
Abstract
The structure of microbial cellulose (MC) produced by Acetobacter xylinum was studied in presence of Fluorescent Brightener, Direct Blue 1, 14, 15, 53, Direct Red 28, 75 and 79, as probe. X-ray diffraction pattern of the product showed that it was a crystalline complex of dye and cellulose. The product has the structure in which the monomolecular layer of the dye molecule is included between the cellulose sheets corresponding to the (\( 1\bar{1}0 \)) planes of microbial cellulose. As a result of dye inclusion, d-spacing of lower angle plane (100) of products becomes 8.0–8.8 Å instead of 6.1 Å of MC. The d-spacing for the higher angle plane must be (010) plane due to stronger van der Waals forces between the pyranose rings which reduced 5.3 Å space of (110) plane of MC to 3.9–4.5 Å in the product. However, cellulose regenerated from FB, DR28 products was cellulose I and IV, respectively, and that from each DB1, 14, 15, 53, DR75 and 79 products was cellulose II. Solid state 13C NMR and deuteration-IR showed the product was non-crystalline which was contrasted to X-ray results. The regenerated celluloses were cellulose Iβ, IVI and II, respectively. Thus the structure of the product depends on the characteristics of dye which affects the conformation of cellulose at the nascent stage by the direct interaction with cellulose chains. The different regenerated celluloses as well as different fine structure in the same cellulose allomorph were produced depending mainly on number and position of the sulfonate groups in the dye.
Similar content being viewed by others


Introduction
Cellulose is the most abundant macromolecules produced on earth (Zhao et al. 2007) and most cellulose is produced by vascular plants. Hence, the forest which is the main source of cellulose supply is now on jeopardy. Cellulose produced by an acetic acid bacteria, that is, microbial cellulose (MC) or bacterial cellulose (BC) is expected for an alternative source of cellulose biomass in the near future. MC is an exopolysaccharide produced by various species of bacteria, such as those of the genera Acetobacter, Agrobacterium, Aerobacter, Achromobacter, Azotobacter, Rhizobium, Sarcina, and Salmonella (Chawla et al. 2009). Acetobacter xylinum (or reclassified as Gluconacetobacter xylinus) is one of the best bacterial species for the production of bacterial cellulose in large-scale and utilizes a wide variety of substrate (Moosavi-Nasab and Yousefi 2011). However, four major types of cellulose allomorphs have been reported so far, cellulose I, II, III and IV on the basis of X-ray diffraction patterns (Isogai 1994; Sarko 1978; Marchessault and Sarko 1967; Walton and Blackwell 1973; Marchessault and Sundararajan 1985). Each polymorph yields a recognizable diffraction patterns. Crystal structures of cellulose allomorphs, such as unit cell sizes, packing modes of cellulose chains, conformations at Cl, C4 and C6 (rotational angles ϕ, φ and χ, respectively) have been studied from as early as 1920s by X-ray diffractometry, electron microscopy, IR spectroscopy and Raman spectroscopy, and other techniques.
The mechanisms of formation of cellulose I microfibril in nature, and the control of cellulose I microfibril formation by living organisms have been of great interest to the scientists. The microfibril produced in A. xylinum is crystallize cellularly (Brown et al. 1976; Zaar 1977, 1979). Arguments for and against various relationships between polymerization and crystallization in microfibril formation in plants and bacteria were based largely on theoretical considerations and indirect evidence until 1979, when it was discovered that some chemicals could be used to alter cellulose microfibril assembly in vivo (Haigler et al. 1980; Haigler and Benziman 1982; Brown et al. 1983). These alternating chemicals, such as fluorescent brightening agents, direct dyes and cellulose derivatives, allowed direct testing of the temporal relationship between polymerization and crystallization in cellulose biogenesis, and provided inside into how organisms regulate the size of microfibrils. The present article will discuss elaborately into how direct dyes have been used to investigate the control of cellulose biogenesis in A. xylinum which was recognized as a model organism to date. These alternating agents have therefore allowed elucidation of basic principles that govern the biogenesis of bacterial cellulose as well as its formation mechanisms.
Only a few studies relating to the structure of nascent fibril have been reported to date. Among these investigators, two working groups Kai et al. and Brown et al. are mentionable. Brown and his groups (Haigler et al. 1980; Brown et al. 1983; Haigler and Chanzy 1988) judging from the electron microscopy of products from Acetacter-culture with direct dyes reported that the dyes adhered to the surface of cellulose protofibril in a stact state formed non-crystalline product thus, this product formed cellulose I upon dye-extraction. On the other hand, Kai and his groups (Kai et al. 1990, 1994; Kai 1984a) found that the products from the incubated medium with Fluorescent Brightener, Congo Red and Direct Red 80 showed the characteristic X-ray diffraction diagrams indicating the formation of crystalline complexes, and these complexes reproduced celluloses Iβ, IV and II, respectively, after dye-extraction. However, direct dyes have different affinities for cellulose under the experimental conditions, which would be affected by the kind, number and position of substituents on the planar molecular backbone (Kai and Mondal 1996, 1997; Mondal and Kai 1998, 2001).
In this paper, we specially investigate the structure of the nascent fibril produced by A. xylinum bacteria using direct dyes as alternating agents. As these alternating agents allow direct testing of the temporal relationship between polymerization and crystallization in cellulose biogenesis, this is very important for the clarification of the mechanism of formation of cellulose fibril. The direct dyes considered in this paper are Fluorescent Brightener (FB), Direct Blue 1, 14, 15, 53 (DB1, DB14, DB15 and DB53) Direct Red 28, 75 and 79 (DR28, DR75, DR79). DR28, DB 1, 14, 15 and 53 have same biphenylene bis(azo) skeletal backbone, and DR75 and 79 have same ureylenebis(naphthylazo) benzene skeletal backbone. Although each group contains same skeletal backbone, their substituent groups are different. Hence, it is possible to encounter their effect on the product structure as well as on the regenerated cellulose. The structure of the product from Acetobacter-culture in the presence of direct dyes and its regenerated cellulose was characterized by X-ray diffractometer, solid state 13C NMR and infrared spectroscopies.
