
Abstract
Background
Methylmalonic acidemia with homocystinuria is caused by a rare inborn error of vitamin B12 (cobalamin) metabolism. There are four complementation classes of cobalamin defects cblC, cblD, cblF, and cblJ that are responsible for combined methylmalonic acidemia and homocystinuria.
Case presentation
We report a case of a Pakistani family composed of six children diagnosed with methylmalonic acidemia and homocystinuria (MMA + HCU). Mutation analysis of siblings revealed a variable combination of c.394C>T mutation in the MMACHC gene and c.262_264del in CD320 gene. All siblings had variable age of onset, initial symptomatology, the severity of disease, and response to treatment. The maximum age of presentation was 6.5 years and the minimum age was at birth. The spectrum of symptoms ranged from a subtle learning disability to global developmental delay within the same family. None of them had a seizure disorder, megaloblastic anemia, visual disturbance, and vascular events.CD320 defect itself is very rare, and only 12 cases have been reported so far. We report six cases, four of them had homozygous MMACHC variant c.394C>T and the other two had heterozygous MMACHC mutations in c.394C>T and c.262_264del in CD 320 genes. To date, neither such case has been reported in the literature or this compound heterozygous mutation.
Conclusion
Our case report emphasizes that the diagnosis of inherited metabolic disorder in a child obviates the need to screen all siblings for the same disorder.
Similar content being viewed by others


Background
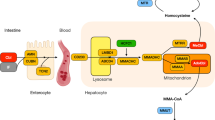
Cobalamin is vitamin B12, an essential water-soluble vitamin, which has a vital role in the functioning of several enzymes in our body [1]. After ingestion, cobalamin binds to intrinsic factors and enters the enterocytes. In circulation, it is complexed with transcobalamin and taken into the cells by the transcobalamin receptor. This receptor is encoded by CD320 located on the short arm of chromosome 19 [2]. Defect in transcobalamin receptor and intracellular cobalamin metabolism results in methylmalonic acidemia and homocystinuria [3]. The MMACHC protein is responsible for the intracellular trafficking of cobalamin. It is involved in the decyanation of cyanocobalamin as well as in the dealkylation of alkylcobalamins through a glutathione transferase activity, leading to the formation of adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl) [4]. Adenosylcobalamin (AdoCbl) is a cofactor in the metabolism of methyl malonyl co A into succinyl co A which is the final step in the metabolism of amino acids (valine, leucine, threonine, and methionine) odd chain fatty acids and cholesterol. Methylcobalamin (MeCbl) acts as a cofactor in the re-methylation of homocysteine to methionine [5].
We discussed a family of 6 siblings having symptomatic methylmalonic acidemia and homocystinuria with a novel combination of defects in the MMACHC and CD 320 gene. All siblings presented with variable initial symptomatology, age of onset, and severity of symptoms and showed variable response to treatment.
Case presentation
This family first sought medical attention when their eldest daughter suffered from behavioral changes at 6 years of life. At that time she became aphasic, isolated, non-interactive, lost interest in her surroundings, poor appetite, and lost weight. She was worked up for tuberculous meningitis which was normal, and she was discharged home on multivitamins. Her symptoms deteriorated further, her gait became ataxic, and gradually she lost ambulation at 10 years of age. Her magnetic resonance imaging of the thoracolumbar spine performed was normal, but MRI brain revealed bilateral symmetrical hyper-intensities in periventricular and peritrigonal areas suggestive of demyelination due to a metabolic disorder. She was worked up for metabolic disorder; her laboratory parameters showed compensated metabolic acidosis, mild ketoacidosis, normal serum ammonia and lactate level. Her urinary gas chromatography mass spectrometry (GCMS) revealed a peak of methylmalonic acid (MMA). She was started on oral vitamin B12 (methylcobalamin), carnitine, and protein restriction with little improvement. When she visited us she was on this treatment for the last 6 months.
Her parents were first cousins; she was born at term with normal antenatal, natal, and postnatal periods. She achieved all her milestones appropriately according to age and was a student of grade 2 when she started to have the first symptom. Furthermore, she has left school because of illness. Currently she is 11 years of age.
Her family history revealed that her younger sister (sibling 2) has a global developmental delay. She was born full-term with normal antenatal natal and postnatal history. She did not achieve any of her motor, language, and communication skills. Likewise, she is hypotonic, microcephalic, and bedridden. The family has been told that she has cerebral palsy due to some adverse event at birth, but the parents denied any such adverse event. Currently, she is 8 years of age; her clinical and metabolic details are shown in Table 1.
3rd sister (sibling 3) developed symptoms at 6.5 years of age in the form of psychiatric illness. She was phobic about unseen things, had bouts of laughter and cries; otherwise, she was well, walking around having normal hearing and speech.
