
Abstract
Arginine side-chains are often key for enzyme catalysis, protein–ligand and protein–protein interactions. The importance of arginine stems from the ability of the terminal guanidinium group to form many key interactions, such as hydrogen bonds and salt bridges, as well as its perpetual positive charge. We present here an arginine 13Cζ-detected NMR experiment in which a double-quantum coherence involving the two 15Nη nuclei is evolved during the indirect chemical shift evolution period. As the precession frequency of the double-quantum coherence is insensitive to exchange of the two 15Nη; this new approach is shown to eliminate the previously deleterious line broadenings of 15Nη resonances caused by the partially restricted rotation about the Cζ–Nε bond. Consequently, sharp and well-resolved 15Nη resonances can be observed. The utility of the presented method is demonstrated on the L99A mutant of the 19 kDa protein T4 lysozyme, where the measurement of small chemical shift perturbations, such as one-bond deuterium isotope shifts, of the arginine amine 15Nη nuclei becomes possible using the double-quantum experiment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Of the twenty standard proteinogenic amino acids that are found in nature few are as important as arginine. This essential amino acid is found at many protein interaction surfaces (Crowley and Golovin 2005; Rohs et al. 2010) and has been identified in numerous enzymatic active sites (Casey et al. 2014; Friedt et al. 2014; Zeymer et al. 2016) and substrate-binding pockets (Goldschen-Ohm et al. 2011; Gargaro et al. 1996). The importance of the arginine side-chain for protein functions stems from its terminal guanidinium group that is the most basic moiety of the three positively charged amino acids. The high pKa (~ 14) (Fitch et al. 2015) of the guanidinium group renders the arginine side-chain positively charged at all physiologically relevant pHs (Harms et al. 2011) and thereby provides nature with a reliable means of placing a positive charge at virtually any point within a protein structure. An important feature of the arginine guanidinium group is that the positive charge is delocalised and the arginine side-chain is therefore capable of an impressive range of hydrogen bonds and ionic interactions. These interactions include bidentate salt bridges with carboxylates and phosphates, cation–π interactions with aromatic rings and hydrogen bonding with the side-chain groups of aspartic and glutamic acids as well as with backbone carbonyl oxygens (Borders et al. 2008; Nieto et al. 1997).
Solution state nuclear magnetic resonance (NMR) spectroscopy is well positioned to characterise the interactions formed by arginine side-chains due to the atomic resolution the technique can provide. Protein side-chains and their interactions are inherently dynamic and whilst NMR spectroscopy is considered suitable for the study of such systems, it is often the case that dynamic processes obscure the underlying information (Kleckner and Foster 2011). For example, the NMR signals of interest are often severely broadened when states are interconverting with rates that are comparable to the difference in chemical shift between the exchanging states. A specific example is encountered for the terminal –NηH2 amines of the arginine side-chain (Yamazaki et al. 1995; Yoshimura et al. 2017; Henry and Sykes 1995). The partial double-bond character of the Cζ–Nε bond in the guanidinium group causes a decrease in the rotational frequency about this bond, which leads to significant exchange-broadening of the NMR signals associated with the 15Nη nuclei (Nieto et al. 1997). Moreover, the –NηH2 amine protons often exchange rapidly with the bulk solvent at physiological pH (Henry and Sykes 1995). Thus, the interconversion of the two 15Nη nuclei combined with the exchange of the amino protons with the bulk solvent often result in 1Hη–15Nη NMR correlations that are so broad that very limited information can be gleaned.
The line broadening induced by chemical exchange is reduced when the difference in chemical shift between the exchanging sites, Δω, is reduced (McConnell 1958; Hansen and Led 2003). Many NMR experiments have been developed over the last few decades to manipulate the effective line broadening of chemically exchanging nuclei in a quantitative manner by reducing the effective chemical shift difference (Palmer 2014; Korzhnev et al. 2008; Farber and Mittermaier 2015; Hansen et al. 2008; Zhuravleva et al. 2008; Carr and Purcell 1954; Palmer and Massi 2006). Also, different line broadenings are generally observed for zero-, single-, double- and triple-quantum coherences of a spin system, because these coherences have different precession frequencies and therefore different Δω (Yuwen et al. 2016; Orekhov et al. 2004; Pervushin et al. 1999). A major motivation of the presented work is to characterise the interactions formed by arginine side-chains by generally allowing for an observation of the 15Nη chemical shifts. Rather than quantifying the chemical exchange, as has been done previously (Henry and Sykes 1995; Gerecht et al. 2017), our focus here is to eliminate the effects of chemical exchange in order to obtain NMR correlation spectra of arginine 15Nη nuclei.
Below we describe an experiment based on 13C detection and the evolution of a double-quantum 15Nη coherence that overcomes the hurdles associated with the line broadening caused by the rotation about the Cζ–Nε bond and the exchange of the 1Hη protons with the bulk solvent. We also present an application to the 19 kDa protein T4 lysozyme, where the double-quantum experiment allowed the observation of 13Cζ–15Nη(DQ) coherences for all 13 arginine side-chains. The increased resolution provided by this experiment allows the measurement of small chemical shift perturbations of the arginine terminal amines, demonstrated here with a determination of the deuterium isotope shifts (Hansen 2000) which are expected to inform on salt-bridges and hydrogen bonding in biomolecules (Tomlinson et al. 2009; Williamson et al. 2013).