Experimental
Materials
Preparation of product sample
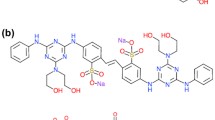
Acetobacter xylinum (IFO 13693), the most studied bacterial cellulose (BC) producer, was cultured in a 15 cm diameter Petridish which contains a 100 ml Hestrin–Schramm medium (Hestrin and Schramm 1954) (pH 6.8) at 28 °C for 3 days in an incubator. Cellulose-free bacterial cell suspension was prepared from a matured Petridish in an ice cooled 120 ml phosphate buffer solution, and 60 ml of the cell suspension was added to 140 ml HS medium (pH 7.0) containing 0.1 wt% direct dye. The mixture was then incubated at 28 °C for 24 h in a static condition or for 12 h in a stirred condition (Kai et al. 1990; Kai and Mondal 1996; Mondal and Kai 1999). The product sample (product) was collected either by centrifuging (6,000 rpm, 5.15 g, 20 min) or by a Tea strainer, and was washed well with 0.1 wt% aqueous sodium hydroxide solution to remove the dye which was not related to dyeing. This was then rinsed well with distilled water and was preserved at about 3 °C in the wet for the next experiments. The name and structure of the direct dyes used in present article are listed in Fig. 1.


Chemical structure and name of direct dyes
Regenerated cellulose from product sample
Extraction of dye from the above product samples was performed by boiling in 70 vol% aqueous EtOH solution for 18 h, and the fresh aqueous ethanol solution was exchanged for every 3 h. This dye extracted sample was again boiled in 1.0 wt% aqueous sodium hydroxide solution for 10 h under N2 atmosphere to remove proteinous matters. This regenerated cellulose sample was neutralized with 1.0 vol% aqueous acetic acid solution and, subsequently, rinsed with distilled water (Kai et al. 1990; Kai and Mondal 1996; Mondal and Kai 1999). The samples were preserved as above.
Preparation of control cellulose sample
The control cellulose I sample was prepared by washing the MC with distilled water to remove the medium components, and then was boiled in 1.0 wt% aqueous sodium hydroxide solution for 10 h under N2 atmosphere. The final sample (cellulose I or MC) was neutralized and rinsed as above. Mercerization of MC was carried out at 20 °C for 25 h to prepare control cellulose II sample. The control cellulose IVII was obtained by which cellulose II was annealed in glycerol at 260 °C for 30 min.
Methods for the measurements
In order to understand the structure of the product from Acetobacter -culture in the presence of each of FB, DB1, 14, 15, 53, DR28, 75 and 79, and the cellulose regenerated from them, the following measurements were performed.
X-ray diffractometry
The X-ray diffraction diagram of an isotropic product sample and its regenerated cellulose was performed by transmission method. In this method, the dried sample was compressed as a pellet into the pore of a sample holder of 1 mm diameter and 1 mm thickness.
Again, an uniplanar oriented membrane of product sample was prepared on a Teflon plate and was dried at room temperature. Its dry size was 3.0 cm × 2.5 cm and dry thickness was not less than 100 mμ. The X-ray diffraction diagram of this uniplanar oriented membrane was measured with the reflection and transmission methods using MXP18 diffractometer (MAC Science Co, Ltd., Tokyo) with Ni-filtered CuKα radiation. The measurement conditions were as follows: divergence slit, 1.0; receiving slit, 0.15; scanning speed, 4.0° min−1; X-ray radiation, 40 kV, 200 mA.
Solid state 13C NMR spectroscopy
Solid state 13C NMR experiments of the wet samples were performed at room temperature by a JEOL JNM EX 270 spectrometer operating at 6.8 MHz under a static magnetic field of 6.35 T. The mass rates were 5.0–5.5 kHz, and the contact time for the CP process was 1,000 ms throughout this work. The chemical shifts relative to tetramethyl silane were determined by using the crystalline peak at 17.3 ppm of hexamethyl benzene as an internal standard. The non-dried samples were packed in a cylinder type MAS rotor with an O-ring sealed to avoid the loss of water during NMR measurements The spectrum of the crystalline component was measured selectively by taking NMR signals after a relaxation of the non-crystalline component. Therefore, to determine the crystalline component of the control and regenerated celluloses, signals obtained after τ = 50 s were measured by using T1CP pulse sequence with CP process as the spectrum of the cxystalline component (Torchia 1978).
Deuteration-IR spectroscopy
In order to obtain IR spectra of the sample, a membrane suitable for IR spectra (about 10 μm in dry thickness) was prepared on a Teflon plate from non-dried samples and was dried at room temperature. The deuteration of the sample was performed by the method described in previous article (Kai and Mondal 1997). The sample membrane was set inside a glass cell so that the IR beam was perpendicular to the membrane surface. Before deuteration, the glass cell with the sample was dried under reduced pressure (10−3 torr) for 3 h, and then vapour phase deuteration of the sample was performed by D2O (99.8 %, Aldrich Chemical Co. USA) for a given time at room temperature. In this experiment, the deuteration times used in all the samples were 0, 1, 3, 7 20, 80 and 1,000 min. The deuteration sample was again dried under reduced pressure for the same time of deuteration up to 20 min, and over 20 min deuteration the drying time was fixed to 20 min, and then IR spectrum was measured. FT-IR systems 800 (Nicolet) was used for IR spectroscopy. The amount of OH and OD groups is calculated according to the method described in the previous paper (Kai and Mondal 1997; Mondal and Kai 1998, 1999).
Results and discussion
Overview of the mechanism of cellulose biosynthesis
Model system for the study of cellulose biosynthesis
No single system has emerged as ideal for the study of cellulose biosynthesis. Very few genera of bacteria synthesize cellulose, but the gram-negative bacterium A. xylinum secretes large quantities of cellulose as microfibrils for a row of synthetic sites along the longitudinal axis of the cell (Ross et al. 1991). The microfibrils formed each synthetic sites merge to form a large ribbon of cellulose in the growth medium. These ribbons and associated cells tangle, and form a floating pellicle that allows the non-motile strickly aerobic bacteria to grow in the higher oxygen tension at the surface. A. xylinum is easily maintained in culture, and conditions of synthesis are readily manipulated; therefore, this bacterium is an excellent model system for studying the molecular mechanisms of cellulose polymerization.
