
Abstract
The present work systematically investigated the deactivation of sulfated titania in the esterification of acetic acid and n-butanol. Under the set conditions, sulfated titania was used 20 times and for 10 h accumulatively. The catalyst had the largest catalytic activity of 95.69 % at the second cycle and then the catalytic activities gradually dropped, and the last-cycle conversion of acetic acid was 46.15 %, suggesting a serious deactivation of the catalyst of sulfated titania after use in 20 cycles. XPS, FTIR, XRD, BET, TG–DSC, TEM and NH3-TPD were employed to characterize the new and deactivated catalysts, to systemically study the deactivation phenomena of sulfated titania and to find some reasons for the considerable deactivation of sulfated titania in the esterification. Some deactivation phenomena were observed on the deactivated catalyst when compared with the new one. (1) The deactivated catalyst decreased its acidity, based on the IR and NH3-TPD characteristic results. (2) The deactivated catalyst increased its surface specific area and its pore volume, and decreased its pore diameter, based on the BET characteristic results. (3) Some carbon deposits appeared on the surface of the deactivated catalyst, and some originally active sulfate groups may have been lost, poisoned and turned into less-catalytic and non-catalytic sulfur species based on the XPS and TG–DSC results. (4) The deactivated catalyst decreased its crystallinity based on the XRD results. (5) The deactivated catalyst diminished in particle aggregation, based on the SEM results. Thus, a deactivation mechanism is tentatively proposed. Namely, some originally active surface sulfate groups may have been gradually turned into the undesired free sulfuric acid and organic sulfate esters (OR)–(O=S=O)–O or (OR)–(O=S=O)–(OR) [R = C4H9, etc.] due to Ti4+ cations’ hydrolysis by H2O and their alcoholysis by n-butanol, which may lead to a gradual acidity degradation of the catalyst so as to lead to a gradual deactivation of sulfated titania. All the characteristic results more or less support the proposed deactivation mechanism.
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Nowadays, many researchers are looking for new solid acid catalysts [1–8] for replacing traditional liquid acid catalysts, accompanied by such problems as corrosions and serious side reactions. Sulfated metal oxides [1–8] are attracting more and more attention in recent years and are becoming a type of very promising catalysts, because they are less corrosive and environmentally friendly. Besides, they also exhibit high catalytic activities in many kinds of acid catalyzed organic reactions. However, there is a fatal problem that they can be readily deactivated despite their initial high activities [9, 10]. Some possible deactivation reasons [9, 10] can be summarized as follows for sulfated zirconia: (1) acidity degradation; (2) surface sulfur species can be reduced from S6+ to a lower valance state; (3) coke formation; (4) sulfur loss; (5) phase transformation from catalytically active tetragonal ZrO2 to catalytically inactive monoclinic ZrO2. However, there are still many debates and obscurities on the exact deactivation reasons of sulfated metal oxides. Additionally, the deactivation of sulfated metal oxides is mainly focused on alkane reactions, the deactivation in other reactions such as esterification is studied much less than in alkane conversion reactions. So, some progressive studies should also be made to find out the exact reasons for their rapid deactivation in other types of acid-catalyzed reactions rather than only to be limited to alkane conversion reactions.
The present work systematically investigated the deactivation of sulfated titania in the esterification of acetic acid and n-butanol. X-ray photoelectron spectroscopy (XPS), FTIR, XRD, BET, TG–DSC, transmission electron microscopy (TEM) and NH3-temperature programmed desorption (TPD) were employed to characterize the new and deactivated catalysts, to study the deactivation phenomena of sulfated titania systemically and to find some reasons for the considerable deactivation of sulfated titania in the esterification. It is deduced by the corresponding characteristic results that a kind of re-hydroxylation of Ti4+ cations, hydrolysis by H2O and alcoholysis by butanol, may have taken place on the deactivated catalyst, and correspondingly some originally active surface sulfate groups may have been turned into free sulfuric acid and organic sulfate esters (OR)–(O=S=O)–O or (OR)–(O=S=O)–(OR), which leads to a gradual acidity degradation of the catalyst so as to lead to a gradual deactivation of sulfated titania. All the characteristic results more or less support the proposed deactivation mechanism.
Experiments
Materials
Chemical reagents used in the present work, such as titanium tetrachloride (TiCl4), ammonia (25 % NH3·H2O), anhydrous ethanol (C2H5OH), concentrated sulfuric acid (98 % H2SO4), n-butanol (C4H9O10), and acetic acid (CH3COOH), are all analytical reagent grade and purchased from SCRC of china.
Preparations of the catalysts
82 g of TiCl4 was added into 1,617 g ice with violent stirring, a lot of white smoke was observed instantly, and a mixture (1) was obtained. Subsequently, an extra 1,407 g deionized water and 748 g ice were added to the above mixture (1) with violent stirring, and a white milky mixture (2) was obtained. By adding ammonia dropwise to the above mixture (2) to obtain a final pH of 9–10, a hydrous Ti(OH)4 precipitate (3) is produced. The precipitate (3) was then filtered in a Buchner funnel and the precipitate cake was washed with deionized water until there was no Cl− detectable by AgNO3 (0.01 M) in the last-patch water solution filtered out of the Buchner funnel.