Materials and methods
Protein preparation
Uniformly labelled [13C, 15N]-T4 Lysozyme L99A was overexpressed and purified from Escherichia coli. BL21 (DE3) cells were grown at 37 °C in M9 minimal media supplemented with 1 g/L 15NH4Cl and 3 g/L [13C6]-glucose as the sole nitrogen and carbon sources. The expression and purification was performed as described previously (Vallurupalli et al. 2009) with minor modifications. Cells were induced with 1 mM IPTG at OD600 of ~ 1.0 before protein expression was allowed to proceed at 16 °C. Cells were harvested by centrifugation after 16 h. The resulting cell pellet was re-suspended and lysed by sonication before purification by ion-exchange (IEX) and gel-filtration (GF) chromatography. ESI-MS confirmed the sample was isotopically enriched to a level beyond 99.7%. The sample was exchanged into NMR buffer (50 mM sodium phosphate, 25 mM NaCl, 2 mM EDTA, 2 mM NaN3, pH 5.5, 1% D2O) and concentrated to ~ 2 mM. Samples for the measurement of deuterium isotope shifts were prepared by spiking the above samples with additional D2O to a final concentration of 10 vol.%.
NMR spectroscopy
All NMR experiments were carried out at 25 °C on Bruker Avance III(HD) spectrometers with 1H operating frequencies of 500, 700 and 800 MHz and equipped with helium-cooled TCI (700, 800 MHz) or nitrogen-cooled Prodigy (500 MHz) inverse cryoprobes. The 1H–15N HSQC spectrum (Fig. 1b) was acquired as a 1024 × 512 complex matrix with spectral widths of 16 ppm (1H) and 80 ppm (15N). Adiabatic 13C decoupling was applied during t 1 and suppression of the H2O resonance was achieved using a water-selective flip-back pulse (Andersson et al. 1998) immediately after the first INEPT element. Four scans were collected for each t 1 increment with a recycle delay of 1 s resulting in a total experiment time of 1 h 20 min. Both the 13Cζ–15Nη HSQC (Fig. 1c) and HDQC (Fig. 2b) spectra were acquired as 512 × 48 complex matrices with spectral widths of 11 ppm (13C) and 12 ppm (15N). 256 scans were collected for each t 1 increment with a recycle delay of 3 s resulting in a total acquisition time for each experiment of 23 h. The 13Cζ–15Nε HSQC (Fig. 4a) spectrum was acquired as a 512 × 48 complex matrix with spectral widths of 11 ppm (13C) and 10 ppm (15N). 128 scans were collected for each t 1 increment with a recycle delay of 3 s resulting in a total experiment time of 12 h. The 3D 13Cζ–15Nε–15Nη experiment (Fig. 4b) was acquired as a 1024 × 32 × 32 complex cube with spectral widths of 40 ppm (13C) and 10 ppm (15Nε and 15Nη). 16 scans were collected for each t 1/t 2 increment with a recycle delay of 3.5 s resulting in a total experiment time of 71 h. The spectra in Fig. 5 were collected as above using pulse sequences modified to include 2H decoupling in the indirect dimension. The 2H signal of the lock solvent was preserved by flanking the decoupling sequence with a pair of opposite phase 2H pulses (y,− y), both of which were orthogonal to the decoupling field (x). NMR data was processed using NMRPipe (Delaglio et al. 1995) and subsequently analysed using the Analysis module of the CCPNMR package (Vranken et al. 2005).

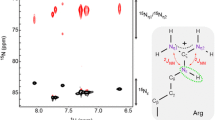
a Chemical structure of the arginine side-chain. The arrow highlights the Cζ–Nε bond, about which rotation is partially restricted. b 1H–15N HSQC spectrum with focus on the arginine side-chains of T4L99A recorded at 11.7 T. Whilst the majority of the 15Nε resonances are detectable, the peaks associated with the amine 15Nη are broad and overlap significantly. c 13Cζ–15Nη HSQC spectrum of T4L99A recorded at 16.4 T. Arginine residues that exhibit slow rotation about the Cζ–Nε bond give rise to two sharp peaks (R52, R95, R145 and R148). For many of the arginine side-chains, the rotational frequency about the Cζ–Nε bond approaches the chemical shift difference between the two 15Nη causing a substantial line broadening of the NMR signals

a Pulse sequence for obtaining 13Cζ–15Nη double-quantum correlation (HDQC) spectra of arginine side-chains in proteins. The carrier positions are 13C: 156 ppm, 15N: 71 ppm (78 ppm decoupling) and 1H: 7 ppm. Narrow and wide bars represent 90° and 180° pulses, respectively, and are applied at maximum power. The delay Δ is 1/(4J CN) = 12.5 ms. Shaped pulses are represented by bell shapes with letters indicating the shape of the pulse [E: E-BURP-2, R: RE-BURP (Geen and Freeman 1991)] and are applied with durations of 1.5 ms (E) and 4.5 ms (R) at 16.4 T. Pulses are applied with x phase unless stated otherwise. The phase cycle used is φ1: x, − x, φ2: 2(x), 2(y), 2(− x), 2(− y), φrec: x, 2(− x), x. Decoupling sequences are represented by striped boxes indicating the type of decoupling: WALTZ64 (Shaka et al. 1983; Zhou et al. 2007) (4 kHz), GARP4 (Shaka et al. 1985) (0.7 kHz). Gradient pulses of 1 ms are represented by black rectangles and applied with strengths of g1: 19.8 G/cm, g2: 5.9 G/cm, g3: 12.3 G/cm, g4: 16.6 G/cm, g5: 7.0 G/cm. b 13Cζ–15Nη HDQC spectrum of T4L99A recorded at 16.4 T. The indirect 15N chemical shift is encoded by the double-quantum components \(4C_{z}^{\zeta }N_{+}^{{{{{\eta}1}}}}N_{+}^{{{{{\eta}2}}}}\) and \(4C_{z}^{\zeta }N_{ - }^{{{{{\eta}1}}}}N_{ - }^{{{{{\eta}2}}}}\) and processed such that the cross peaks appear at the average chemical shift of the two contributing 15Nη frequencies
Results and discussion