Biosynthesis of cellulose I microfibrils
The biogenesis of native cellulose is a complex process requiring the biosynthesis of high molecular weight 1, 4-glucans and the formation of highly crystalline metastable cellulose I microfibrils; and in many cases, requiring the organization and orientation of these microfibrils into cell walls (Colvin 1964). Proposed mechanisms of cellulose biosynthesis have historically been intimately related to the contemporary understanding of the physical structure of cellulose (Ranby 1952). It is evident that any proposed mechanism of cellulose biosynthesis must be consistent with the ultimate physical properties of macromolecules. Since almost all native cellulose is fibrillar cellulose I, it is probable that there is a common biosynthetic mechanism among all organisms that produce crystalline cellulose I microfibrils (Sarko and Muggli 1974).
Native cellulose I is generally thought to exist in a crystalline lattice of extended parallel glucan chains. It has been proposed that cellulose II, which is infrequently found in nature, is composed of antiparallel glucan chains, and is more thermodynamically stable than cellulose I (Sarko and Muggli 1974; Gardner and Blackwell 1974). For example, cellulose II polymorph mutants of Acetobacter lacked organised particles in the lipopolysaccharide (LPS) envelope. They have a functional cellulose synthase, which affect “export” component (Brown 1989). Hence, loss of this control means loss of native cellulose I polymorph synthesis. An understanding of the molecular mechanisms by which organisms synthesize and assemble crystalline metastable cellulose I fibrils is not yet complete. No hypothesis has heretofore been advanced that adequately unifies knowledge of the crystal structure of cellulose, the substructure of microfibrils, and the biosynthetic mechanism.
Fluorescent brightening agents disrupts ribbon assembly by associating with the subunits of the ribbon called elementary fibrils, and preventing their normal aggregation, when it is added to active cultures of A. xylinum. The planar calcofluor molecule binds along the glucan chains through hydrogen bonding (Maeda and Ishida 1967) and dipolar interactions (Rattee and Breuer 1974). In the presence of fluorescent brightening agents and direct dyes, the bacteria synthesize broad bands that are extruded perpendicular to the longitudinal cell axis instead of normal twisting ribbon (Haigler et al. 1980; Haigler and Chanzy 1988; Kai et al. 1994; Kai 1984a; Kai and Kitamura 1985; Kai and Koseki 1985; Benziman et al. 1980). Hence, this is the only direct method for the discussion of mechanism and crystallization of cellulose.
Regulation by a unique mechanism
Acetobacter xylinum has long been considered the most successful prokaryotic model for studies on the mechanism of cellulose biogenesis in intact cell (Zaar 1979; Schramm and Hestrin 1954; Brown Jr 1979; Delmer et al. 1982; Aloni and Benziman 1982). The largest enzyme in the regulation of cellulose synthesis is the membrane bound cellulose synthase. Regulation of the enzyme involves an unusual system of “fine control” based on a novel nucleotide, cyclic diguanylic acid (c-di-GMP) and the regulatory enzymes maintaining its intracellular turnover (Ross et al. 1985, 1986, 1987). A conceptual scheme for the mechanism of regulation of cellulose synthesis, which accounts for the results obtained from the study of the cell-free preparations from A. xylinum, is outline by Ross et al. (Ross et al. 1987). Diguanylate cyclase catalyzes the synthesis of c-di-GMP from two molecules of guanosine triphosphate (GTP), via linear dinucleotide triphosphate pppGpG, in two distinct PPi-releasing steps. Within the cell, PPi is rapidly cleaved to yield Pi (Benziman and Palgi 1970). The pathway of c-di-GMP degradation is initiated by a c-di-GMP-specific Ca2+ sensitive phosphodiesterase PDE-A, which cleaves a single phosphodiester in the cyclic structure yielding linear dimer pGpG and simultaneously inactivates the molecule. The mononucleotide residues of the inactive dimer structure are then recovered as free 5′-GMP units through the action of a second phosphodiesterase, PDE-B. The Ca2+ inhibition of PDE-A represents an additional locus of regulatory control. According to this feature of the system fluctuations in Ca2+ levels may modulate the rate of cellulose synthesis as they influence the persistence of activator in the system.
Mechanism of dye-cellulose complex formation and its model structure
It is well known that fluorescent brightener and direct dyes alter normal ribbon of cellulose fibrils produced when A. xylinum cultures in HS medium (Hestrin and Schramm 1954). The X-ray diffraction diagrams of the product in the presence of direct dyes, such as FB, DB1, DB14, DB15, DB53, DR28, DR75 and DR79 indicate good agreement with the alteration concept of cellulose assembly. The 2θ values of native MC observed at 14.5, 16.8 and 22.7° correspond to the (\( 1\bar{1}0 \)), (110) and (020) planes respectively (Table 1). These data indicated that the BC samples were the typical crystalline forms of cellulose I (Oh et al. 2005; Yan et al. 2008). These three diffraction peaks of MC appear as two peaks in the products: one of which occurs at lower angle side in the range 2θ = 10.1–10.9° and the other in the range 2θ = 19.9–22.9° (higher angle side) (Table 1). However, 2θ values at lower and higher angle sides differ somewhat from each other depending on the dye used. These new diffraction patterns of product do not overlap either with the dye powder or with MC means the product is a complex of cellulose and a dye (dye-cellulose complex). The 2θ values at lower angle side mostly remain unchanged in all cases, whereas those at higher angle side vary a wide range depending on dye characteristics. Although the experimental results were reproducible, it is assumed that the dye molecule is included in the lower angle plane of X-ray diffraction as its space become broader. From the electron microscopy, Brown et al. (1983) reported that fluorescent brightener was included between the (020) planes of MC. On the other hand, Kai (1984a, b), from their X-ray measurements results, suggested that fluorescent brightener was included corresponding to the (\( 1\bar{1}0 \)) plane of MC and thereby formed complex. In this section we will discussed a model structure which may clear the diffraction plane where the dye is actually included.