The washed precipitate was dried at 100 °C for 12 h to remove most of water. The dried hydroxide was grounded and sieved by a 100-mesh sieve. The sieved hydroxide was then sulfated for 24 h by adding H2SO4 (1 mol/l) on the ratio of 15 ml solution to 1 g hydroxide. Furthermore, the obtained mixture was filtered and the filter cake was dried at 100 °C for 12 h, and was ground and was sieved by a 100-mesh sieve. The obtained powder was calcined at 500 °C in static air atmosphere in a muffle furnace for 3 h. Then, the calcined sample was naturally cooled to room temperature, obtaining the final sulfated titania.
Stability of sulfated titania in the esterification
The n-butanol and acetic acid esterification was used as a model reaction to evaluate the stability of the prepared sulfated titania samples. The reaction was performed at atmospheric pressure in a three-necked flask, which was equipped with a condenser and a water separator and heated by a temperature-controlled oil-bathed pot. The reactant ratio of acetic acid/butanol was 20:50 ml (0.3494:0.5462 mol), and the catalyst amount (mcat) was 3 g, which was 4.88 wt% of the total mass of reagents including acetic acid and n-butanol.
The detailed experimental strategies were as follows: (1) the reactant system was first heated up to the refluxing temperature (98–112 °C) and reacted for 0.5 h; (2) when the reaction ended, the liquid product was distilled out of the reaction system until the temperature of the reaction system reached to 116 °C and there seemed to be little liquid left on the catalyst, and the corresponding condensed liquid product was analyzed by the gas chromatography; (3) the catalyst, still kept in the flask without any further processing, was continued to be used for the next-cycle esterification by adding a new reaction mixture of the acetic acid (20 ml) and n-butanol (50 ml). It is necessary to note that the above reaction conditions are chosen based on our previous studies on the optimized reaction conditions of the esterification.
The new and deactivated sulfated titania are denoted as Ti0 and Ti20 (after 20-times use), respectively.
The used gas chromatograph is SP-6890 (ShanDongLuNanRuiHong Chemical Instrument Corporation) equipped with a capillary chromatographic column of FFTP (50 m × 0.32 m × 1 μm). The FID detector is used, and the column temperature is 80 °C and the injection temperature is 180 °C.
Characterization of the catalysts
The XPS analyses were conducted using Thermofisher ESCALAB 250 system with Al Kα radiation under ultrahigh vacuum (UHV), calibrated internally by the carbon C 1s binding energy (BE) at 284.6 eV.
X-ray powder patterns were recorded on a Brucker D8 diffractometer, with a copper tube as radiation source (λ = 0.15405 nm) and operated at 40 kV and 20 mA, all the profiles were recorded at 1° (2θ)/min. The scan range was from 2θ = 10° to 80°.
FT-IR spectra were acquired using the transmission mode on a Bruker TENSOR 27 FT-IR spectrometer with a MCT detector. The samples were mixed with KBr and compacted into thin pellets under 8 kPa. The spectra in the range of 4,000–600 cm−1 were recorded at room temperature.
The BET surface area was calculated by measuring N2 adsorption–desorption isotherms at liquid N2 temperature at 77 K on Sorptomatic 1990. The pore size distribution was obtained from the desorption isotherm branches by BJH methods.
TEM experiments were performed on an H-800 electron microscope with an acceleration voltage of 200 kV.
The TG–DSC measurements were performed on an STA409PC apparatus, and the procedures for heating rate were recorded at a rate of 10 °C/min, the heating range was from room temperature to 1,000 °C, containing high-purity Ar at 30 ml/min. For every test, 13.0–13.5 mg of sample was used.
TPD of NH3 was performed on CHEMBET-3000 (American Quantachrome Company) equipped with a TCD detector. The sample of about 0.2 g catalyst was treated at 400 °C for 1 h in flowing helium (20 cm3/min) prior to NH3 adsorption at room temperature. After the sample was subsequently purged at 100 °C for 1 h in flowing helium (20 cm3/min) to remove physisorbed NH3, the TPD spectrum was recorded at a heating rate of 10 K/min from 100 to 800 °C.
Results and discussion
Stability of sulfated titania in the esterification
The esterification and some well-known side reactions of the esterification are seen as below.
Besides, butene may progressively crack into propene and ethene, and these alkenes may progressively polymerize into alkene polymers. All the unwanted side reactions are bound to be disadvantageous to the activity and stabilities of sulfated titania, because they can produce carbon deposits on the catalyst which can decrease the catalytic activity of the catalyst by poisoning it and decreasing the effective concentration of active sulfate groups and active sites on the catalyst. The stability of sulfated titania is shown in Fig. 1. Sulfated titania was used 20 times and 10 h accumulatively. The catalyst has a largest catalytic activity of 95.69 % at the second cycle and then the catalytic activities gradually drop. The last-cycle conversion of the acetic acid is 46.15 %, suggesting a considerable deactivation of the catalyst of sulfated titania after using in 20 cycles.