The conventional approach to probe the chemical shift of an amine or an amide 15N within a biomolecule involves the collection of 1H–15N correlation experiments, which make use of the 1 J HN scalar coupling between the 15N nucleus and the directly bound proton (Bodenhausen and Ruben 1980). This inverse-type experiment employs 1H detection, which affords high sensitivity as well as an additional chemical shift dimension that reduces spectral crowding and subsequent signal overlap. However, for arginine side-chains, two chemical exchange processes lead to severe line broadenings of the 1H–15N correlations, in particular at physiological pH and at ambient temperature. Firstly, the directly bound 1Hε and 1Hη protons of the guanidinium group undergo a rapid chemical exchange with the solvent, which lead to a line broadening in the 1H dimension and an attenuation of the signals in the NMR experiment. In samples prepared at neutral and high pH, the line broadening and loss of signal intensity is so severe that it often prevents a detection of the signal, unless the proton in question is involved in a strong hydrogen bond (Zeymer et al. 2016). In samples prepared at lower pH (< 6.5), the exchange is sufficiently slowed so that 1Hε–15Nε HSQC correlation spectra can be obtained (Morgan et al. 1999; Trbovic et al. 2009; Iwahara and Clore 2006). For example, for the 19 kDa L99A mutant of T4 Lysozyme (T4L99A) at pH 5.5 and at 298 K all of the 13 arginine side-chains can be observed in 1Hε–15Nε HSQC correlation spectra (Fig. 1b, ϖ1(15N) ~ 85 ppm). Nonetheless, even with the favourable 1H exchange conditions, the 1Hη–15Nη correlations largely remain significantly broadened and of low intensity (Fig. 1b, ϖ1(15N) ~ 71 ppm). This is a manifestation of a second exchange process resulting from the restricted rotation about the Cζ–Nε bond.
With adaptations to a recently published NMR experiment (Gerecht et al. 2017; Werbeck et al. 2013), carbon-detected 13Cζ–15Nη HSQC can be employed to overcome the line broadenings associated with the exchange of the 1Hη protons with bulk solvent (Yoshimura et al. 2017). In the previous 13Cζ–15Nε HSQC experiment (Werbeck et al. 2013), 13Cζ equilibrium magnetisation is selectively excited using an Eburp-2 pulse (Geen and Freeman 1991) and transferred to the two-spin order longitudinal spin density matrix element, \(2C_{z}^{\zeta }N_{z}^{\varepsilon }\), using an INEPT (Morris and Freeman 1979) sequence of length 1/(2J CN) (approx. 25 ms) with selective inversion of 15Nε. Anti-phase transverse \(2C_{z}^{\zeta }N_{{{{x,y}}}}^{\varepsilon }\) magnetisation is subsequently evolved and transferred back to transverse \(C_{x}^{\zeta }\) for detection. Two changes were made in order to obtain the 13Cζ–15Nη HSQC spectra: (1) change the inversion pulse in the INEPT blocks to be selective for 15Nη and (2) change the length of the INEPT to 1/(4J CN). Thus, after the first INEPT, the resulting magnetisation of interest is proportional to \(2C_{z}^{\zeta }N_{z}^{{{{{\eta}1}}}}+2C_{z}^{\zeta }N_{z}^{{{{{\eta}2}}}}\). As the 13Cζ–15Nη HSQC experiment relies on the chemical shift evolution of 13Cζ and 15Nη as well as the 1 J CN scalar coupling between them, the exchange of 1Hη with the bulk solvent does not affect the obtained spectrum. It should be noted that because the experiment is based on 13C-excitation and 13C-detection there is an intrinsic sensitivity penalty owing to the lower gyromagnetic ratio of 13C compared to 1H. However, in the case of 15Nη, this is outweighed by the elimination of the exchange with the bulk solvent, thus resulting in a clear improvement over the 1H–15N spectrum (Fig. 1c). In spite of that many of the 13Cζ–15Nη correlations are substantially broadened in the 15N dimension and consequently overlapped.
Owing in part to the ability to form salt-bridges with negatively charged side-chains such as aspartic and glutamic acids, a range of Cζ–Nε bond rotational rates are typically observed for arginine residues in proteins (Nieto et al. 1997; Gerecht et al. 2017). How these rates affect the NMR spectra depends on both the rate of rotational exchange and the absolute chemical shift difference between the two exchanging sites; in this case 15Nη1 and 15Nη2. In the slow-exchange regime (k ex \( \ll \; {\big |}\)ω(15Nη1) − ω(15Nη2)\( {\big |}\)), and for arginine side-chains involved in strong hydrogen bonds or salt bridges, a sharp signal is observed for each of the two 15Nη (e.g. R52, R148, R95 in T4L99A), whilst in the fast-exchange regime (k ex \( \gg \; {\big |}\)ω(15Nη1) − ω(15Nη2)\( {\big |}\)) a single sharp signal is observed at the average chemical shift (e.g. free arginine at elevated temperature). In the intermediate exchange regime, as the rate of exchange approaches the chemical shift difference (k ex ≈ \( {\big |}\)ω(15Nη1) − ω(15Nη2)\( {\big |}\)), the signals coalesce and result in a broad resonance that is often at the limit of detection. This exchange-broadening is apparent for R14 and several signals around 157.0 ppm (13C) in T4L99A (Fig. 1c). An arginine single-quantum 13Cζ–15Nε/η experiment has been published recently (Yoshimura et al. 2017), where the effect of chemical exchange is minimised by a combination of cross-polarisation and 13C detection. Although the cross-polarisation quenches the exchange-broadening caused by the exchange with the solvent and the rotation about the Cζ–Nε bond during transfer steps, the resulting single-quantum 13Cζ–15Nη spectrum still suffers from significant overlap. As described below a double-quantum coherence can be created that is insensitive to the rotation about the Cζ–Nε bond and thus unaffected by the line broadening resulting from this exchange process.