The diffraction diagram of the uniplanar oriented membrane of MC obtained by reflection method is strong at low angle side (near 14.4°), whereas those at high angle side (near 16.8°) are very weak. The opposite diffraction pattern is found in the case of transmission method (Table 2) (Takai et al. 1975). However, the diffraction diagram of the FB and DR28 products measured by reflection method shows that the diffraction at lower angle side becomes strong whereas that at higher angle side becomes very weak like MC. In contrast, in the diffraction diagram by the transmission method the diffraction peak at lower angle side becomes very weak, and that at high angle side becomes strong. The result of FB and DR28 is a good agreement with the result of Mondal and Kai (2001) and Kai and Mondal (1997). Although the diffraction peak at low angle side of each DB1, DB14, DB15, DB53, DR75 and DR79 products obtained by reflection method is somewhat stronger than that obtained by transmission method, the strength of the diffraction peak at high angle side does not change remarkably. Furthermore, the diffraction diagrams of each DB1, DB14, DB15, DB53, DR75 and DR79 products differ somewhat from each other.
From comparison of X-ray diagrams of the products with MC, it is clear that the structure of the products in the presence of a direct dye differs from that of MC. According to Takai et al. (1975) the (\( 1\bar{1}0 \)) plane at low angle side of oriented membrane of MC is preferentially parallel to the membrane surface, and the (110) plane is perpendicular to (\( 1\bar{1}0 \)) plane. As the membrane of FB and DR28 products were prepared on a Teflon plate and the diffraction nature are closely similar to MC, the diffraction plane at low angle side is parallel to its surface, whereas that at high angle side is perpendicular to it. In contrast, although the sample preparation method is similar in all cases for the membranes of DB1, DB14, DB15, DB53, DR75 and DR79 products, the diffraction planes do not attain such orientation like MC, FB or DR28 product. However, the stronger diffraction by reflection at low angle side of these membranes indicates that it has a trend to become parallel to its surface (Takai et al. 1975). The diffraction plane at high angle side is not necessarily perpendicular to the diffraction plane at low angle side. The aspect of the plane orientation of these product membranes is similar to that of cellulose II membrane (Kai and Mondal 1997). The model of the reflecting planes of MC with inter-molecular hydrogen bonding, complex loss of the inter-molecular hydrogen bonding due to the inclusion of dye layer between the cellulose sheets, and generalized model structure of MC (cellulose I) and dye-cellulose complex are shown in the Figs. 2, 3 and 4 respectively. As shown in the Fig. 3, if a dye molecule is included in the form of monomolecular layer between the cellulose chains corresponding to the (\( 1\bar{1}0 \)) plane of MC, the X-ray measurement result can be explained satisfactorily. The diffraction plane of the FB or DR28 product near 2θ = 10.4° is parallel to the surface of the membrane similar to the (\( 1\bar{1}0 \)) plane of MC. As a result of inclusion of dye molecule in the form of a monolayer between the (100) planes of product (Fig. 4), the d-spacing of product at low angle side must be become broader by at least 2.0 Å than that of 6.1 Å of MC. However, the diffraction plane of the product near 22.9° must be the (010) plane of the product, because van der Waals forces between pyranose rings of the cellulose chains in the sheets become more stronger due to which 5.3 Å space between the (110) planes of MC become narrowed to 1.4 Å. Kai (1984b) reported from lattice space calculation with X-ray diffraction of the cellulose produced in the presence of fluorescent brightener that the spacing become (\( 1\bar{1}0 \)) planes of MC was available to the dye inclusion rather than (020) planes which Brown et al. (1983) assumed. As the (\( 1\bar{1}0 \)) plane of MC shifted to the broader d-spacing in the product, and (\( 1\bar{1}0 \)) and (110) planes are perpendicular to one another, there are no observed effect of dye inclusion between the (020) plane, and this is a good agreement with that in Kai et al. reports. The structure of cellulose regenerated from the product differs from that of MC means a dye is able to affect the conformation of cellulose chain by the direct interaction of cellulose chain with the dye molecule through the formation of complex product. If a dye is stacked only on the surface of a protofibril as Haigler et al. (Haigler and Chanzy 1988) suggested, it becomes very difficult for it to affect the conformation of all cellulose chains directly, therefore, it is not possible to explain the fact that structures of the product and cellulose regenerated from it differ from each other by the dye used.

Schematic model of the reflecting planes of MC with inter-molecular hydrogen bonding

Schematic model of reflecting planes of dye-cellulose complex. Dye molecule is included between the (\( 1\bar{1}0 \)) planes in the form of monomolecular layer and intermolecular hydrogen bonding is not found like MC

Generalized model structure of MC (cellulose I) and dye-cellulose complex. Resulting from X-ray diagrams of the product samples, the dye molecule seems to be included between the (\( 1\bar{1}0 \)) planes of MC or (100) planes in the complex, respectively dotted lines and solid lines: Configuration of cellulose chains in MC and complex, respectively
MC is extruded from the cell in the shape of the cellulose sheet (or microfibril) corresponding to the (\( 1\bar{1}0 \)) plane (Kai 1984a, b) and the hydrogen bonding is not recognized in the nascent cellulose sheet. Thus, the structure has the cellulose I formative ability which is maintained by the hydrogen bonding or van der Waals forces (Kai and Koseki 1985; Kai et al. 1989). Due to the inclusion of dye between nascent cellulose sheets, the inter-molecular hydrogen bonding between the sheets is prevented, and it is, therefore, suggested that the cellulose chain distance in the sheet will be changed from 5.3 Å to lower value at 3.9 Å as shown in Fig. 4. On the other hand, as the dye is included between the (\( 1\bar{1}0 \)) planes, the d-spacing of low angle plane of product become 8.3 Å instead of 6.1 Å of MC. This variation of d-spacing in the complex depends on the size and structure of dye, and their interactions with cellulose in the nascent stage.
If the structure of the product has the unit cell shown in the Fig. 4, infrared and solid state 13C NMR results can also be explained easily. Solid state 13C NMR spectra of cellulose component of product samples are broader than those of the MC. Moreover, the upfield C4 and C6 resonance lines for all the products appear at 84.7 and 62.5 ppm, respectively (Table 3), which are the non-crystalline regions of the cellulose component of MC. No remarkable resonance lines of dye powder overlap onto lines of the product sample. This study suggest that the whole spectra of cellulose components of each dye product are very similar to that of the non-crystalline component of MC, and hence, the hydrogen bonding are not found as in MC. Although the product shows the crystalline X-ray diffraction pattern, these NMR results indicate that all products are in the non-crystalline state. This non-crystalline structure is due to inclusion of a dye layer between the (100) planes of the product where hydrogen bonding of MC is hindered.