Stability of sulfated titania
The present work tries to find the reasons for the deactivation of sulfated titania by employing some characteristic methods as below, to try to find some reasons for the considerable deactivation of sulfated titania in the esterification.
XPS analyses for the new and deactivated catalysts
Fig. 2a–d shows the S 2p, Ti 2p, O 1s, C 1s of the new catalyst Ti0 and the seriously-deactivated catalyst Ti20.

a BE of S 2p in the new and deactivated catalysts. b BE of Ti 2p in the new and deactivated catalysts. c BE of O 1s in the new and deactivated catalysts. d BE of C 1s in the new and deactivated catalysts
Fig. 2a shows the XPS spectrum in the S 2p BE region for Ti0 and Ti20, and the BE of S 2p was observed at about 168.6 eV corresponding to the typical S6+ of sulfate [11–13]. The spin–orbit (Ti 2p3/2) signals at about 459.2 eV, seen in Fig. 2b, indicated that Ti element mainly existed as the chemical state of Ti(IV) [11–13]. The O 1s BE, seen in Fig. 2c, of the two catalysts observed at about 530.4 eV is attributed to the presence of a S–O–Ti linkage[11–13]. The BE of about 530.2 eV can be attributed to matrix oxygen in TiO2, and the BE of about 531.7 eV reveals the presence of other two different oxygen species in the sample, and this could be due to the presence of an OH− group, S–O bonds, or a chemisorbed water molecule [11–13]. It is emphasized that there are observable differences of these binding energies of the new and the seriously deactivated catalysts (e.g., the observable difference of O 1s of them can be seen in Fig. 2c), possibly arisen from the weakening of the bonding interaction (between the surface sulfate groups and metal Ti4+ cations) and also possibly arisen from the re-hydroxylation of Ti4+ cations on the deactivated catalyst. The C 1s BE, seen in Fig. 2d, of the two catalysts observed at about 284.6 eV.
Based on the XPS results, the surface atomic compositions of the new and deactivated catalysts are shown in Table 1. Apparently, the carbon content in Ti0 is only arisen from unavoidable and ubiquitous carbon contaminants, but the carbon content in Ti20, by far larger than that in Ti0, is sure to arise from a combination of the above so-called carbon contaminants and extra carbon deposits originated from the side reactions accompanying the esterification. The increased 41.62 % of the carbon content in Ti20, referred to that in Ti0, implies that extra considerable carbon deposits appear on the surface of the deactivated catalysts. It is well known that carbon deposits are considered an important reason for the deactivation of sulfated metal oxides due to their poisoning sulfate species [1, 9, 10]. So, we consider that the increased carbon deposits may be partly responsible for the deactivation. Some carbon deposits may be heavy alkene polymers from unwanted side reactions, and some carbon deposits may be organic sulfate esters (including organic groups), transformed from sulfate group poisoning by organic groups (based on the next deactivation mechanism proposed by us). After many cycles of uses, carbon deposits must accumulate on the catalyst’s surface. Emphatically, the surface S content in Ti20 only decreases 13.45 %, slightly lower than that in Ti0, signifying that most of the surface sulfate groups still stand on the deactivated catalyst surface. However, the surface sulfur species on the deactivated catalyst are not active enough to generate strong Lewis acidity or Brønsted acidity any more, so we think that these originally active surface sulfate groups may have been poisoned due to their being turned into some less-catalytic or non-catalytic sulfur species including organic groups.
It is necessary to note that the reason why we do not consider the possible adsorption of the acetic acid, n-butanol and n-butyl acetate is that these substances are not accumulative and are easily distilled away from the surface of the catalyst and the weight percentage of the heavy carbon deposits, which can accumulate with the continual use of the catalyst, are bound to be much larger than that of the above small molecules. When we immersed the deactivated catalyst in ethanol overnight, there were no discernible signals of the above small molecules and any carbon deposits in the immersed solution by the gas chromatography, displaying that the above small molecules are distilled out of the deactivated catalyst and carbon deposits may be heavy alkenes polymers or poisoned sulfur species including organic groups, still kept on the deactivated catalyst.
Besides, there appeared an increase of 4.56 % of the surface O atom on the deactivated catalyst, displaying that the catalyst can adsorb water, or metal Ti4+ cations can catch OH groups from the reaction system so as to form Ti–OH, which is supported by the IR results as below.