A 15Nη double-quantum experiment
The basic crux of our approach to characterise 15Nη chemical shifts is to create a double-quantum coherence that is insensitive to the rotation about the Cζ–Nε bond. Firstly, it is noted that the nature of the exchange between 15Nη1 and 15Nη2 means that the population of the two exchanging sites is identical. Secondly, the double-quantum coherences \(4C_{z}^{\zeta }N_{+}^{{{{{\eta}1}}}}N_{+}^{{{{{\eta}2}}}}\) and \(4C_{z}^{\zeta }N_{ - }^{{{{{\eta}1}}}}N_{ - }^{{{{{\eta}2}}}}\) evolve under the free precession Hamiltonian with frequencies of ±(ω(15Nη1) + ω(15Nη2)), respectively, where \(C_{i}^{\zeta }\), \(N_{i}^{{\eta}1}\) and \(N_{i}^{{{{{\eta}2}}}}\) denote 13Cζ, 15Nη1 and 15Nη2 standard spin density operator matrix elements, respectively (Sørensen et al. 1984). A chemical exchange that interchanges 15Nη1 and 15Nη2, therefore leaves the double-quantum precession frequencies unchanged. Consequently, the rotation about the Cζ–Nε bond does not affect the evolution of the double-quantum coherences \(4C_{z}^{\zeta }N_{+}^{{{{{\eta}1}}}}N_{+}^{{{{{\eta}2}}}}\) and \(4C_{z}^{\zeta }N_{ - }^{{{{{\eta}1}}}}N_{ - }^{{{{{\eta}2}}}}\), and thus no exchange-broadening arising from this rotation is expected to be observed.
The pulse sequence that was developed here to evolve the double-quantum coherences \(4C_{z}^{\zeta }N_{+}^{{{{{\eta}1}}}}N_{+}^{{{{{\eta}2}}}}\) and \(4C_{z}^{\zeta }N_{ - }^{{{{{\eta}1}}}}N_{ - }^{{{{{\eta}2}}}}\) of arginine side-chains is shown in Fig. 2a. Briefly, magnetisation is selectively transferred from 13Cζ to 15Nη via the one bond scalar coupling 1 J CN using an INEPT element that incorporates selective 13Cζ excitation and a selective 15Nη inversion pulse. A density element proportional to the three-spin order longitudinal density element \(4C_{z}^{\zeta }N_{z}^{{{{{\eta}1}}}}N_{z}^{{{{{\eta}2}}}}\) is obtained at point a by allowing \(C_{y}^{\zeta }\) to evolve for 2Δ = 1/(2J CN) under the scalar coupling Hamiltonian. Subsequently a \({\text{90}}_{{\upphi {{2~=~{x}}}}}^{\circ }\) 15N pulse generates the multiple-quantum coherence \(4C_{z}^{\zeta }N_{y}^{{{{{\eta}1}}}}N_{y}^{{{{{\eta}2}}}}=Z{Q_x} - D{Q_x}\), where:
The double-quantum component is selected by phase-cycling the 15N excitation pulse (x,y) with a concomitant inversion of the receiver phase. Subsequently the selected double-quantum coherence, \(D{Q_{{x}}}=0.5{\text{(4}}C_{z}^{\zeta }N_{x}^{{{{{\eta}1}}}}N_{x}^{{{{{\eta}2}}}} - {\text{4}}C_{z}^{\zeta }N_{y}^{{{{{\eta}1}}}}N_{y}^{{{{{\eta}2}}}})\), is allowed to evolve between a and b during the variable delay, t 1, where the evolutions under the one-bond 1H–15N scalar couplings are suppressed with a 1H WALTZ decoupling scheme (André et al. 2007; Shaka et al. 1983). Coupling to the 13Cζ nucleus is refocused by a 180° 13C pulse in the middle of the t 1 period. The evolution proceeds according to,
where the DQ x coherence evolves with the sum of the two underlying 15N frequencies (ΩNη1 + ΩNη2) during t 1, and \( DQ_{{{y}}} = 0.5(4C_{z}^{\zeta } N_{x}^{{\eta 1}} N_{y}^{{\eta 2}} + ~4C_{z}^{\zeta } N_{y}^{{\eta 1}} N_{x}^{{\eta 2}} ) \). The \({\text{90}}_{x}^{\circ }\) 15N pulse followed by the gradient pulse g4 at point b selects for the \( 4C_{z}^{\zeta } N_{y}^{{\eta 1}} N_{y}^{{\eta 2}} \) component of the double-quantum coherence, which is transferred back to transverse in-phase carbon magnetisation, \(C_{y}^{\zeta }\), for detection via a retro-INEPT between b and c, again incorporating a selective 15Nη inversion pulse. Frequency discrimination in the indirect dimension is achieved by incrementing the \({\text{90}}_{{\upphi {\text{2}}}}^{\circ }\) 15N pulse by 45° (Bax et al. 1981). It is important to note that in order to eliminate the effect of the exchange between the two 15Nη, the experiment has been designed such that the magnetisation of interest does not at any point exist as transverse single-quantum 15N magnetisation.
