physiol Plant 141:56–70
Zhang W, Gao S, Zhou X, Xia J, Chellappan P, Zhou X, Zhang X, Jin H (2010) Multiple distinct small RNAs originate from the same microRNA precursors. Genome Biol 11:R81
Zhang W, Gao S, Zhou X, Chellappan P, Chen Z, Zhou X, Zhang X, Fromuth N, Coutino G, Coffey M, Jin H (2011) Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol Biol 75:93–105
Zhao B, Ge L, Liang R, Li W, Ruan K, Lin H, Jin Y (2009) Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol Biol 10:29
Acknowledgments
We thank Penn State Genomics Core Facility, University Park, PA, for performing the sequencing. This project was supported by MPD Programme of Foundation for Polish Science, cofinanced from European Union, Regional Development Fund (Innovative Economy Operational Programme 2007-2013): MPD/2010/3 (IM and EK), by National Science Centre grant no. 2013/09/N/NZ1/01037 (EK), by a scholarship within the project “Scholarship support for Ph.D. students specializing in majors strategic for Wielkopolska’s development,” Sub-measure 8.2.2 Human Capital Operational Programme, co-financed by European Union under the European Social Fund (EK) and by START programme from the Foundation for Polish Science (MS).
Conflict of interest
The authors declare that they have no competing interests.
Authors’ contributions
Izabela Makałowska, Elżbieta Kaja, Philip J. Jensen, Michael J. Axtell, and Timothy McNellis conceived and designed the experiments; Elżbieta Kaja performed the experiments; Michał W. Szcześniak, Elżbieta Kaja, and Michael J. Axtell analyzed the data; and Elżbieta Kaja, Michał W. Szcześniak, and Izabela Makałowska wrote the paper. All authors read and approved the final manuscript.
Data archiving statement
Raw and processed small RNA-seq data are submitted to the Gene Expression Database at the National Center for Biotechnology Information. Accession numbers for all biological replicates data are provided in Table 1.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by A. M. Dandekar
Elżbieta Kaja and Michał W. Szcześniak contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
(DOCX 58 kb)
Online Resource 2
(DOCX 53 kb)
Online Resource 3
Comparison of abundance of redundant and nonredundant reads in 12 apple libraries. (PPTX 1286 kb)
Online Resource 4
Length and 5′-nucleotides distribution of redundant reads in 12 apple libraries. (PPTX 1205 kb)
Online Resource 5
(DOCX 15 kb)
Online Resource 6
(DOCX 13 kb)
Online Resource 7
(PDF 207 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Kaja, E., Szcześniak, M.W., Jensen, P.J. et al. Identification of apple miRNAs and their potential role in fire blight resistance. Tree Genetics & Genomes 11, 812 (2015). https://doi.org/10.1007/s11295-014-0812-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-014-0812-3