FT-IR spectra of the new and deactivated catalysts
As seen in Fig. 3a, b, the bands at about 1221, 1135, 1046, 945 cm−1 can be attributed to S–O and S=O asymmetric and symmetric vibrations in the new and deactivated sulfated titania [14–24]. As seen in the new and the deactivated sulfated titania and their precipitate counterpart, the broad peaks at around 3,423 cm−1 correspond to the O–H stretching vibration mode of the surface hydroxyls and the planar water, and the peaks at 1,628 cm−1 can be attributed to the OH stretching vibration of water adsorbed on their surfaces [14–24]. The band at 1,402 cm−1 can be attributed to increased hydroxyl groups [25], possibly arisen from the combination of Ti4+ cations with hydroxyl groups from water and n-butanol by us. The band at 1,112 cm−1 may be free sulfuric acid, possibly arisen from the transformation of sulfate groups originally coordinated to Ti4+ cations during the hydrolysis of Ti4+ cations by water or alcoholyzed by butanol (based on the as-below deactivation mechanism proposed by us). For the deactivated sulfated titania, the band at 1,221 cm−1 is less intense on the deactivated catalysts than on the new catalyst Ti0, displaying that the deactivated catalyst decrease their acidity. However, the band at 1,402 cm−1 is much more intensive on the seriously deactivated catalyst Ti20 than on the new one, disclosing a possible re-hydroxylation of Ti4+ on the deactivated catalyst. Besides, the band at 1,151 cm−1 is more intense on the deactivated catalyst than on the new one. The above band variations, before and after deactivation, show a considerable acidity degradation of the deactivated sulfated titania, possible arisen from the hydrolysis of Ti4+ cations by water or alcoholyzed by butanol and corresponding to some transformation of active sulfate groups into non-active sulfur species such as free sulfuric acid.

a IR profiles of the precipitate Ti (OH)4 and sulfated titania from 4,000 to 600 cm−1. b IR profiles of the precipitate Ti (OH)4 and sulfated titania from 1,700 to 800 cm−1
TG–DSC profiles of the new and deactivated catalysts
The TG–DSC profiles of the new and deactivated sulfated titania are shown in Fig. 4a, b. These TG–DSC profiles are analyzed by referring to the literatures [14, 15, 26–28]. Below 200 °C, the weight loss can be mainly attributed to the removal of water (in hydration or structural), corresponding endothermic peaks seen in DSC profiles. At about 300 °C, there come endothermic peak due to the evaporation or decomposition of free sulfuric acid, showing a type of transformation of sulfate groups on the catalysts. The peak temperature 315 °C for the deactivated catalyst is lower than the peak temperature 363 °C for the new catalyst, disclosing that there are more sulfate groups turn into free sulfuric acid on the deactivated catalyst. In the region of 200–500 °C, the weight loss of the new sulfated titania can be mainly attributed to dehydroxylation for the new catalyst. However, the weight loss of the deactivated sulfated titania can be less attributed to dehydroxylation, and more attributed to a possible desorption or decomposition of its surface carbon deposits such as alkene polymers and organic sulfate esters (based on the next deactivation mechanism proposed by us), which is disclosed by the obvious exothermal stage seen in the DSC profile of the deactivated sulfated titania in the region of 200–500 °C. This much more exothermic than that of the new sulfated titania. Besides, the exothermal crystallization process is obvious at about 400 °C seen on the DSC profile of the deactivated catalyst, disclosing a weakening of strong coordination between sulfate species and Ti4+ cations for the deactivated catalyst.

a TG profiles of the new and deactivated catalysts. b DSC profiles of the new and deactivated catalysts
From around 500 °C on, there appears a gradual evolution of SOx (SO3, SO2) due to the remaining sulfate esters and active sulfate groups, with an weakly endothermic stage seen in DSC profiles of the deactivated sulfated titania. From around 500 °C on, there appears a gradual evolution of SOx (SO3, SO2) due to of active sulfate groups, with an considerably-endothermic stage seen in DSC profiles of the new sulfated titania. The above fact discloses there are much less active sulfate groups on the deactivated catalyst than on the new one.
Based on the above TG results, the estimated water contents are 1.60 wt% for the new and 2.91 wt% for the deactivated) and the estimated content of the decomposed carbon deposits is 4.12 wt% for the deactivated catalyst. The estimated sulfate group contents are 4.55 wt% for the new and 2.49 wt% for the deactivated, respectively. Therefore, a surface sulfated group loss of 2.06 wt% is seen on the deactivated sample, which may be partly due to the transformation of some sulfate groups into free sulfuric acid or sulfate esters (based on the next deactivation mechanism proposed by us). Emphatically, in the region of 500–630 °C, a gradual decreasing is seen for the new, but a platform appears for the deactivated, which may be arisen from the absence of the transformed sulfate groups.
Emphatically, there are still some remaining sulfur species of 2.49 wt% on the deactivated catalyst. In the view of its considerable deactivation, we consider that most of these remaining sulfur species are poisoned or turned into non-catalytic sulfate esters. A deactivation mechanism will be proposed in section “NH3-TPD profiles of the new and deactivated catalysts” of this paper.