Two-dimensional Fourier transformation of the interferogram results in a signal for each arginine residue with the 13Cζ frequency along the direct dimension and the sum of the two coupled 15Nη frequencies, \({\Omega _{{{\text{N}{\eta}1}}}}+{{{{\Omega}}}_{{{\text{N}{\eta}2}}}},\) along the indirect dimension. The exchange of the two 15Nη sites with one another has no effect on the double-quantum frequency and thus the broad, featureless signals in the 13Cζ–15Nη HSQC are rendered substantially sharper (Fig. 2b). In the spectrum in Fig. 2b, a single peak is observed for each arginine residue and the data is processed such that the indirect chemical shift reflects the average of the two contributing 15Nη nuclei. The double-quantum experiment is particularly useful to probe flexible arginine side chains, where in the case of T4L99A all eight signals around 157.0 ppm (13C) are well resolved. A disadvantage of the double-quantum experiment compared to the single-quantum experiment is the faster (ca. twofold) transverse relaxation during the indirect chemical shift evolution period, since the spin density matrix elements evolved, \( 4C_{z}^{\zeta } N_{{x,y}}^{{\eta 1}} N_{{x,y}}^{{\eta 2}} ~ \), are transverse with respect to both 15Nη nuclei, which in turn relax with the two directly bound protons. The faster relaxation, which leads to lower signal-to-noise, only becomes significant for arginine side-chains that are rigid, for example, R95 and R148 (Werbeck et al. 2013) (Fig. 2b). However, the rigid side-chains are typically less affected by the exchange process and so the data obtained from the 13Cζ–15Nη HSQC experiment is often useful for these residues (Fig. 1c). The substantially better resolution provided by the double-quantum experiment adequately compensates for the associated loss of signal for less rigid residues.
The rapid transverse relaxation of the 15Nη nuclei of rigid residues in medium-to-large proteins can be mitigated by preparing the sample in a 100% D2O buffer. Substitution of the 1Hη protons with deuterium leads to slower 15Nη transverse relaxation and thus sharper lines in the indirect dimension of the double-quantum experiment. However, the longitudinal relaxation time of the 13Cζ nucleus also increases, which limits the permitted recycle rate of the experiment and increases the overall acquisition time. Whilst we have not observed a significant sensitivity gain per unit time using a 100% D2O buffer, recording spectra in 100% D2O could be useful in applications where experimental time is not a concern. Such an approach may enable the study of even larger proteins.
A route for chemical shift assignments of the 13Cζ–15Nη(DQ) spectrum
In favourable circumstances, an existing 13Cζ–15Nε assignment can be transferred to the 13Cζ–15Nη(DQ) spectrum based on the 13Cζ chemical shift alone. However spectral overlap of the arginine 13Cζ is not uncommon in even modestly-sized proteins. The 15Nη double-quantum experiment described above can be embedded within the existing 13Cζ–15Nε HSQC sequence (Werbeck et al. 2013) to provide a three-dimensional experiment for chemical shift assignment; Fig. 3. Briefly, magnetisation proportional to \(2C_{z}^{\zeta }N_{z}^{\varepsilon }\) is generated via an INEPT block with 13Cζ and 15Nε selective pulses. This magnetisation is then allowed to evolve during the first chemical shift evolution period t 1, between a and b, to encode the 15Nε chemical shift. One-bond scalar couplings to 1Hε and 13Cζ/δ are refocused with a 1H WALTZ decoupling scheme and a 13C adiabatic inversion pulse, respectively. A second INEPT block, between b and c, with a non-selective high power 180° 15N pulse, cleanly converts the \(2C_{z}^{\zeta }N_{z}^{\varepsilon }\) longitudinal two-spin order element to the three-spin order element \( 4C_{z}^{\zeta } N_{z}^{{\eta 1}} N_{z}^{{\eta 2}} \). As shown above, the double-quantum component is selected with a phase-cycle and allowed to evolve, with 1H and 13C decoupling, during the second chemical shift evolution period t 2. Finally, the magnetisation is returned to in-phase carbon, \(C_{y}^{\zeta }\), for detection between d and e, using a third INEPT block that is selective only for 15Nη. Figure 4 demonstrates how the resulting three-dimensional dataset is used to unambiguously assign the 15Nη resonances R137 and R148 in T4L99A, both of which have a 13Cζ frequency of 156.95 ppm.