Infrared spectra of the product membrane in the presence of direct dyes show that the absorptions due to intra- and inter-molecular hydrogen bondings, which are observed in the MC (Moosavi-Nasab and Yousefi 2011; Oh et al. 2005), are not appeared. This means that due to the effect of a direct dye, the intra- and intermolecular hydrogen bond formation is prevented in the product sample. Thus existence of hydrogen bonding or not will be in the deuteration-IR. When MC is deuterated for about 80 min, all accessible OH groups (non-crystalline OH groups) changes to OD groups with the peak value near to 2,530 cm−1 (Kai and Mondal 1997). Even if the degree of deuteration is different from product to product, this 80 min time is sufficient to deuterate most of the accessible OH groups. Hence, after 80 min deuteration, the absorption spectrum of the product shows the spectrum of only crystalline structure. In 80 min deuteration, around 77–80 % of OH groups of FB and DR28 products transform to OD, respectively, and the spectrum of the deuterated sample is too broad with the peak value at about 3,405 cm−1 (inter-molecular hydrogen bonding) of the MC (Table 4). In this case, if experimental error is taken into consideration, all OH groups in the complex may be deuterated in 80 min. In contrast to these, the absorption spectra of OH groups of deuterated DB1, DB14, DB15, DB53, DR75 and DR79 products indicate that in these dye products, there exist some intra- and inter-molecular hydrogen bonding. Although the degree of deuteration is lower than that of FB and DR28 products and characteristically depends on the dye used, the observed band near to 3,478–3,480, 3,438–3,442 and 3,339–3,342 cm−1 indicate that the structure of these products has conceived cellulose II crystallinity. In this case we may consider two causes: (a) dye-cellulose interaction does not occur in all region of microfibril; and (b) during washing loosely bound dye molecules are washed off, as a result some of the product may be regenerated.
The region where a dye molecule is dropped out seem to occur cellulose II which crystallize during rinsing or drying process, as a result the diffusion of D2O is very difficult in that region. Judging deuteration results of the product, it can suggested that the direct binding of the cellulose sheets as well as intra- and inter-molecular hydrogen bonding of the cellulose sheet in the product do not exist as it does in the MC. Increased diffusion of D2O in the product structure due to lack of hydrogen bonding between the (100) planes supports that the model structure (Fig. 4) is ideal. The easy deuteration of accessible OH groups in the product reveals that the amount of accessible OH groups is always higher than that of MC or cellulose II as well as of regenerated cellulose from the product. The prevention of hydrogen bond formation by the dye molecule by the inclusion of the dye between cellulose sheets of the product corresponding to the (100) plane in the X-ray diffraction, also support the X-ray model structure of complex formation.
Mechanism of crystalline structure formation in the regenerated cellulose
It is clear from the X-ray diffraction, solid state 13C NMR and IR spectroscopies results that the products obtained in the presence of direct dyes, such as FB, DB1, DB14, DB15, DB53, DR28, DR75 and DR79, are crystalline, and the dye molecule is included between the monolayer of cellulose sheets corresponding to the (\( 1\bar{1}0 \)) plane of X-ray diffraction. If the dye is extracted from the product by EtOH and NaOH procedure, the structure of the regenerated cellulose is dependent on the dye used in the culture medium. Tables 5 and 6 show that celluloses Iβ and IVI are regenerated from the FB and DR28 products respectively, and cellulose II is regenerated from each of the DB1, DB14, DB15, DB53, DR75 and DR79 products, but the fine structure of the latter products is different from one another. Furthermore, the crystallinity of these regenerated celluloses are lower than that of the corresponding control celluloses (Table 7).
X-ray diffraction pattern of FB and DR28 celluloses gives the characteristic crystals of celluloses I and IV, respectively. Although X-ray diffraction of FB and DR28 celluloses are two typical celluloses I and IV, it is very difficult to characterize their family without IR and 13C NMR. In our previous article, we have reported that FB cellulose is cellulose Iβ (Mondal and Kai 2001). Hayashi et al. (1987) suggested that chemical shifts of C4 and C6 at about 66.3–67.0 and 63.0–64.0 ppm are due to cellulose IVI and IVII, respectively. Thus, the 13C NMR spectra of DR28 cellulose and its chemical shifts confirms that it is cellulose IVI crystal (Table 6) which is also supported by the IR results. Assigned IR spectra of the hydrogen bonding near 3,425 and 3,341 cm−1 support the 13C NMR results that the DR28 cellulose is cellulose IVI (Kai and Koseki 1985). The development of intra- and inter-molecular hydrogen bondings assigned by IR, and the splitting of Cl and C4 lines, and the chemical shift of C6 line at 63.0–64.0 ppm characterized by 13C NMR (Table 6) in each DB1, DB14, DB15, DB53, DR75 and DR79 celluloses are in a good agreement with the X-ray results of cellulose II. Although the regenerated cellulose from these products are cellulose II, the weaker downfield peak of Cl carbon indicates that the hydrogen bonding pattern in DB1, DB14, DB15 and DB53 celluloses are different to that of DR75 and DR79 celluloses. According to Horii et al. (1982), the downfield peak of the corresponding carbon indicate the condition of hydrogen bonding. The weaker downfield peak of Cl carbon of DB1, DB14, DB15 and DB53 celluloses than that of DR75 and DR79 celluloses suggests that the hydrogen bonding pattern in the former celluloses is weaker than that of latter celluloses, which have similar pattern to cellulose II. Higher crystallinity in X-ray results and lower accessible OH groups (i.e. higher non-accessible OH groups) in deuteration-IR results strongly support the stronger hydrogen bond formation in DR75 and DR79 celluloses (Table 8). On the other hand, Hayashi et al. (1987) reported from their solid state 13C NMR and IR results that bands or signals related to the chain conformation were singular (little or no splitted) in cellulose I family and doublets (splitted into two peaks) in the cellulose II family. This is consistent with two chain conformations, i.e. “bend” for I family and “bend and twisted” for II family, that have one and two types of glucosidic linkage. Thus, the single or little splitted peaks of FB and DR28 celluloses suggest that the chain conformation of FB and DR28 celluloses is “bend” and more symmetric. Similarly, the doublets or two peaks of DB1, DB14, DB15, DB53, DR75 and DR79 celluloses suggest that the chain conformation of these celluloses is “bend and twisted” and asymmetric. This discussion confirms that structures of FB and DR28 celluloses are cellulose Iβ and IVI, respectively, and that of other celluloses are cellulose II.