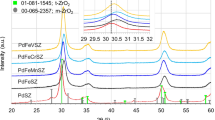
X-ray diffraction of the new and deactivated catalysts
The wide-angle XRD patterns of the new catalyst Ti0, the deactivate catalyst Ti20 and their precipitate counterpart are shown in Fig. 5. All diffraction lines of the catalyst corresponded well to those for the tetragonal anatase phase of TiO2, JCPDS Card No: 21-1272 (JCPDS Catalogue). The peaks at 2θ ≈ 25°, 38°, 48°, 54°, 62° are all uniquely attributable to the anatase phase of TiO2 [16, 17]. There is no discernible rutile phase, indicating that the existence of sulfate groups can effectively retard the metastable anatase phase to the rutile phase by Ti–O–S linkages. The deactivated catalyst has diffraction peaks more intense than those of the new catalyst, which means the former has a larger crystallinity than the latter, disclosing a possible weakening of Ti–O–S linkages on the deactivated catalyst. If only the highest diffraction peaks of the new and deactivated catalysts compared, the estimated crystallinity of the deactivated catalyst are ca. 20.69 % higher than the new one. Besides, the transformed sulfate species including free sulfuric acid and sulfate esters are not to inhibit the crystallization of metal oxides any more.

XRD patterns of the new and deactivated catalysts and Ti(OH)4
Specific surface areas and pore distributions of the catalysts
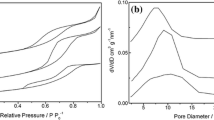
The specific surface areas, pore volumes, and pore diameters of the new and the seriously deactivated sulfated titania are summarized in Table 2. For the deactivated catalyst when compared with the new one, the surface specific area, the pore volume and the pore diameter increase by 16.17, 17.73 and −13.50 %, which means there are more and smaller pores in the deactivated catalyst than in the new one. The adsorption–desorption isotherm of Fig. 6a shows typical type IV isotherms with hysteresis loop from about 0.4 on caused by capillary condensation in mesopores, showing that the two catalysts has both a lot of mesopores. It should be noted that the two adsorption–desorption isotherms of the two catalysts differentiate in their places, reflecting that their respective internal pores differentiate in pore diameter distribution (namely pore diameter and pore quantity). These differences can be apparently seen in Fig. 6b. The aforementioned differences of the new and seriously deactivated catalysts may be partly related to the re-hydroxylation of Ti4+ cations, which can increase hydrophilicity of the metal oxide so as to lead to the its decreased particle diameters and its increased specific surface area. Besides, when the bonding interaction between sulfate groups and Ti4+ metal cations is weakened or destructed, the crystallization of the catalyst and the loss of sulfate groups may be accelerated, which can bring about some variations in the specific surface area, the pore volume, and the pore diameter of the deactivated catalyst.

a Adsorption–desorption isotherms of the new and the deactivated catalysts. b Pore distribution of the new and the deactivated catalysts
SEM of the new and deactivated catalysts
Fig. 7 gives the SEM images of the two catalysts. The amorphous phase of TiO2, which is well-defined spheres, is the main phase of the two catalysts. When combined with the XRD results, there must be the anatase phase of crystalline TiO2 in the two catalysts, which cannot be observed by the SEM photos and may be stay at the surface of the amorphous TiO2 spheres as small particles of several nanometers. The deactivated catalyst is smaller in diameter and has less aggregation than the new one, which corresponds to the improved surface specific area of the deactivated catalyst. The above phenomena may be related to the re-hydroxylation of Ti4+ cations into Ti–OH, because the increased –OH groups can increase the hydrophily of metal oxides.

SEM photos of the new and the deactivated catalysts (the left is Ti0 and the right is Ti20)
NH3-TPD profiles of the new and deactivated catalysts
Different NH3-TPD profiles are shown for the two catalysts in Fig. 8. It is well agreed [29, 30] that NH3 desorption peak temperature can stand for the acidities of the samples below the decomposition temperatures of sulfate species, if the reduction of sulfate groups is omitted, the larger the desorption peak temperatures, the larger the acid strength of tested samples, the larger the integral area of the desorption curves, and the larger the concentration of the acidic active sites in the tested samples. The new catalyst has a NH3-desorption peak around 569 °C indicating the formation of superacid sites. The deactivated catalyst has two NH3-desorption peaks at around 537 and 619 °C, showing the decreasing acidity of the deactivated catalyst, possibly arisen from the weakening of the bonding interaction between the sulfate groups and the metal Ti4+ cations. Besides, below the temperature of 469 °C for the new catalyst, and the temperature of 424 °C for the deactivated catalyst, there are some acidic sites of weak and mediate strength. Based on the corresponding integral areas of these desorption peaks, which stands for the concentration of acidic sites, the deactivated catalyst decrease its concentration of acidic sites largely after the continual use, only about 30 % of that of the new catalyst.

NH3-TPD profiles of the new and deactivated catalysts
The acidity degradation of the deactivated catalyst may be related to the re-hydroxylation of Ti4+ cations of strong Lewis acidity which turn them into Ti–OH of weak Lewis acidity, and the transformed Ti–OH species are not to generate strong Brønsted acidity as well as initial Ti4+ cations of strong Lewis acidity.