Pulse sequence to obtain the intra-residue correlation between 15Nε and 15Nη double-quantum chemical shifts. The carrier positions are 13C: 156 ppm, 15N: 84 ppm (Rε), 78 ppm (square 180° and decoupling), 71 ppm (Rη) and 1H: 7 ppm. Narrow and wide bars represent 90° and 180° pulses respectively and are applied at maximum power. The delay Δ is 1/(4J CN) = 12.5 ms. Shaped pulses are represented by bell shapes with letters indicating the shape of the pulse [E: E-BURP-2, Rε: 15Nε selective RE-BURP, Rη: 15Nη RE-BURP41, C: smoothed CHIRP (Ermakov et al. 1993)] and are applied with durations of 1.5 ms (E), 6 ms (Rε, Rη) and 500 μs (C) at 11.74 T. Pulses are x phase unless stated otherwise. The phase cycle used is φ1: x, − x, φ2: 2(x), 2(y), 2(− x), 2(− y), φrec: x, 2(− x), x. Decoupling sequences are represented by striped boxes indicating the type of decoupling: WALTZ64 (Zhou et al. 2007) (4 kHz), GARP4 (Shaka et al. 1985) (0.7 kHz). Gradient pulses of 1 ms are represented by black rectangles and applied with strengths of g1: 19.8 G/cm, g2: 5.9 G/cm, g3: 12.3 G/cm, g4: 16.6 G/cm, g5: 7.0 G/cm, g6: 21.9 G/cm, g7: 25.2 G/cm, g8: 9.1 G/cm

a 13Cζ–15Nε HSQC spectrum of T4L99A recorded at 16.4 T. The overlap of R137 and R148 in the 13C dimension is highlighted by red dashed line. This ambiguity hampers the chemical shift assignment of the double-quantum spectrum in Fig. 2b. Chemical shift assignments are taken from Werbeck et al. (2013). b 15Nε–15Nη(DQ) 2D-plane extracted at 13Cζ = 156.95 ppm (red line in a) from the 3D experiment (Fig. 3) recorded at 11.74 T. The 3D spectrum allows unambiguous chemical shift assignment of the 15Nη double-quantum spectrum in Fig. 2b
Accessing small and residue-specific chemical shift perturbations
The observed chemical shift of a particular nucleus in an NMR experiment is very sensitive to the local molecular environment. For many years, localised chemical shift changes in NMR spectra have been used to measure side-chain pKas (Wang et al. 1996; Tollinger et al. 2002), investigate ligand binding (Williamson 2013) and assess protein folding (Calzolai and Zahn 2003). More recently, the magnitude of the deuterium isotope shift of lysine amines (Tomlinson et al. 2009; Williamson et al. 2013) has been used to infer the presence of solution-state salt-bridges in proteins. The technique relies on the detection of a small 15N chemical shift difference observed for an amine when one or more of the bound 1H are exchanged with deuterium (D) and thus highly resolved NMR spectra are essential. Arginine is very well suited to the formation of salt bridges and the deuterium isotope shift of the terminal –NηH2 amines is likely to be a useful parameter to characterise salt-bridge formation in solution. It has previously not been possible to measure these potentially very small and useful isotope shifts using standard proton-detected NMR experiments due to the shortcomings of these experiments described above. However, the double-quantum experiment presented above opens up a possible route for determining the isotope shifts, as well as other chemical shift perturbations, of the –NηH2 amines of arginine side-chains.
The − 15NηH j D2−j , j = 0,1,2 isotopomers of arginine side-chains are generated by dissolving the protein sample in a buffer containing a suitable percentage of D2O (10–30 vol.%). The conventional carbon-detected 13Cζ–15Nη HSQC experiment is applicable to measure the deuterium isotope shift for arginine side-chains that are slowly exchanging about the Cζ–Nε bond and give rise to two separate 13Cζ–15Nη resonances, for example R96 in T4L99A. Unfortunately, the addition of D2O increases the number of signals observed in an already overcrowded spectral region. For a significant number of residues in T4L99A, intermediate exchange of the two 15Nη sites combined with spectral overlap of isotopomers makes a quantification of residue-specific isotope shifts nearly impossible using the 13Cζ–15Nη HSQC experiment (Fig. 5a). The increased resolution afforded by the double-quantum experiment means that it is very well suited for the measurement of small chemical shift perturbations, such as the isotope shift, for the vast majority of arginine residues; particularly those that are in intermediate exchange regimes (Fig. 5b; Table 1). Even at a temperature of 278 K, where the rotational correlation time of T4L99A approaches 20 ns, the majority of these flexible arginine side-chains are still well resolved (Figure S1).

a Extreme signal overlap observed in the 13Cζ–15Nη HSQC spectrum of T4L99A recorded at 18.8 T in a buffer containing 10 vol.% D2O. b 13Cζ–15Nη HDQC spectrum recorded on the same sample. The resolution is high enough to quantify the residue-specific deuterium isotope shifts of all but one of the arginine side-chain amines contributing to the very broad signal in (a). The marked peaks indicate the –15Nη 2H4 isotopomer and the red arrow indicates the location of the corresponding –15Nη 2H3D isotopomer. Only two peaks are overlapped in this spectrum, that is –15Nη 2H3D signal for R8 overlaps with the –15Nη 2H4 signal of R154. The isotope shifts of the remaining residues are easily identified from this data. The pulse sequences used to obtain the spectra in a and b have been modified to include 2H decoupling (WALTZ16, 1 kHz) during the indirect evolution period
The obtained isotope shifts for the flexible arginine residues of T4L99A correlate very closely with the value measured for free arginine. This suggests that the solvent-exposed and flexible residues are not involved in any significant interactions. It should be noted that due to the nature of the double-quantum experiment, the individual isotope shift of 15Nη1 and 15Nη2 cannot be distinguished using this sequence. Nonetheless, an important experimental parameter that reports on interactions of the arginine side-chain can be obtained. Further NMR experiments combined with theoretical approaches to further characterise the hydrogen-bonding and salt-bridging behaviour of arginine residues are on-going.
Summary and conclusion
In summary, we presented pulse schemes to characterise arginine side-chain 15Nη amines in solution. The preparation and subsequent evolution of a double-quantum 15Nη coherence eliminates the line broadenings associated with slow-to-intermediate rotation about the Cζ–Nε partial double bond thus leading to dramatically sharpened peaks in the NMR spectrum. The double-quantum experiment is complementary to the 13Cζ–15Nη single-quantum experiment because the double-quantum experiment is ideally suited to characterise arginine side-chains whose 13Cζ–15Nη single-quantum resonances are severely broadened because of exchange, whilst the single-quantum experiment provides (15Nη1,15Nη2) site-specific information for the well-resolved resonances. An application to the 19 kDa T4L99A protein demonstrated the strengths of the double-quantum experiment and allowed the quantification of small deuterium isotope shifts to provide information on the interactions of the arginine side-chain guanidinium group. The magnitude of the detected isotope shifts showcases the high resolution of this experiment and suggests its potential application to many other applications where residue-specific chemical shift perturbations are of interest. The presented experiments add to a growing list of methods for characterising functional protein side-chains, which ultimately will allow a quantification of the structure, dynamics, and interactions of side-chains in solution to a level where their specific contribution to enzymatic function and protein interactions can be elucidated.