Influence of direct dyes on the nascent structure
A number of direct dye are selected to clarify the effect on the nascent structure of MC. These are FB, DB1, DB14, DB15, DB53, DR28, DR75 and DR79. When A. xylinum was cultured in the presence of each of them, the normal ribbon formation become hindered (Haigler and Chanzy 1988) and produced an altered product in which dye molecule was included between the monolayer of cellulose sheet corresponding to the (\( 1\bar{1}0 \)) plane (Haigler and Chanzy 1988; Kai and Mondal 1996, 1997; Kai 1984b). However, cellulose regenerated from the altered product was a cellulose allomorph other than native MC whose unit cell type was Iα. Celluloses Iβ and IVI were regenerated from FB and DR28 products, respectively, and cellulose II was regenerated from each DB1, DB14, DB15, DB53, DR75, DR79 product depending on the characteristic of direct dye used. These results are due to the easy of dye-cellulose interactions in the nascent stage and rearrangement of intra- and inter-molecular hydrogen bondings produced during dye extraction. Which factors of dye is mainly responsible for the transformation of Iα rich MC to other cellulose allomorphs will be discussed in latter.
The structure of FB is different among the direct dyes used. It has two sulfonate groups in its stilbene skeletal backbone and two hydroxyethyl amino groups attached to triazinyl group in each side of stilbene (Table 9). The molecular weight of FB is 960.9 and the cellulose regenerated from FB product is cellulose Iβ. Again, all direct dyes DR28, DB1, DB14, DB15 and DB53 have same biphenylenebis (azo) skeletal backbone structure of DR28, but contain different substituent groups and have different position of the same substituent groups in the backbone. The smallest direct dye among the dyes used is DR28 having two sulfonate (–SO3Na) and two amine (–HN2) groups, and its molecular weight is 696.67. On the other hand, DB1 has two sulfonate, two hydroxyl (–OH) and two methoxy (–OCH3) groups more than that of DR28 and its molecular weight is 992.82. The regenerated cellulose from each DR28 and DB1 product are cellulose IVI and II, respectively. If the interaction of sulfonate and amine groups with cellulose microfibril produces cellulose IVI from DR28 product, then interaction of additional groups in DB1 produces cellulose II from DB1 product. There are no differences between the structure of DB1 and DB53 except the position of methoxy groups in DB1. Instead of methoxy groups, there are methyl groups in the same position in DB53. However, the structure of DB1 and DB53 products is similar, and cellulose regenerated from each of them is cellulose II. As the regenerated celluloses from both DB1 and DB53 are cellulose II, it is assumed that methoxy and methyl groups have no remarkable effect on the product as well as on regenerated celluloses from them. Similar effect is observed in the case of DB14 and DB15. Now we may compare the effect of DB1 and DB15 on the nascent MC. Between these dyes, there are no other differences except only the position of sulfonate groups. That is, positions of sulfonate groups are, at 5 and 7 in the napthalene nucleus in DB1, and at 3 and 6 in the naphthalene nucleus in DB15. As positions of sulfonate groups in these dyes are different, the uniplanar orientation of DB1 product membrane measured by X-ray diffraction is different to that of DB15 product membrane. Although the cellulose regenerated from these dyes is cellulose II, the different absorptions due to the hydrogen bonding observed by deuteration-IR suggest that their fine structures are not same. Similar effect is observed in the case of DB14 and DB53. On the other hand, as the sulfonate groups position of DB1 and DB53 are same, but their molecular weights are different (992.8 for DB1 and 960.82 for DB53), there are no observed effect of these dyes on the product structure as well as their regenerated celluloses. The molecular weight of DR79 is 1,050.89 and is maximum among the dyes used. The regenerated cellulose from DR79 product is cellulose II. From these facts, it has been suggested that only molecular weight has no effect on the nascent MC.
From the Table 9, it can be seen that DR75 and DR79 have common four sulfonate, two hydroxyl and –NHCONH– groups, whereas DR75 has two uncommon amine groups and DR79 has two uncommon methoxy and two methyl groups. The skeletal backbone structure of both DR75 and DR79 is ureylenebis(naphthylazo)benzene, but its effect on the nascent MC is closely similar to each other. So, it can be taken into account that uncommon groups, such as methyl, methoxy and amine groups have no extra effect on the nascent MC. But these dyes have different effect on the uniplanar orientation of the product membrane as well as on the fine structure of their regenerated celluloses compared to dyes having four sulfonate groups in biphenylenebis(azo) skeletal structure. It is assumed that these different effects of DR75 and DR79 are due to the interaction of ureylene group of dyes with nascent MC. However, among FB, DB1, DB14, DB15, DB53, DR28, DR75 and DR79, two sulfonate and two amino groups in FB and DR28 are responsible for producing celluloses Iβ and IVI, respectively, while four sulfonate and two hydroxyl and two amino groups in DB1, DB14, DB15 and DB53 are responsible for cellulose II, although the effect of amino groups is not clear.
