
Abstract
The room temperature study of polycrystalline iron, Fe0.99Si0.01 and Fe0.97Si0.03 alloys, was performed using the 57Fe transmission Mössbauer spectroscopy (TMS). The TMS measurements allow us to determine changes of chemical composition and content of iron oxides in the studied samples during exposure to air at 570, 870 and 1070 K. The results indicate that the chemical composition of samples changes significantly and it is strongly dependent on the temperature at which the studied samples are exposed to air. Moreover, it was shown that the addition of few atomic percent of Si impurities to iron leads to a significant slowdown of the internal oxidation process. The iron and silicon internal oxidation kinetics for Fe0.99Si0.01 and Fe0.97Si0.03 alloys were calculated using the Deal-Grove model. The activation energies E A for the internal oxidation of iron atoms in studied Fe–Si alloys were estimated: E A = 1.46 (3) eV for Fe0.99Si0.01 and E A = 1.62 (6) eV for Fe0.97Si0.03.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Corrosion of iron has been intensively studied for years due to the fact that iron as well as their alloys is still one of the most important construction materials for the modern industry, and their inevitable contact with oxygen (atmospheric for instance) often leads to changes in their mechanical, magnetic, structural and thermodynamic properties [1–6]. The oxidation behaviour of Fe–Si alloys at high temperatures in different atmospheres has been investigated by several researchers [7–11]. One of the main conclusions which comes from these works is that the addition of silicon has a significant effect on the oxidation resistance of iron in O2 or in air up to 1300 K. In this work, the polycrystalline iron and Fe0.99Si0.01 and Fe0.97Si0.03 alloys, which were exposed to air at 570 K, 870 K and 1070 K, were investigated using the 57Fe transmission Mössbauer spectroscopy (TMS). These measurements allowed the comparison of the oxidation process in three different materials under the same oxidation conditions. Base on TMS data, it is possible to calculate the iron and silicon internal oxidation kinetics and the activation energies for the internal oxidation of iron atoms in studied Fe–Si alloys.
Experimental Procedures
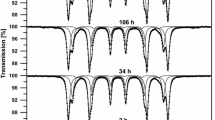
Samples of pure Fe (99.98 %) were prepared by iron rod cold-rolling to the final thickness of about 0.035 mm. At the same time, the samples of Fe0.99Si0.01 and Fe0.97Si0.03 alloys were prepared in an arc furnace. Appropriate amounts of the 99.98 % pure iron and 99.95 % pure silicon were melted into a water-cooled copper crucible in argon atmosphere and then quickly cooled to room temperature. Solidified alloys were remelted twice to ensure homogeneity. The weight losses during the melting process were below 0.2 % of the original weight, so the compositions of the obtained ingots were close to the nominal ones. Resulting ingots were cold-rolled to the final thickness of about 0.035 mm. In the next step, iron and Fe0.99Si0.01 and Fe0.97Si0.03 foils were annealed in the vacuum at 1270 K for 2 h. The base pressure during the annealing procedure was lower than 10−6 mbar. To obtain homogeneous and defect-free samples [12], after annealing process, the foils were slowly cooled to room temperature for 6 h. The annealed samples were exposed to air at 570, 870 and 1070 K for several hours. Each step of the sample processing was followed by TMS measurements, which were performed at room temperature by means of a constant-acceleration POLON spectrometer of standard design, using a 3.7 GBq 57Co-in-Rh standard source with a full width at half maximum (FWHM) of 0.22 mm/s. Each measured TMS spectrum was analysed in terms of a sum of different number of single lines and six-line patterns, corresponding to various isomer shifts IS, quadrupole splitting QS and hyperfine fields B at 57Fe nuclei generated by different chemical states of 57Fe Mössbauer probes. The fitting procedure was done under the thin absorber approximation, and it was assumed that the shape of each line could be fitted by Voigt function (the convolution on Lorentzian and Gaussian curves). For each six-line pattern, the two-line area ratio I 16/I 34 is constant and equal to 3/1. The ratio I 25/I 34 and three linewidths Γ 16, Γ 25 and Γ 34 are free parameters. Some of the obtained fits are shown in Figs. 1, 2, 3 and 4.

