
Abstract
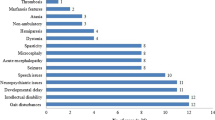
The most common inborn error of cobalamin (cbl) metabolism in China is the cblC type characterized by combined methylmalonic acidemia and hyperhomocysteinemia. The clinical presentation is relatively nonspecific, such as feeding difficulty, recurrent vomiting, hypotonia, lethargy, seizures, progressive developmental delay, and mental retardation, together with anemia and metabolic acidosis. More specific biochemical findings include high levels of propionylcarnitine (C3), free carnitine (C3/C0), and acetylcarnitine (C3/C2) measured by tandem mass spectrometry (MS/MS), elevation of methylmalonic acid (MMA) measured by gas chromatography–mass spectrometry (GC-MS), and increased total homocysteine with normal or decreased methionine. We report on 50 Chinese patients with combined methylmalonic acidemia and hyperhomocysteinemia. Forty-six belonged to the cblC complementation group. Mutation analysis of the MMACHC gene was performed to characterize the mutational spectrum of cblC deficiency, and 17 different mutations were found. Most were clustered in exons 3 and 4, accounting for 91.3% of all mutant alleles. Two mutations were novel, namely, c.315 C>G (p.Y105X) and c.470 G>C(p.W157S). In terms of genotype–phenotype correlation, the c.609 G>A mutation was associated with early-onset disease when homozygous. Unlike previous reports from other populations, c.609 G>A (p.W203X) was the most frequent cblC mutation detected in our study of Chinese patients, affecting 51 of 92 MMACHC alleles (55.4%). The high prevalence of this nonsense mutation could have potential therapeutic significance for Chinese cblC patients. Besides traditional approaches consisting of hydroxocobalamin injections, carnitine, betaine, and protein restriction, novel drugs that target premature termination codons may have a role in the future.
Similar content being viewed by others

Abbreviations
- AdoCbl:
-
adenosylcobalamin
- cblC:
-
cobalamin C
- C3:
-
propionylcarnitine
- C0:
-
free carnitine
- C2:
-
acetylcarnitine
- C24:
-
tetracosane
- GC-MS:
-
gas chromatography–mass spectrometry
- MeCbl:
-
methylcobalamin
- MGA:
-
margaric acid
- MMA:
-
methylmalonic acid
- MS/MS:
-
tandem mass spectrometry
- PCR:
-
polymerase chain reaction
- TA:
-
DL-tropic acid
- VitB12 :
-
vitamin B12 cyanocobalamin
References
Ben-Omran TI, Wong H, Blaser S et al (2007) Late-onset cobalamin-C disorder: a challenging diagnosis. Am J Med Genet A 143:979–984
Carrillo-Carrasco N, Sloan J, Valle D et al (2009) Hydroxocobalamin dose escalation improves metabolic control in cblC. J Inherit Metab Dis 32:728–731
Coelho D, Suormala T, Stucki M et al (2008) Gene identification for the cblD defect of vitamin B12 metabolism. N Engl J Med 358:1454–1464
Cusmano-Ozog K, Lorey F, Levine S et al (2007) Cobalamin C disease identified and expanded newborn screening: the California experience. J Investig Med 55:S90
den Dunnen JT, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15:7–12
Froese DS, Zhang J, Healy S et al (2009) Mechanism of vitamin B12-responsiveness in cblC methylmalonic aciduria with homocystinuria. Mol Genet Metab 98:338–43
Han LS, Gao XL, Ye J et al (2005) Application of tandem mass spectrometry in diagnosis of organic acidemias. Zhonghua Er Ke Za Zhi 43:325–330
Han LS, Ye J, Qiu WJ et al (2007) Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: a four-year report. J Inherit Metab Dis 30:507–514
Han LS, Ye J, Qiu WJ et al (2008) Diagnosis of inborn errors of metabolism using tandem mass spectrometry and gas chromatography mass spectrometry. Zhonghua Yi Xue Za Zhi 88:2122–2126
Hannibal L, Kim J, Brasch NE et al (2009) Processing of alkylcobalamins in mammalian cells: A role for the MMACHC (cblC) gene product. Mol Genet Metab 97:260–266
Kerem E, Hirawat S, Armoni S et al (2008) Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 30(372):719–727
Kuhara T (2002) Diagnosis and monitoring of inborn errors of metabolism using urease-pretreatment of urine, isotope dilution, and gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 781:497–517
Kim J, Gherasim C, Banerjee R (2008) Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci USA 105:14551–14554
Lerner-Ellis JP, Anastasio N, Liu J et al (2009) Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat 30:1072–1081
Lerner-Ellis JP, Tirone JC, Pawelek PD et al (2006) Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet 38:93–100
Morel CF, Lerner-Ellis JP, Rosenblatt DS (2006) Combined methylmalonic aciduria and homocystinuria (cblC): phenotype–genotype correlations and ethnic-specific observations. Mol Genet Metab 88:315–321
Nogueira C, Aiello C, Cerone R et al (2008) Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab 93:475–480
Rosenblatt DS, Aspler AL, Shevell MI et al (1997) Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (cblC). J Inherit Metab Dis 20:528–538
Rossi A, Cerone R, Biancheri R et al (2001) Early-onset combined methylmalonic aciduria and homocystinuria: Neuroradiologic findings. AJNR Am J Neuroradiol 22:554–563
Rutsch F, Gailus S, Miousse IR et al (2009) Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat Genet 41:234–239
Smith SE, Kinney HC, Swoboda KJ et al (2006) Subacute combined degeneration of the spinal cord in cblC disorder despite treatment with B12. Mol Genet Metab 88:138–145
Thiele J, Van Raamsdonk JM (2006) Gene discovery in methylmalonic aciduria and homocystinuria. Clin Genet 69:402–403
Wang F, Han LS, Hu YH et al (2009) Analysis of gene mutations in Chinese patients with methylmalonic acidemia and homocysteinemia. Zhonghua Er Ke Za Zhi 47:189–193
Welch EM, Barton ER, Zhuo J et al (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature 447:87–91
Yuen YP, Lai CK, Chan YW et al (2007) DNA-based diagnosis of methylmalonic aciduria and homocystinuria, cblC type in a Chinese patient presenting with mild developmental delay. Clin Chim Acta 375:171–172
Zhang Y, Song JQ, Liu P et al (2007) Clinical studies on fifty-seven Chinese patients with combined methylmalonic aciduria and homocysteinemia. Zhonghua Er Ke Za Zhi 45:513–517
Zytkovicz TH, Fitzgerald EF, Marsden D et al (2001) Tandem mass spectrometry analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem 47:1945–1955
Acknowledgments
We are very grateful to the patients, their families, and the pediatricians from the Peking University First Hospital and Guangdong Children’s Hospital who sent patient data and DNA samples. This study was supported by Technology Pillar Program in the Eleventh Five-year Plan Period (2006AB105A05, 2006BA105A07); National High Technology Research and Development Program (2007AA02Z447); Key Subject of Shanghai Municipality (2008ZD001); Scientific Research Foundation Project of Shanghai Municipality (2006043).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Matthias Baumgartner
Competing interests: None declared.
Rights and permissions
About this article
Cite this article
Wang, F., Han, L., Yang, Y. et al. Clinical, biochemical, and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia (cblC type) in China. J Inherit Metab Dis 33 (Suppl 3), 435–442 (2010). https://doi.org/10.1007/s10545-010-9217-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9217-0