References
Andersson P, Gsell B, Wipf B, Senn H, Otting G (1998) HMQC and HSQC experiments with water flip-back optimized for large proteins. J Biomol NMR 11:279–288
André I, Linse S, Mulder FAA (2007) Residue-specific pKa determination of lysine and arginine side chains by indirect 15N and 13C NMR spectroscopy: application to apo calmodulin. J Am Chem Soc 129:15805–15813
Bax A, Freeman R, Frenkiel TA, Levitt MH (1981) Assignment of carbon-13 NMR spectra via double-quantum coherence. J Magn Reson 43:478–483
Bodenhausen G, Ruben DJ (1980) Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem Phys Lett 69:185–189
Borders CL, Broadwater JA, Bekeny PA, Salmon JE, Lee AS, Eldridge AM, Pett VB (2008) A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci 3:541–548
Calzolai L, Zahn R (2003) Influence of pH on NMR structure and stability of the human prion protein globular domain. J Biol Chem 278:35592–35596
Carr HY, Purcell EM (1954) Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev 94:630–638
Casey AK, Hicks MA, Johnson JL, Babbitt PC, Frantom PA (2014) Mechanistic and bioinformatic investigation of a conserved active site helix in α-isopropylmalate synthase from Mycobacterium tuberculosis, a member of the DRE-TIM metallolyase superfamily. Biochemistry 53:2915–2925
Crowley PB, Golovin A (2005) Cation-π interactions in protein-protein interfaces. Proteins Struct Funct Bioinforma 59:231–239
Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A, (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Ermakov VL, Bohlen JM, Bodenhausen G (1993) Improved schemes for refocusing with frequency-modulated chirp pulses. J Magn Reson Ser A 103:226–229
Farber PJ, Mittermaier A (2015) Relaxation dispersion NMR spectroscopy for the study of protein allostery. Biophys Rev 7:191–200
Fitch CA, Platzer G, Okon M, Garcia-Moreno EB, McIntosh LP (2015) Arginine: its pKa value revisited. Protein Sci 24:752–761
Friedt J, Leavens FMV, Mercier E, Wieden H-J, Kothe U (2014) An arginine-aspartate network in the active site of bacterial TruB is critical for catalyzing pseudouridine formation. Nucleic Acids Res 42:3857–3870
Gargaro AR, Frenkiel TA, Nieto PM, Birdsall B, Polshakov VI, Morgan WD, Feeney J (1996) NMR detection of arginine-ligand interactions in complexes of Lactobacillus casei dihydrofolate reductase. Eur J Biochem 238:435–439
Geen H, Freeman R (1991) Band-selective radiofrequency pulses. J Magn Reson 93:93–141
Gerecht K, Figueiredo AM, Hansen DF (2017) Determining rotational dynamics of the guanidino group of arginine side chains in proteins by carbon-detected NMR. Chem Commun 53:10062–10065
Goldschen-Ohm MP, Wagner DA, Jones MV (2011) Three arginines in the GABAA receptor binding pocket have distinct roles in the formation and stability of agonist- versus antagonist-bound complexes. Mol Pharmacol 80:647–656
Hansen PE (2000) Isotope effects on chemical shifts of proteins and peptides. Magn Reson Chem 38:1–10
Hansen DF, Led JJ (2003) Implications of using approximate Bloch–McConnell equations in NMR analyses of chemically exchanging systems: application to the electron self-exchange of plastocyanin. J Magn Reson 163:215–227
Hansen DF, Vallurupalli P, Kay LE (2008) An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J Phys Chem B 112:5898–5904
Harms MJ, Schlessman JL, Sue GR, Garcia-Moreno EB (2011) Arginine residues at internal positions in a protein are always charged. Proc Natl Acad Sci USA 108:18954–18959
Henry GD, Sykes BD (1995) Determination of the rotational dynamics and pH dependence of the hydrogen exchange rates of the arginine guanidino group using NMR spectroscopy. J Biomol NMR 6:59–66
Iwahara J, Clore GM (2006) Sensitivity improvement for correlations involving arginine side-chain Ne/He resonances in multi-dimensional NMR experiments using broadband 15N 180° pulses. J Biomol NMR 36:251–257
Kleckner IR, Foster MP (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim Biophys Acta Proteins Proteomics 1814:942–968
Korzhnev DM, Kay LE, (2008) Probing invisible, low-populated states of protein molecules by relaxation dispersion nmr spectroscopy: an application to protein folding. Acc Chem Res 41:442–451
McConnell HM (1958) Reaction rates by nuclear magnetic resonance. J Chem Phys 28:430–431
Morgan WD, Birdsall B, Nieto PM, Gargaro AR, Feeney J (1999) 1H/15N HSQC NMR studies of ligand carboxylate group interactions with arginine residues in complexes of brodimoprim analogues and lactobacillus casei dihydrofolate reductase. Biochemistry 38:2127–2134
Morris GA, Freeman R (1979) Enhancement of nuclear magnetic resonance signals by polarization transfer. J Am Chem Soc 101:760–762
Nieto PM, Birdsall B, Morgan WD, Frenkiel TA, Gargaro AR, Feeney J (1997) Correlated bond rotations in interactions of arginine residues with ligand carboxylate groups in protein ligand complexes. FEBS Lett 405:16–20