The room-temperature TMS spectra of the Fe sample exposed to air at 570 and 1070 K for several hours h, fitted with a sum of various numbers of six-line patterns corresponding to different chemical states of iron atoms

The room-temperature TMS spectra of the Fe sample exposed to air at 870 K for several hours h, fitted with a sum of various numbers of single lines and six-line patterns corresponding to different chemical states of iron atoms

The room-temperature TMS spectra of the Fe0.99Si0.01 sample exposed to air at 570, 870 and 1070 K for several hours h, fitted with a sum of various numbers of six-line patterns corresponding to different chemical states of iron atoms

The room-temperature TMS spectra of the Fe0.97Si0.03 sample exposed to air at 570, 870 and 1070 K for several hours h, fitted with a sum of various numbers of six-line patterns corresponding to different chemical states of iron atoms
Results and Discussion
Results for Pure Iron
As one can see in Figs. 1 and 2, the measured TMS spectra for pure iron samples change significantly with exposure to air. For samples exposed to air at 570 K for 2 and 16 h, each spectrum can be described with one six-line pattern only having the hyperfine parameters similar to those for metallic iron with b.c.c. structure (α-Fe). At longer exposure times (40 and 100 h), the additional magnetically splitted components were observed. These subspectra were accounted for by adding to the model of the spectrum three sextets. The estimated, during fitting procedure, hyperfine parameters for the samples mentioned above correspond to α-iron and two different iron oxides. One sextet with B ≈ 51.7 T, IS ≈ 0.37 mm/s and QS ≈ −0.2 mm/s could be a result of the α-Fe2O3 presence in the sample [13]. The last two sextets with B ≈ 48.9 and 46.1 T, IS ≈ 0.24 mm/s and 0.68 mm/s and QS < 0.1 mm/s correspond to the two different sites of Fe atoms in Fe3O4 compound [14]. At higher temperatures (870 and 1070 K), the obtained spectra consist of a various number of sextets which correspond to the existence of α-Fe and α-Fe2O3 and Fe3O4 compounds. Here it is worth noting that changes in relative intensities of two Fe3O4 sextets were observed. This fact suggests that stoichiometric and non-stoichiometric Fe3O4 were formed [15, 16]. Moreover, in spectra obtained for iron exposed to air at 870 K for 2 and 4 h, the additional non-magnetic components were observed. Two singlets with IS ≈ 1.05 and 0.55 mm/s could be assigned to stoichiometric and non-stoichiometric FeO [17]. At the same time, in all measured TMS spectra, there are no other components which could be connected with FeOOH or iron carbides [18, 19]. The above findings are important evidence that in polycrystalline Fe exposed to air at elevated temperatures, only three types of compounds are formed: FeO, α-Fe2O3 and Fe3O4. Moreover, as a result of the analysis, the relative areas I(x) of components for each spectrum were determined. Assuming that the relative ratio of the Lamb–Mössbauer factors f α-Fe: f FeO: f α-Fe2O3: f Fe3O4 is equal to 1: 1: 1.08: 1.05 [1], the fraction c(x) of absorbing atoms corresponding to each component can be easily calculated using the I(x) values as follows:
The computed c(x) values were used to find parameters c(Fe), c(FeO), c(Fe2O3) and c(Fe3O4) which are the total intensities of spectrum components related to the existence of 57Fe nuclear probes in α-Fe, FeO, α-Fe2O3 and Fe3O4, respectively. These parameters are listed in Table 1. Finally, to obtain the chemical composition of studied samples, it is necessary to taking into account that the relative quantities of iron atoms in observed compounds are different and equal to 1: 1: 2: 3 for α-Fe, FeO, α-Fe2O3 and Fe3O4, respectively. The chemical composition estimated under the above-mentioned assumptions is shown in Fig. 5. The data show that exposure of polycrystalline iron to air at elevated temperatures results in the formation of three different iron oxides, which are quite similar to those obtained in previous works concerning the oxidation of iron in air [5].