A novel deactivation mechanism for sulfated titania in the esterification
Yamaguchi [1] points out (O=S=O)(OR)2 characteristic IR bands are [1350–1460, 1150–1230, 975–1000 cm−1] (for ν3), and 910 cm−1 (for ν1), all of which overlap with those characteristic bands of surface sulfate groups. So, we consider there may be some organic sulfate esters (OR)–(O=S=O)–O or (OR)–(O=S=O)–(OR) produced on the deactivated catalyst’s surface, based on our IR results.
Besides, there found the existence of free sulfuric acid, and the possibility of Ti4+ cations hydrolysis by H2O and alcoholysis by butanol.
So, we deduce that these so-called organic sulfate esters are possibly arisen from the hydrolysis of surface sulfate groups by H2O and alcoholysis by n-butanol. A possible deactivation mechanism of sulfated titania in the esterification, is proposed in Scheme 1.

A possible deactivation mechanism of sulfated titania in the esterification (M stands for Ti)
Scheme 1 actually shows a strong trend of re-hydroxylation of Ti4+ cations: (1) Ti–O–(SO2)–O–Ti moieties are turned into Ti–OH and Ti–O–(SO2)–O–C4H9 and even C4H9–O–(SO2)–O–C4H9 after the alcoholysis of surface sulfate groups by n-butanol. (2) Ti–O–(SO2)–O–Ti moieties are turned into Ti–OH and Ti–O–(SO2)–OH after the hydrolysis of surface sulfate groups by H2O. Subsequently, Ti–O–(SO2)–OH moieties can be turned into Ti–O–(SO2)–O–C4H9 and even C4H9–O–(SO2)–O–C4H9 by n-butanol. Certainly, free sulfuric acid can also be obtained by the subsequent hydrolysis of Ti–O–(SO2)–OH by H2O. So, the originally active sulfate groups are turned into free sulfuric acid and organic sulfate esters during the re-hydroxylation of Ti4+ cations by their hydrolysis by H2O and alcoholysis by butanol.
Additionally, the above organic sulfate esters may be subsequently turned into 1-butene and 2-butene (seen in Scheme 1) and other side products [31]. Cited by the literature [31], Glencoe et al. had proposed an oxidizing mechanism of reactions of alkanes on sulfated zirconia, thinking that the high activity of sulfated metal oxides in alkane conversion is due to their one-electron oxidizing ability, leading to ion radicals and then to surface sulfate esters (observed in their experiments), which are the active intermediates in the mechanism. These surface sulfate esters either ionize generating carbocations or eliminate forming olefins, both by carbocationic reactions with no requirement of superacidity [31].
Due to the so-called hydrolysis and alcoholysis, Ti4+ gets more and more weakly bonded to the single-bond O atom in S–O of surface sulfate species and even possibly completely loses it, and subsequently is easier to combine with H2O or hydroxyls so as to low its Lewis acidity, partly supported by the intensified band at 1,402 cm−1 for the deactivated catalyst. If only a single-bond O atom is partly lost, the Ti···(OR)–(O=S=O)–O–Ti is produced. If two single-bond O atoms are both partly lost, the Ti···(OR)–(O=S=O)–(OR)···Ti is produced. Additionally, due to the OR groups are ones attracting electrons strongly, the S=O bond order is decreased and its IR wavenumbers are correspondingly shifted to lower ones, leading to the weakening of the band at 1,221 cm−1 and the intensifying of the band at 1,151 cm−1 for the deactivated sulfated titania. The band at 1,112 cm−1 also give a support for the hydrolysis of Ti4+ by H2O.
Therefore, some originally catalytically active surface sulfate species may have been gradually turned into the undesired free sulfuric acid and organic sulfate esters (OR)–(O=S=O) or (OR)–(O=S=O)–(OR), as carbon deposits on the deactivated catalyst. Though some of the produced free sulfuric acid and organic sulfate esters can still partly combined on TiO2 surface but the original strong bonding interaction (between the sulfate groups and metal Ti4+ cations) is weakened or destructed partially or completely.
Emphatically, in our proposed mechanism, the water produced from the esterification may play a key role in the deactivation, because it can directly hydrolyze strongly Lewis acidic Ti4+ cations on the catalyst to form weakly Lewis acidic Ti–OH species and turn surface sulfate groups into free sulfate groups, and simultaneously it can also accelerate and facilitate the alcoholysis of strongly Lewis acidic Ti4+ cations of the catalyst to form weakly Lewis acidic Ti–OH species, leading to the production of sulfate esters.
Based on the paper of Suwannakarn et al. [32], some other workers consider that water can wash away the sulfate groups from the catalyst surface. As reported previously, ionic sulfur species supported on the sulfated zirconia catalyst surface can be modified and successively transformed into H2SO4, HSO4 −, and SO4 2− by the presence of free water in the liquid phase, leading to the loss of active sites from the solid surface. Cited by the literature [32], Omota et al. had reported that after contacting fresh sulfated zirconia catalyst samples with water, a rapid drop in the pH of the solution occurred, indicating that acid species likely were being leached out into solution. Other authors also have documented the capability of water to leach out the active catalytic species in SZ [32].