4th sibling was a 5-year-old boy and according to his parents, he was well except for his disinterest in studies and learning disability.
The youngest of all were identical twin boys aged 3.5 years. One of them has developed symptoms one month back with regression in motor and language skills.
There was history of the death of one sibling at 15 days of life as sudden infant death and one early trimester miscarriage.
The common thing in all siblings was that none of them had a single episode of metabolic crisis, blood transfusion, seizures, visual loss despite the variable onset of symptomatology, age of onset, and severity of symptoms.
Detailed examination, laboratory parameters, and genetic mutation of all siblings are shown in Table 1.
Baseline investigations for all revealed hypochromic microcytic anemia with mean Hb of 10.1 ± 2.01 g/dl, mean platelets of 250,000 ± 25,000/ul, and TLC of 4500 ± 213/ul. Liver function tests, serum ammonia, renal function tests, serum calcium, magnesium, and CSF were normal. The metabolic screen revealed raised serum methylmalonic level (mean 7100 ± 205 nmol/L normal = less than 600 nmol/L) along with raised levels of homocysteine levels (mean 109.33 ± 42.35 umol/L in normal = less than 15 umol/L) in urine confirming the diagnosis of methylmalonic acidemia with homocystinuria. Mean serum level of cobalamin (695 ± 82 umol/l; reference range 150–600) and cysteine (9.1 ± 2.1; reference range 4.7–14.1) was in the normal range. Magnetic resonance imaging (MRI) scan of the brain showed variable type and severity of brain involvement in siblings 1, 2, 5 as shown in Fig. 1.

Magnetic resonance imaging and magnetic resonance spectroscopy of sibling 1. a Sibling 1: 12 year female. Axial FLAIR and T2W images. Bilateral symmetrical white matter hyperintensities in peri-ventricular and peritrigonal white matter. b Sibling 2. 06 year female: Axial T2W image—bilateral increased white matter intensities in centrum semi-ovale and peritrigonal white matter. Ventricular and extra-ventricular CSF spaces are prominent. c Sibling 5: 2.5 years male—axial T2W images: bilateral symmetrical increased white matter intensities with mild hydrocephalus. MRS multi-voxel technique. Increased Choline peak with abnormal Choline creatine ratio favors demyelinating disease
Methylmalonic acidemia advanced panel for all siblings and mother (father has expired) was performed at Centogene Germany, which included the entire coding region of ABCD4, ACSF3, LMBRD1, MCEE, MLYCD, MMAA. MMAB, MMACHC, MMADHC, MTR, MTRR genes. Four siblings have homozygous pathogenic MMACHC variant c.394C>T P.(Arg132*) which creates a premature stop codon and causes cobalamin c disease (cblC), plus they were carrier for pathogenic variant CD320,c.262_264del p.(glu88del) which creates an in-frame deletion of three base pairs in exon2 and is responsible for transcobalamin receptor defect (Fig. 2).

Pedigree of family
Two siblings and mother had a heterozygous mutation for both above-mentioned pathogenic variants. Mother aged 40 years was found to have forgetfulness and insomnia when inquired about her symptoms in detail. Her systemic examination was normal including the central nervous system. We performed her metabolic workup afterward which revealed elevated Methylmalonic level (mean 625 nmol/L) along with raised levels of homocysteine levels (240 umol/L) in serum confirming the diagnosis of methylmalonic acidemia with homocystinuria.
They are being managed with Inj. hydroxocobalamin 1 mg intramuscular weekly, tablet Betaine oral daily, oral carnitine, and folic acid. Detailed parental counseling was done regarding the diagnosis, progression of disease, outcome, and adherence to treatment. The initial response to our treatment is shown in Table 1. All siblings are on our close follow-up and post-treatment levels of MMA showed marked improvement (Table 1). Mother sleep has improved after treatment.
Discussion
Methylmalonic acidemia with homocystinuria (MMA with HCU) is caused by defects in the intracellular metabolism of cobalamin that interfere with the formation of methylcobalamin and adenosylcobalamin [6]. There are seven defects of intracellular cobalamin metabolism, among them cblC, cblD, cblF, cblJ, cblX causes MMA with HCU. Cobalamin c (cblC) defect is the most common inborn error of intracellular cobalamin metabolism with over 550 patients has been diagnosed so far [7]. Transcobalamin receptor defect causing MMA and HCU has recently been described in the literature and 12 patients have been reported so far [8]. This is the first-ever case of a family having dual pathogenic MMACHC and CD320 variants responsible for MMA and HCU. The MMACHC variant c.394C>T has been described previously as causing late-onset disease [9]. In our case four of among six affected kids were homozygous for this mutation, and they developed symptoms beyond one year of life which is consistent with the literature. The other two symptomatic siblings were heterozygous for MMACHC and CD320 variants. All previously reported CD320 variants were asymptomatic and diagnosed on newborn screening except one reported by Karth et al. [10] and all of the reported cases are homozygous for c.262_264delGAG except one reported by Anastasio et al. [11]. In our case these two siblings were carrying one mutation for cblC and one for transcobalamin receptor defect, having carrier status for two different genes responsible for the same disease. The mother also had the same combination and had mild symptoms.