The chemical composition of Fe, Fe0.99Si0.01 and Fe0.97Si0.03 samples exposed to air at 570, 870 and 1070 K as a function of exposure time. The standard uncertainties for the presented quantities do not exceed 3 %
Results for Fe0.99Si0.01 and Fe0.97Si0.03 Alloys
The TMS spectra analysis revealed the existence of components which could be assigned to single-phase Fe–Si alloy with b.c.c. structure [20], α-Fe2O3 and stoichiometric and non-stoichiometric Fe3O4 compounds with hyperfine parameters similar to those obtained for pure iron spectra. In the next step, based on the procedure proposed in Sect. 3.1, the total intensities of TMS spectrum components c(alloy), c(Fe2O3) and c(Fe3O4) and the chemical composition for the studied alloys were calculated (Tables 2, 3 and Fig. 5).
As one can expect, a few atomic percent of Si impurities dissolved in the iron matrix have a significant influence on oxidation process of iron atoms in studied materials. Firstly, in contrast to pure iron oxidation where exposure to air at elevated temperatures results in the formation of three different iron oxides, in Fe–Si alloys the dominant type of iron oxide is Fe2O3. FeO does not form at all and the intensity of Fe3O4 component in measured spectra does not exceed 5 %. Secondly, the comparison of data, as presented in Tables 1, 2 and 3 leads to a conclusion that the internal oxidation of iron atoms slows with increasing Si content.
Oxidation Kinetics and Activation Energy
To analyse thermal oxidation of iron in Fe–Si alloys, the Deal–Grove model was applied [21]. This model requires that the oxidation of Fe follows a mechanism whereby a thin layer of oxide is formed by place exchange after adsorption has occurred, with further oxidation occurring due to an electric field mechanism. The electric field is formed by electrons tunnelling from the metal to the adsorbed oxygen, and is able to support ion transport through the place exchange and formed thin oxide layer enabling the oxide layer to increase in thickness. Due to the fact that TMS technique is not sensitive to thin oxide layer (below 100 nm), the parabolic regime (for thick films) was applied:
where FeOX is a fraction of oxidised iron atoms in studied alloys at time t, and k denotes the quadratic reaction rate constant which is expressed as follows:
where E A is activation energy for iron oxidation, kB stands for Boltzmann constant, k 0 is pre-exponential factor and T denotes temperature. The parabolic iron oxidation kinetic curves obtained for the Fe0.99Si0.01 and Fe0.97Si0.03 alloys exposed to air at 870 K and 1070 K, respectively, are shown in Fig. 6. The knowledge of the reaction rate k for two different temperatures, T 1 = 870 K and T 2 = 1070 K, combined with Eq. (3) enabled the determination of the activation energy E A for the oxidation of iron atoms in the temperature interval of 870–1070 K, using the following equation:

This equation is valid assuming the pre-exponent factor k 0 in Eq. (3) is temperature independent. The estimated quadratic reaction rate constants and activation energies of iron oxidation process for studied Fe–Si alloys were presented in Table 4. The obtained E A values for Fe–Si alloys are much higher compared to the activation energy for the oxidation reaction of pure Fe, which was determined to be 32 (6) kJ/mol [22]. This is in accord with the observation that the iron oxidation process in Fe–Si alloys was much slower compared to the one in pure iron sample. Moreover, the higher EA value for Fe0.97Si0.03 alloy also agrees with the previous conclusion that the internal oxidation of iron atoms slows with increasing Si content.