Suwannakarn et al. [32] think alcohols can also wash away catalysts’ surface sulfate groups despite the their far weaker washing functionality than that of water. Suwannakarn et al. observed monoalkyl hydrogen sulfate and dialkyl sulfate in the methanol filtrate solution after washing sulfated zirconia by 1H NMR studies, and deduced that the removal of sulfate ions from the catalyst surface as sulfuric acid in alcohols (methanol, ethanol, and butanol), which subsequently reacts with alcohol to form monoalkyl hydrogen sulfate and dialkyl sulfate.
Indeed, the weakened IR band at 1,221 cm−1 and the intensified IR bands at 1,151 cm−1 on the deactivated sulfated titania, disclose some possible existence of surface sulfate esters, partly corresponding to the above viewpoints of Suwannakarn et al. Above all, the intensified band at 1,402 cm−1 and the increased percentage of the surface O atom on the deactivated sulfated titania, give some direct evidence of the re-hydroxylation of Ti4+. Moreover, our interpretations emphasize that some of the decayed surface sulfate species, which have been transformed into organic sulfate esters, can keep standing on the catalyst surface based on our XPS analyses. Our interpretations clarify how surface sulfate groups are gradually hydrolyzed or alcoholyzed into free sulfuric acid and organic sulfate moieties, and subsequently how the Lewis acidity of Ti is gradually decreased, and subsequently how the catalyst deactivation gradually takes place. The deactivation mechanism can also partly explain the catalyst surface carbon deposits observed in our XPS and TG–DSC analyses for the deactivated sulfated titania, because these organic sulfate esters including alkyls (C4H9 etc.) also being a type of main carbon deposit of the deactivated catalyst.
Besides, the deactivation mechanism can interpret well the former characteristic results such the larger crystallinity and the variation of specific surface area and the acid degradation of the deactivated catalyst.
It is necessary to note that although the above characteristic results and some literatures [31, 32] more or less support the tentatively proposed deactivation mechanism, there still need more distinct evidences such as the study of the carbon deposits on the deactivated catalyst by mass spectrometry to prove it further.
Conclusions
Some deactivation phenomena were observed on the deactivated catalyst when compared with the new one. (1) The deactivated catalyst decreased its acidity, based on the IR and NH3-TPD characteristic results. (2) The deactivated catalyst increased its surface specific area and its pore volume, and decreased its pore diameter, based on the BET characteristic results. (3) Some carbon deposits appeared on the surface of the deactivated catalyst, and some originally active sulfate groups may have been lost, poisoned and turned into less-catalytic and non-catalytic sulfur species, based on the XPS and TG–DSC results. (4) The deactivated catalyst decreased its crystallinity, based on the XRD results. (5) The deactivated catalyst diminished in particle aggregation, based on the SEM results.
Thus, a deactivation mechanism is tentatively proposed, namely some originally active surface sulfate groups may have been gradually turned into the undesired free sulfuric acid and organic sulfate esters (OR)–(O=S=O)–O or (OR)–(O=S=O)–(OR) [R = C4H9, etc.] due to the hydrolysis of Ti4+ cations by H2O and their alcoholysis by n-butanol, which may lead to a gradual acidity degradation of the catalyst so as to lead to a gradual deactivation of sulfated titania. All the characteristic results more or less support the proposed deactivation mechanism.
References
Yamaguchi T (1990) Recent progress in solid superacid. Appl Catal 61:1
Yamaguchi T (2001) Alkane isomerization and acidity assessment on sulfated ZrO2. Appl Catal A 222:237
Reddy BM, Patil MK (2009) Organic syntheses and transformations catalyzed by sulfated zirconia. Chem Rev 109:2185
Huang YY, McCarthy TJ, Sachtler WMH (1996) Preparation and catalytic testing of mesoporous sulfated zirconium dioxide with partially tetragonal wall structure. Appl Catal A 148:135
Garg S, Soni K, Kumaran GM, Bal R, Gora-Marek K, Gupta JK, Sharma LD, Murali Dhar G (2009) Acidity and catalytic activities of sulfated zirconia inside SBA-15. Catal Today 141:125
Vijay S, Wolf EE (2004) A highly active and stable platinum-modified sulfated zirconia catalyst 1. Preparation and activity for n-pentane isomerization. Appl Catal A 264:117
Sert E, Atalay FS (2010) Kinetic study of the esterification of acetic acid with butanol catalyzed by sulfated zirconia. Reac Kinet Mech Cat 99:125