Orekhov VY, Korzhnev DM, Kay LE (2004) Double- and zero-quantum NMR relaxation dispersion experiments sampling millisecond time scale dynamics in proteins. J Am Chem Soc 126:1886–1891
Palmer AG III, Massi F (2006) Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev 106:1700–1719
Palmer AG (2014) Chemical exchange in biomacromolecules: past, present, and future. J Magn Reson 241:3–17
Pervushin KV, Wider G, Riek R, Wuthrich K (1999) The 3D NOESY-[1H,15N,1H]-ZQ-TROSY NMR experiment with diagonal peak suppression. Proc Natl Acad Sci USA 96:9607–9612
Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS (2010) Origins of specificity in protein-DNA recognition. Annu Rev Biochem 79:233–269
Shaka AJ, Keeler J, Frenkiel T, Freeman R (1983) An improved sequence for broadband decoupling: WALTZ-16. J Magn Reson 52:335–338
Shaka AJ, Barker PB, Freeman R (1985) Computer-optimized decoupling scheme for wideband applications and low-level operation. J Magn Reson 64:547–552
Sørensen OW, Eich GW, Levitt MH, Bodenhausen G, Ernst RR (1984) Product operator formalism for the description of NMR pulse experiments. Prog Nucl Magn Reson Spectrosc 16:163–192
Tollinger M, Forman-Kay JD, Kay LE (2002) Measurement of side-chain carboxyl pKa values of glutamate and aspartate residues in an unfolded protein by multinuclear NMR spectroscopy. J Am Chem Soc 124:5714–5717
Tomlinson JH, Ullah S, Hansen PE, Williamson MP (2009) Characterization of salt bridges to lysines in the protein G B1 domain. J Am Chem Soc 131:4674–4684
Trbovic N, Cho J-H, Abel R, Friesner RA, Rance M, Palmer AG (2009) Protein side-chain dynamics and residual conformational entropy. J Am Chem Soc 131:615–622
Vallurupalli P, Hansen DF, Lundström P, Kay LE (2009) CPMG relaxation dispersion NMR experiments measuring glycine 1Hα and 13Cα chemical shifts in the ‘invisible’ excited states of proteins. J Biomol NMR 45:45–55
Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins Struct Funct Bioinforma 59:687–696
Wang Y-X, Freedberg DI, Yamazaki T, Wingfield PT, Stahl SJ, Kaufman JD, Kiso Y, Torchia DA (1996) Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272 †. Biochemistry 35:9945–9950
Werbeck ND, Kirkpatrick J, Hansen DF (2013) Probing arginine side-chains and their dynamics with carbon-detected nmr spectroscopy: application to the 42 kDa human histone deacetylase 8 at high pH. Angew Chem Int Ed 52:3145–3147
Williamson MP (2013) Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc 73:1–16
Williamson MP, Hounslow AM, Ford J, Fowler K, Hebditch M, Hansen PE (2013) Detection of salt bridges to lysines in solution in barnase. Chem Commun 49:9824–9826
Yamazaki T, Pascal SM, Singer AU, Forman-Kay JD, Kay LE (1995) NMR pulse schemes for the sequence-specific assignment of arginine guanidino 15N and 1H chemical shifts in proteins. J Am Chem Soc 117:3556–3564
Yoshimura Y, Oktaviani NA, Yonezawa K, Kamikubo H, Mulder FAA (2017) Unambiguous determination of protein arginine ionization states in solution by NMR spectroscopy. Angew Chem Int Ed 56:239–242
Yuwen T, Vallurupalli P, Kay LE (2016) Enhancing the sensitivity of CPMG relaxation dispersion to conformational exchange processes by multiple-quantum spectroscopy. Angew Chem Int Ed 55:11490–11494
Zeymer C, Werbeck ND, Zimmermann S, Reinstein J, Hansen DF (2016) Characterizing active site conformational heterogeneity along the trajectory of an enzymatic phosphoryl transfer reaction. Angew Chem Int Ed 55:11533–11537
Zhou Z, Kümmerle R, Qiu X, Redwine D, Cong R, Taha A, Baugh D, Winniford B (2007) A new decoupling method for accurate quantification of polyethylene copolymer composition and triad sequence distribution with 13C NMR. J Magn Reson 187:225–233
Zhuravleva A, Orekhov VY, (2008) Divided evolution: a scheme for suppression of line broadening induced by conformational exchange. J Am Chem Soc 130:3260–3261
Acknowledgements
Dr Angelo Figueiredo is acknowledged for help with recording NMR spectra, Dr Lisbeth Andersen and Karola Gerecht are acknowledged for help with the preparation of isotopically labelled T4L99A. The Wellcome Trust (ref 101569/z/13/z) and the Engineering and Physical Sciences Research Council (EPSRC) are acknowledged for supporting the ISMB NMR facility at University College London and the Francis Crick Biomedical NMR centre is acknowledged for access to high-field NMR spectrometers. HM acknowledges UCB pharma for a PhD studentship. This research is supported by the Leverhulme Trust (RPG-2016-268).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mackenzie, H.W., Hansen, D.F. A 13C-detected 15N double-quantum NMR experiment to probe arginine side-chain guanidinium 15Nη chemical shifts. J Biomol NMR 69, 123–132 (2017). https://doi.org/10.1007/s10858-017-0137-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-017-0137-2