Indirect Observation of Si Oxidation Process
The Si oxidation process cannot be directly observed using the 57Fe Mössbauer spectroscopy. This is because this technique is not sensitive to presence of Si oxides in measured samples. Taking this into account, the indirect method for the above-mentioned observation is proposed. The analysis of TMS spectra, obtained for the Fe0.99Si0.01 and Fe0.97Si0.03 samples, reveals the existence of two components (sextets) which could be assigned to single-phase Fe–Si alloy with b.c.c. structure. The first sextet (the iron component), with B ≈ 32.9 T and IS ≈ 0.00 mm/s, corresponds to the presence of Fe atoms without any Si atoms in their first coordination shell. The second one (the satellite component), with B ≈ 30.5 T and IS ≈ 0.05 mm/s, corresponds to Fe atoms with one Si atom in their first coordination shell [20]. Assuming that the single-phase Fe–Si alloy with b.c.c. structure is quite homogenous and in annealed alloys, before exposure to air, all Si atoms are dissolved in iron matrix, it is possible to calculate atomic concentration of Si atoms cSi which are not oxidised and located in the single-phase Fe–Si alloy as follows:
where I(iron) and I(satellite) denote the intensities of the iron and the satellite components, respectively. Z is the first coordination number which for b.c.c. structure is equal to 8. The obtained values presented in Table 5 show a slight decrease of Si concentration in single-phase Fe–Si alloy during exposure to air. Having in mind that the 57Fe Mössbauer spectroscopy is not sensitive to the presence of Si oxides, this result could be explained by faster oxidation of Si atoms in studied Fe–Si alloys. In other words, the oxygen atoms which diffuse into Fe–Si matrix at elevated temperatures prefer to form silicon oxides, and after their formation these compound cannot be directly observed using 57Fe Mössbauer spectroscopy. This observation combined with the results obtained in Sects. 3.2 and 3.3 could give an answer as to why the addition of a few atomic percent of Si impurities to iron leads to a significant slowdown of the internal oxidation process. The most plausible explanation of this phenomenon is connected with the surface segregation process of Si atoms in Fe–Si alloys. An increased concentration of Si atoms on the surface of grains in polycrystalline Fe–Si alloy could form a stable silicon oxide passivation layer which significantly slows down the internal oxidation process. The segregation of Si on Fe–Si alloy surface was reported in several papers [23–25]. Unfortunately, due to the fact that these studies were performed on single crystal, it is necessary to confirm the segregation process in polycrystalline Fe–Si alloys.
In the next step, using Eq. (1), the fraction of iron atoms with one Si impurity in their first coordination shell c(satellite) was computed. These values give information about the fraction of Si atoms which are not oxidised (dissolved in iron matrix), and 1-c(satellite) values denote the fraction of oxidised silicon atoms SiOX. The obtained SiOX values for Fe0.99Si0.01 and Fe0.97Si0.03 samples as a function of exposure time are shown in Fig. 6. These results were used to estimate the parabolic iron oxidation kinetic curves for studied alloys which were exposed to air at 870 K. The computed quadratic reaction rate constants are k = 2.17 (5) 10−3 h−1 for Fe0.99Si0.01 and k = 1.45 (3) 10−3 h−1 for Fe0.97Si0.03. During analysis of the silicon oxidation kinetic curves, it was found that parabolic regime is not valid for samples oxidised at 1070 K. In that case, the logarithmic oxidation kinetics was much more plausible:
where k denotes a logarithmic reaction rate constant and C is a constant. The computed logarithmic rate constants are k = 2.2(9) for Fe0.99Si0.01 and k = 0.39 (3) for Fe0.97Si0.03. This result suggests that in diluted Fe–Si alloys, the silicon oxidation mechanism could be temperature-dependent.
Conclusions
The results reported in this paper can be concluded as follows:
-
1.
The TMS measurements of pure iron and Fe0.99Si0.01 and Fe0.97Si0.03 alloys allowed us to determine changes of chemical composition and the presence of newly formed compounds in studied materials during exposure to air at 570, 870 and 1070 K.
-
2.
As it was shown, a few atomic percent of Si impurities dissolved in the iron matrix have a significant influence on oxidation process of iron atoms in studied materials. In contrast to pure iron oxidation where exposure to air at elevated temperatures results in the formation of three different iron oxides, in Fe–Si alloys the dominant type of iron oxide is Fe2O3. FeO does not form at all and the intensity of Fe3O4 component in measured spectra does not exceed 5 %.
-
3.
The comparison of data obtained for pure iron and Fe0.99Si0.01 and Fe0.97Si0.03 leads to a conclusion that the internal oxidation of iron atoms slows with increasing of Si content.
-
4.