Chen L, Wang JX, Meng DW, Pan H, Su GH, Xiao Y, Yan ZF (2013) S2O8 2−/Al–O–MCM-41 catalysts for the esterification of acetic acid with n-butanol: influences of the preparation conditions on catalytic performances. Reac Kinet Mech Cat 109:575
Li BH, Gonzalez RD (1998) An in situ DRIFTS study of the deactivation and regeneration of sulfated zirconia. Catal Today 46:55
Fogash KB, Hong Z, Kobe JM, Dumesic JA (1998) Deactivation of sulfated-zirconia and H-mordenite catalysts during n-butane and isobutane isomerization. Appl Catal A 172:107
Wang B, Mao W, Ma HZ (2009) A mild simple method for liquid-phase selective catalytic oxidation of toluene with ozone over CeO2 promoted sulfated TiO2. Ind Eng Chem Res 48:440
Kruse N, Chenakin S (2011) XPS characterization of Au/TiO2 catalysts: binding energy assessment and irradiation effects. Appl Catal A 391:367
Joseph Antony Raj K, Viswanathan B (2009) Single-step synthesis and structural study of mesoporous sulfated titania nanopowder by a controlled hydrolysis process. ACS Appl Mater Interfaces 1:2462
Noda LK, Gonçalves NS, de Borba SM, Silveira JA (2007) Raman spectroscopy and thermal analysis of sulfated ZrO2 prepared by two synthesis routes. Vib Spectrosc 44:101
Fu BS, Gao LJ, Niu L, Wei RP, Xiao GM (2009) Biodiesel from waste cooking oil via heterogeneous superacid catalyst SO4 2−/ZrO2. Energy Fuels 23:569
Mao W, Ma HZ, Wang B (2010) Mild ring-opening coupling of liquid-phase cyclohexane to diesel components using sulfated metal oxides. J Hazard Mater 176:361
Li ZL, Wnetrzak R, Kwapinski W, Leahy JJ (2012) Synthesis and characterization of sulfated TiO2 nanorods and TiO2/TiO2 nanocomposites for the esterification of biobased organic acid. ACS Appl Mater Interfaces 4:4499
Föttinger K, Zorn K, Vinek H (2005) Influence of the sulfate content on the activity of Pt containing sulfated zirconia. Appl Catal A 284:69
Biró K, Figueras F, Békássy S (2002) Acylation of B15C5 crown ether by acetic anhydride in the absence of solvent, on sulfated zirconias prepared in different conditions. Appl Catal A 229:235
Smith YR, Joseph Antony Raj K, Subramanian V, Viswanathan B (2010) Sulfated Fe2O3–TiO2 synthesized from ilmenite ore: a visible light active photocatalyst. Colloids Surf A 367:140
Mishra MK, Tyagi B, Jasra RV (2003) Effect of synthetic parameters on structural, textural, and catalytic properties of nanocrystalline sulfated zirconia prepared by sol–gel technique. Ind Eng Chem Res 42:5727
Wang ZC, Hu XF, Käll Per-Olov, Helmersson Ulf (2001) High Li+-ion storage capacity and double-electrochromic behavior of sol–gel-derived iron oxide thin films with sulfate residues. Chem Mater 13:1976
Deutsch J, Trunschke A, Müller D, Quaschning V, Kemnitz E, Lieske H (2004) Acetylation and benzoylation of various aromatics on sulfated zirconia. J Mol Catal A 207:51
Klose BS, Jentoft FC, Schlögl R (2005) In situ diffuse-reflectance infrared spectroscopic investigation of promoted sulfated zirconia catalysts during n-butane isomerization. J Catal 233:68
MiLin Zhang, Jun Wang, ChangSong Mei, XiaoYan Jing, Xue Duan (2002) Synthesis, characterization and properties of magnetic nanometer-sized solid superacid. Chem J Chin Univ 23(7):1347–1351
Srinivasan R, Keogh RA, Davis BH (1995) Activation and characterization of Fe–Mn–SO4 2−/ZrO2 catalysts. Appl Catal A 130:135
Jiménez-Morales I, Santamaría-González J, Maireles-Torres P, Jiménez-López A (2011) Calcined zirconium sulfate supported on MCM-41 silica as acid catalyst for ethanolysis of sunflower oil. Appl Catal B 103:91
Rezaei M, Alavi SM, Sahebdelfar S, Yan ZF, Teunissen H, Jacobsen JH, Sehested J (2007) Synthesis of pure tetragonal zirconium oxide with high surface area. J Mater Sci 42:1228
Jong Rack S, Jong-Geol K, Tae-Dong K, Eun Hee P (2002) Characterization of titanium sulfate supported on zirconia and activity for acid catalysis. Langmuir 18:1666
Srinivasan R, Keogh RA, Davis BH (1995) Activation and characterization of Fe–Mn–SO4 2−/ZrO2 catalysts. App Catal A 130:135
Ghenciu A, Fărcaşiu D (1996) The mechanism of conversion of hydrocarbons on sulfated metal oxides. Part II. Reaction of benzene on sulfated zirconia. J Mol Catal A 109:273
Suwannakarn K, Lotero E, Goodwin JG Jr, Lu CQ (2008) Stability of sulfated zirconia and the nature of the catalytically active species in the transesterification of triglycerides. J Catal 255:279
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Shi, W., Li, J. A deactivation mechanism of sulfated titania in the esterification of acetic acid and n-butanol. Reac Kinet Mech Cat 111, 215–233 (2014). https://doi.org/10.1007/s11144-013-0628-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-013-0628-4