From the above discussion, it is clear that the reactivity of sulfonate groups in a dye is the most important for the formation of form dye–cellulose complex (product) as well as for the structure of cellulose regenerated from it. In our experiment, cellulose II was obtained from the product of dyes having four sulfonate groups, while celluloses Iβ and IVI were obtained from product of dyes (FB and DR28) having two sulfonate groups. In the former case, higher sulfonate groups of dye increased mainly the solubility toward water, while hydroxyl, amine, etc. groups increased the affinity toward the cellulose chains. During dye extraction from the products, higher hydrophilicity due to four sulfonate groups may affect the polarity as well as the conformation of the cellulose chains and, as a results, cellulose II is regenerated through the conformational change from DB1, DB14, DB15, DB53, DR75 and DR79 products. Similarly, lower hydrophilicity due to two sulfonate groups in FB and DR28 affects the conformation which produces celluloses Iβ and IVI (cellulose I family) from FB and DR28 products. Thus the results suggest that the interaction of nascent microfibril with two sulfonate groups containing direct dyes have cellulose I family formative ability and that four sulfonate groups containing dyes have cellulose II family formative ability. The other substituent groups in dye have little effect to alter cellulose structure. The order of the reactivity of substituent groups as observed in a dye are: sulfonate > hydroxyl > amine > methoxy ≥ methyl.
Conclusions
The product from Acetobacter-culture in the presence of a direct dye is in the crystalline complex composed of a dye and cellulose. The product has the structure in which the monomolecular layer of the dye molecule is incorporated between cellulose sheets corresponding to the (\( 1\bar{1}0 \)) plane of microbial cellulose. Cellulose regenerated from the product can have any one cellulose allomorph depending on the dye structure. Cellulose regenerated from FB and DR28 products is Cellulose Iβ and IVI, and that from each DB1, DB14, DB15, DB53, DR75 and DR79 product is Cellulose II. The dye molecules may bind along the glucan chains of nascent microbial cellulose through hydrogen bonding, dipolar interactions or van der Waals forces. X-ray diffraction, solid state 13C NMR and deuteration-IR study reveal that the dye molecule hinders to form hydrogen bonding in or between nascent cellulose chains or sheets in the product. During dye extraction, the conformation of cellulose chain and the alignment of cellulose chains in the sheets are rearranged with the development of hydrogen bonding corresponding to the characteristics of dye. It appears that the different interactions between direct dye and nascent cellulose depend mainly on the number as well as position of sulfonate groups in the dye. The nascent cellulose produced by A. xylinum cell is in the form of a microfibril in which hydrogen bonding is absent. But through the conformational change, this nascent cellulose forms the crystalline regenerated cellulose.
References
Aloni Y, Benziman M (1982) Intermediates of cellulose synthesis in Acetobacter. In: Brown RM Jr (ed) Cellulose and other natural polymer systems: biogenesis, structure and degradation. Plenum Press, New York, p 341
Benziman M, Palgi A (1970) Characterization and properties of the pyruvate phosphorylation system of Acetobacter xylinum. J Bacteriol 104:211–218
Benziman M, Haigler CH, Brown RM Jr, White AR, Cooper KM (1980) Cellulose biosynthesis: polymerization and crystallization are coupled processes in Acetobacter xylinum. Proc Natl Acad Sci USA 77:6678–6682
Brown RM Jr (1989) Bacterial cellulose. In: Kennedy JF, Phillips GO, Williams PA (eds) Cellulose structure and functional aspects. Ellis Horwood Ltd, Chichester, p 145
Brown Jr RM (1979) Biogenesis of natural polymer systems with reference to cellulose assembly and deposition. In: Walk WM (ed) Structure and biochemistry of natural biological systems. Proceedings of the third Philip-Morris science symposium, Philip-Morris, Richmond, Virgina, November 9, p 51
Brown RM Jr, Willson JH, Richardson CL (1976) Cellulose synthesis in Acetobacter xylinum: visualization of the sites of synthesis and direct measurements of the in vivo process. Proc Natl Acad Sci USA 73(12):4565–4569
Brown RM, Haigler CH, Suttie J, White AR, Roberts E, Smith C, Itoh T, Cooper K (1983) The biosynthesis and degradation of cellulose. Appl Polym Symp no 37, Wiley & Sons, Inc, New York, p 33
Chawla PR, Bajaj IB, Shrikant AS, Singhal RS (2009) Fermentative production of microbial cellulose. Food Technol Biotechnol 47:107–124
Colvin JR (1964) The biosynthesis of cellulose. In: Zimmermann MR (ed) The formation of wood in forest trees. Academic Press, New York, p 189
Delmer DP, Benziman M, Padan E (1982) Requirements for a membrane partially for cellulose synthesis in intact cells of Acetobacter xylinum. Proc Natl Acad Sci USA 79:5282–5286
Gardner KH, Blackwell J (1974) The structure of native cellulose. Biopolymers 13:1975
Haigler CH, Benziman M (1982) Biogenesis of cellulose I microfibrils occurs by cell-directed self-assembly in Acetobacter xylinum. In: Brown RM Jr (ed) Cellulose and other natural polymer systems: biogenesis, structure and degradation. Plenum Press, New York, p 273
Haigler CH, Chanzy H (1988) Electron diffraction analysis of the altered cellulose synthesized by Acetobacter xylinum in the presence of fluorescent brightening agents and direct dyes. J Ultrastruct Mol Struct Res 98:299–311
Haigler CH, Brown RM, Benziman M (1980) Calcofluor White ST alters the in vivo assembly of cellulose microfibrils. Science 210:903–906
Hayashi J, Kon H, Takai M, Hatano M, Nozawa T (1987) Possible cause of structural irrersibility between cellulose I and cellulose II families. In: Atalla RH (Ed) The structure of cellulose. ACS symposium series no 340, ACS, Washington, p 135
Hestrin S, Schramm M (1954) Synthesis of cellulose by A. xylinum: 2. Preparation of freeze-dried cells capable of polymerizing glucose to cellulose. Biochem J 58:345–352