The estimation of the reaction rate k for two different temperatures, T 1 = 870 K and T 2 = 1070 K, enabled the determination of the activation energy E A for the internal oxidation of iron atoms in studied Fe–Si alloys: E A = 1.46 (3) eV for Fe0.99Si0.01 and E A = 1.62 (6) eV for Fe0.97Si0.03.
-
5.
The indirect observation of Si oxidation process in studied materials was proposed. Using this, the silicon oxidation kinetics for Fe0.99Si0.01 and Fe0.97Si0.03 alloys were presented. The obtained kinetics suggest that in diluted Fe–Si alloys, the silicon oxidation mechanism could be temperature-dependent.
References
D. A. Channing and M. J. Graham, Corrosion Science 12, 1972 (271–289).
D. A. Channing, S. M. Dickerson and M. J. Graham, Corrosion Science 13, 1973 (933–938).
A. P. Grosvenor, B. A. Kobe and N. S. McIntyre, Surface Science 565, 2004 (151–162).
C. Han, C. Zhang, X. Liu, H. Huang, S. Zhuang, P. Han and X. Wu, J Mol Model 21, 2015 (181).
K. Idczak, R. Idczak and R. Konieczny, Physica B 491, 2016 (37–45).
K. Freindl, T. Ossowski, M. Zając, N. Spiridis, D. Wilgocka-Ślęzak, E. Madej, T. Giela, A. Kiejna and J. Korecki, The Journal Of Physical Chemistry C 120, 2016 (3807–3813).
A. R. Lashin, O. Schneeweiss and Y. Houbaert, Corrosion Science 50, 2008 (2580–2587).
M. A. A. Motin, J. Zhang, P. R. Munroe and D. J. Young, Corrosion Science 52, 2010 (3280–3286).
H. Li, J. Zhang and D. J. Young, Corrosion Science 54, 2012 (127–138).
A. R. Lashin, Journal of Alloys and Compounds 567, 2013 (54–58).
L. L. Liu, Q. Q. Guo, S. Liu, C. S. Ni and Y. Niu, Corrosion Science 98, 2015 (507–515).
R. Idczak, R. Konieczny and J. Chojcan, Journal of Applied Physics 115, 2014 (103513).
R. Zboril, M. Mashlan and D. Petridis, Chem. Mater. 24, 2002 (969–982).
B. J. Evans and S. S. Hafner, Journal of Applied Physics 40, 1969 (1411–1413).
J. M. Daniels and A. Rosencwaig, Journal of Physics and Chemistry of Solids 30, 1969 (1561–1571).
F. C. Voogt, T. Hibma, G. L. Zhang, M. Hoefman and L. Niesen, Surface Science 331–333, 1995 (1508–1514).
L. F. Checherskaya, V. P. Romanov and P. A. Tatsienko, Physica Status Solidi 19, 1973 (K177–K182).
F. Van der Woude and A. J. Dekker, Physica Status Solidi 13, 1966 (181–193).
B. Kniep, A. Constantinescu, D. Niemeier and K. D. Becke, Zeitschrift für Anorganische und Allgemeine Chemie 629, 2003 (1795–1804).
R. Idczak, R. Konieczny and J. Chojcan, Acta Physica Polonica A 129, 2016 (367).
B. E. Deal and A. S. Grove, Journal of Applied Physics 36, 1965 (3770–3778).
A. P. Grosvenor, B. A. Kobe and N. S. McIntyre, Surface Science 574, 2005 (317–321).
J. Du Plessis and P. E. Viljoen, Surface Science 131, 1983 (321–327).
H. De Rugy and H. Viefhaus, Surface Science 173, 1986 (418–438).
P. Lejček, V. Paidar, S. A. Firstov, Yu N Ivashchenko and A. V. Krajnikov, Scripta Metallurgica 23, 1989 (2147–2152).
Acknowledgments
This work was supported by the Polish Ministry of Science and Higher Education under the “Iuventus Plus” programme in the years 2015–2017, Project Number IP2014 015573.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Idczak, R. Comparison of the Oxidation Process in Pure Iron and Diluted Fe–Si Alloys: A Transmission Mössbauer Spectroscopy Study. Oxid Met 87, 75–88 (2017). https://doi.org/10.1007/s11085-016-9658-4
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-016-9658-4