The fact that mother and siblings 2, 4 had clinical and biochemical manifestations suggests that double heterozygosity is causing phenotype in them though CD320 biochemical manifestations in humans and mice are mild and CD320 and MMACHC do not interact. There may be another missing intronic or regulatory MMACHC variant that can be detected through whole-genome sequencing. There is a need to search further for compound epigenetic–genetic heterozygosity in patients with typical disease manifestation and genetic heterozygosity in disease-causing genes located in other gene trios [12].
Being an autosomal recessive disorder, there is a 1:4 risk of developing the disease in each pregnancy of carrier parents. Our case is unique in which the whole family is affected with no normal siblings.
All of them developed the disease at different ages, of variable symptoms and severity, progression and outcome, though they had inherited the same genetic mutation since birth and had the same environmental factors. The first affected sibling was sibling 2; currently, she is 8 years of age with global developmental delay. She had an uneventful full-term birth, fed on breast milk, and never had an episode of respiratory distress, unconsciousness, and seizures. But she failed to achieve any of her developmental milestones, had feeding difficulty and poor growth. She was labeled as having cerebral palsy due to some adverse event at birth and never investigated for her condition. Among all siblings, Sibling 2 has classical early-onset Cblc while others have the late-onset disease.
The literature revealed that the patient who presents early-onset disease presents before the first birthday with metabolic crisis and neurological deterioration like MMA while the late-onset disease can present any time after 1st year with neurologic and psychiatric disturbances with or without thrombosis. Early-onset cblC is a much more severe form with the clinical outcome being generally poor despite treatment and metabolic management. The late-onset form has a much more favorable outcome, including reversal of neurological and psychiatric symptoms if treatment is initiated early [13].
The patients with cblC disease display a wide spectrum of clinical manifestations as we described in our patients ranging from subtle learning difficulty to neuropsychiatric symptoms and from neuro-regression to global developmental delay. In contrary to the literature we did not find hematologic, ophthalmologic dermatologic and skeletal abnormalities in our patients. Our case also eluded that early onset of the disease has severe clinical manifestation and rapid worsening as compared to late-onset as the eldest sibling who was first to be diagnosed with the disease had progressive worsening over a period of 5 years [14].
Brain magnetic resonance imaging (MRI) features of methylmalonic aciduria and homocystinuria reveal changes in the basal ganglia along with Hydrocephalus and diffuse supratentorial white matter edema as the main MRI features; this was also seen in our case [14].
Cyanocobalamin (CNC bl) is the most common commercially available form of cobalamin and hydroxocobalamin (OHCbl), methylcobalamin (MeCbl), and 5′-deoxyadenosylcobalamin (AdoCbl) are the naturally occurring forms. In cblC disease, the circulating cyanocobalamin concentration is usually normal with a very low intracellular concentration of methylcobalamin and adenosylcobalamin. A high dose of cyanocobalamin (CNC bl) causes a tenfold increase in c circulating cobalamin with only a small increase in intracellular cobalamin as compared to Hydroxocobalamin (OHCbl). Studies have revealed the efficacious role of OHCbl as supplements in cblC for normal OHCbl within the cellular milieu [15].
Parenteral and oral cobalamin has been used for Transcobalamin receptor defect with the same efficacy resulting in normal serum levels of MMA and HCU [16]. Literature documents poor long-term outcomes in early-onset patients with progression of visual and neurological impairment. We found a good short-term outcome and will follow the family for a long-term outcome.
Conclusion
This is a first case report of a family of six affected siblings with MMA+ HCU with variable phenotype. This case report also describes a novel cobalamin receptor defect CD320 in combination with methylmalonic acidemia type C. We would like to emphasize that the diagnosis of inherited metabolic disorder in a child obviates the need to screen all siblings for the same disorder. Moreover, patients with typical disease manifestation and genetic heterozygosity in disease-causing genes located in other gene trios need further studies for compound epigenetic–genetic heterozygosity.
Availability of data and materials
Not applicable.