Horii F, Hirai A, Kitamaru R (1982) Solid-state high-resolution 13C-NMR studies of regenerated cellulose samples with different crystallinities. Polym Bull 8:163–170
Isogai A (1994) Allomorphs of cellulose and other polysaccharides. In: Gilbert RD (ed) Cellulosic polymers, blends and composites. Hanser Publications, Inc., New York, 1994, p 1
Kai A (1984a) The structure of the nascent fibril produced by Acetobacter xylinum: the X-ray diffraction diagram of cellulose produced in the presence of a fluorescent brightener. Makromol Chem Rapid Commun 5:307–310
Kai A (1984b) The structure of the nascent fibril produced by Acetobacter xylinum: the lattice spacing of cellulose produced in the presence of a fluorescent brightener. Makromol Chem Rapid Commun 5:653–655
Kai A, Kitamura H (1985) The structure of cellulose produced by Acetobacter xylinum in the presence of fluorescent brightener. The influence of concentration of a brightener in the medium on the structure of cellulose. Bull Chem Soc Jpn 58:2860–2862
Kai A, Koseki T (1985) The structure and time evolution of a cellulose sheet in the nascent fibril produced by Acetobacter xylinum. Makromol Chem 186:2609–2614
Kai A, Mondal MdIH (1996) Structure of nascent microbial cellulose and its properties. In: Yalpany M (ed) Biomedical functions and biotechnology of natural and artificial polymers, vol 3. ATL Press, Science Publishers, USA, p 77
Kai A, Mondal MdIH (1997) Influence of substituent of direct dye having bisphenylene bis(azo) skeletal structure on structure of nascent cellulose produced by Acetobacter xylinum. [I]: different influence of Direct Red 28, Blue 1 and 15 on nascent structure. Int J Biol Macromol 20:221–231
Kai A, Kogusuri J, Koseki T, Kitamura N (1989) The nascent structure of bacterial cellulose. In: Schuerch C (ed) Cellulose and wood: chemistry and technology. Wiley and Sons, Inc., New York, p 507
Kai A, Kido H, Ishida N (1990) The effect of a direct dye on the formation process of the structure of bacterial cellulose. Chem Lett 949–952
Kai A, Xu P, Horii F, Hu S (1994) C.p./m.a.s. 13C n.m.r. study on microbial cellulose-fluorescent brightener complexes. Polymer 35:75–79
Maeda H, Ishida N (1967) Specification of binding of hexapyranosyl polysaccharides with fluorescent brightener. J Biochem 62:276–278
Marchessault RH, Sarko A (1967) X-ray structure of polysaccharides. Adv Carbohydr Chem 22:421–482
Marchessault RH, Sundararajan PR (1985) Cellulose. In: Aspinal GO (ed) The polysaccharide, vol 2. Academic Press, New York, p 11
Mondal MdIH, Kai A (1998) Structure of nascent microbial cellulose I. Effects of methyl and methoxy groups of Direct Blue 1 and 53 on nascent microbial cellulose. Polymer J 30:78–83
Mondal MdIH, Kai A (1999) Influence of substitution of direct dye having biphenylenebis-(azo) skeletal structure on nascent cellulose produced by Acetobacter xylinum [II]. J Appl Polym Sci 71:1007–1015
Mondal MdIH, Kai A (2001) Control of the crystal structure of microbial cellulose during nascent stage. J Appl Polym Sci 79:1726–1734
Moosavi-Nasab M, Yousefi A (2011) Biotechnological production of cellulose by Gluconacetobacter xylinus from agricultural waste. Iran J Biotechnol 9:94–101
Oh SY, Yoo DI, Shin Y, Kim HC, Kim HY, Chung YS, Park WH, Youk JH (2005) Crystalline structure analysis of cellulose treated with sodium hydroxide and carbondioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr Res 340:2376–2391
Ranby BG (1952) The mercerization of cellulose. I. A thermodynamic discussion. Acta Chem Scan 6:101–115
Rattee ID, Breuer MM (1974) The physical chemistry of dye adsorption. Academic Press, New York, p 180
Ross P, Aloni Y, Weinhouse H, Michaeli D, Weinberger-Ohana P, Meyer R, Benziman M (1985) An unusual guanyl oligonucleotide regulates cellulose synthesis in Acetobacter xylinum. FEBS Lett 186:191–196
Ross P, Aloni Y, Weinhouse H, Michaeli D, Weinberger-Ohana P, Meyer R, Benziman M (1986) Control of cellulose synthesis in A. xylinum. A unique guanyl oligonucleotide is the intermediate activator of cellulose synthase. Carbohydr Res 149:101–117
Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Meyer R, Braun S, Vroom Ede, van der Marel GA, van Boom JH, Benziman M (1987) Regulation of cellulose synthesis in A. xylinum by a cyclic diguanylic acid. Nature 325:279–281
Ross P, Mayer R, Benzinian M (1991) Cellulose biosynthesis and function in bacteria. Microbiol Rev 55:35–58
Sarko A (1978) What is the crystalline structure of cellulose. TAPPI 61:59–61
Sarko H, Muggli R (1974) Packing analysis of carbohydrates and polysaccharides. III. Valonia cellulose and cellulose II. Macromolecules 7:486–494
Schramm M, Hestrin S (1954) Factors affecting production of cellulose at the air liquid interface of a culture of A. xylinum. J Gen Microbiol 11:123–129
Takai M, Tsuta Y, Hayashi J, Watanabe S (1975) Biosynthesis of cellulose by Acetobacter xylinum. III. X-ray studies of preferential orientation of the crystallites in a bacterial cellulose membrane. Polymer J 7:157–164
Torchia DA (1978) The measurement of proton-enhanced carbon-13 T1 values by a method which suppresses artifacts. J Magn Reson 30:613–616
Walton AG, Blackwell J (1973) Biopolymers. Academic Press, New York, p 468
Yan Z, Chen S, Wang H, Wang B, Wang C, Jiang J (2008) Cellulose synthesized by Acetobacter xylinum in the presence of multi-walled carbon nanotubes. Carbohydr Res 343:73–80
Zaar K (1977) The biosynthesis of cellulose by A. xylinum. Cytobiologie 16:1–15
Zaar K (1979) Visualization of pores (export sites) correlated with cellulose production in the envelope of the gram-negative bacterium A. xylinum. J Cell Biol 80:773–777
Zhao H, Kwak JH, Zhang ZC, Brown HM, Arey BW, Holladay JE (2007) Studying cellulose fiber structure by SEM, XRD, NMR and acid hydrolysis. Carbohydr Polym 68:235–241
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Mondal, M.I.H. Mechanism of structure formation of microbial cellulose during nascent stage. Cellulose 20, 1073–1088 (2013). https://doi.org/10.1007/s10570-013-9880-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-013-9880-z