Abbreviations
- ABCD4:
-
ATP Binding Cassette Subfamily D Member 4
- ACSF3:
-
Acyl-CoA Synthetase Family Member 3
- CD320:
-
Transcobalamin receptor defect 320
- HCU:
-
Homocystinuria
- HB:
-
Hemoglobin
- LMBRD1:
-
Lysosomal cobalamin transport escort protein containing domain 1
- MCEE:
-
Methylmalonyl-CoA Epimerase
- MMA:
-
Methylmalonic acidemia
- MMACHC:
-
MMA DUE TO cblC deficiency: Methylmalonic aciduria type C
- MLYCD:
-
Malonyl-CoA decarboxylase
- MMAA:
-
Methylmalonic aciduria type A
- MMAB:
-
Methylmalonic aciduria type B
- MMADHC:
-
Methylmalonic aciduria type D
- MTR:
-
5-Methyltetrahydrofolate–homocysteine methyltransferase
- MTRR:
-
Methionine synthase reductase
- TLC:
-
Total leukocyte count
References
Tsai AC, Morel CF, Scharer G, Yang M, Lerner-Ellis JP, Rosenblatt DS et al (2007) Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet 143:2430–2434
Keller R, Chrastina P, Pavlíková M, Gouveia S, Ribes A, Kölker S et al (2019) Newborn screening for homocystinurias: recent recommendations versus current practice. J Inherit Metab Dis 42:128–139
Ben-Omran TI, Wong H, Blaser S, Feigenbaum A (2007) Late-onset cobalamin-C disorder: a challenging diagnosis. Am J Med Genet A 143:979–984
Brown KL (2005) Chemistry and enzymology of vitamin B12. Chem Rev 105:2075–2149
Lerner-Ellis JP, Anastasio N, Liu J, Coelho D, Suormala T, Stucki M et al (2009) Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype–phenotype correlations. Hum Mutat 30:1072–1081
Froese DS, Zhang J, Healy S, Gravel RA (2009) Mechanism of vitamin B(12)-responsiveness in cblC methylmalonic aciduria with homocystinuria. Mol Genet Metab 98:338–343
Coelho D, Suormala T, Stucki M, Lerner-Ellis JP, Rosenblatt DS, Newbold RF et al (2008) Gene identification for the cblD defect of vitamin B12 metabolism. N Engl J Med 358:1454–1464
Hannah-Shmouni F, Cruz V, Schulze A, Mercimek-Andrews S (2018) Transcobalamin receptor defect: identification of two new cases through positive newborn screening for propionic/methylmalonic aciduria and long-term outcome. Am J Med Genet A 176:1411–1414
Froese DS, Healy S, McDonald M, Kochan AG, Oppermann BU, Niesen FH et al (2010) Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Genetic analysis of four cases of methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab 1:29–36
Ailan C, Rong Y, Wenjun C, Hong Y (2019) The value of 1H-MRS and MRI in combined methylmalonic aciduria and homocystinuria. J Comput Assist Tomogr 43:559
Zong Y, Liu N, Zhao Z, Kong X (2015) Prenatal diagnosis using genetic sequencing and identification of a novel mutation in MMACHC. BMC Med Genet 16:48
Guéant JL, Chéry C, Oussalah A et al (2018) A PRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat Commun 9:67. https://doi.org/10.1038/s41467-017-02306-5
Karth P, Singh R, Kim J, Costakos D (2012) Bilateral central retinal artery occlusions in an infant with hyperhomocysteinemia. JAAPOS 16:398–400
Carrillo-Carrasco N, Charles P (2012) Methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis 35:10
Quadros EV, Sequeira JM (2013) Cellular uptake of cobalamin: transcobalamin and the TCblR/CD320 receptor. Biochimie 95:1008–1018
Collison FT, Xie YA, Gambin T, Jhangiani S, Muzny D, Gibbs R (2015) Whole exome sequencing identifies an adult-onset case of methylmalonic aciduria and homocystinuria type C (cblC) with non-syndromic Bull’s eye maculopathy. Ophthalmic Genet 36:270–275
Acknowledgements
We thank the family who allows us to share their data for awareness of all such families that have suffered with inherited metabolic diseases without being diagnosed yet.
Funding
None.
Author information
Authors and Affiliations
Contributions
NW conceived, searched, wrote and proofread the manuscript. ZF collected data, analyzed and compiled labs. AI did interpretation and writing of laboratory and radiological data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval has been taken from the Institutional review board, Pakistan Institute of Medical sciences Islamabad. Family has consented for this publication.
Consent for publication
Written consent to publish this information was obtained from study participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Waheed, N., Fayyaz, Z. & Imran, A. Spectrum of clinical manifestation of methylmalonic acidemia and homocystinuria in a family of six siblings: novel combination of transcobalamin receptor defect (CD320) and cblC deficiency (MMACHC). Egypt J Med Hum Genet 22, 75 (2021). https://doi.org/10.1186/s43042-021-00197-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00197-2