
Abstract
Maintenance of cellular homeostasis requires tight and coordinated control of numerous metabolic pathways, which are governed by interconnected networks of signaling pathways and energy-sensing regulators. Autophagy, a lysosomal degradation pathway by which the cell self-digests its own components, has over the past decade been recognized as an essential part of metabolism. Autophagy not only rids the cell of excessive or damaged organelles, misfolded proteins, and invading microorganisms, it also provides nutrients to maintain crucial cellular functions. Besides serving as essential structural moieties of biomembranes, lipids including sphingolipids are increasingly being recognized as central regulators of a number of important cellular processes, including autophagy. In the present review we describe how sphingolipids, with special emphasis on ceramides and sphingosine-1-phosphate, can act as physiological regulators of autophagy in relation to cellular and organismal growth, survival, and aging.
Similar content being viewed by others
Introduction
Autophagy is an intracellular degradation process, which is highly conserved among all eukaryotes. The term itself, meaning self-eating, refers to the fact that cells generate energy and cellular building blocks by degradation of its own components. In that sense autophagy is a pro-survival process, but in unfavorable situations autophagy can contribute to cell death [1]. Sphingolipids were once regarded as being merely structural elements in cell membranes. Now this diverse group of lipids is recognized as important mediators of cellular events, including autophagy, through their roles as functional entities in cellular membranes and as bioactive signaling molecules. Especially three sphingolipid species, ceramide, dihydroceramide (dhCer), and sphingosine-1-phosphate (S1P), have emerged as being important mediators of the autophagic pathway. They are believed to function as a rheostat controlling the balance between sphingolipid-induced autophagy and cell death [2]. Furthermore, sphingolipids are also important regulators of nutrient import from the extracellular environment and autophagic flux [3, 4]. This review aims to describe the interaction between autophagy and sphingolipid metabolism. Furthermore, we address the importance of subcellular localization of the sphingolipid species in this interplay.
Autophagy-machinery and regulation
The term autophagy encompasses several different sub-processes classified according to how the cargo is transported to the lysosome, which include macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy is the most prevalent form, and will accordingly be referred to as autophagy hereafter.
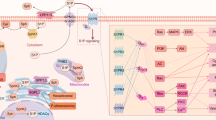
The autophagic degradation pathway overall includes five levels: (1) The formation of a double-membrane structure (also denoted as the isolation membrane), (2) encapsulation of intracellular cargo, (3) formation of the mature autophagosome, (4) fusion with a lysosome, and (5) lysosomal degradation of the cargo (Fig. 1). Each of these critical steps are regulated and effected by a number of AuTophaGy-related proteins (the Atgs), first identified and described in yeast [5, 6], but since shown to have metazoan orthologs [7]. These specific proteins, some of which form complexes, each have their own role in the process of autophagy. The most prominent of these will be discussed briefly in the following section.

Ceramides and other sphingolipids regulate autophagy at multiple levels. mTOR complex 1 (mTORC1) phosphorylates and suppresses the ULK1 complex under nutrient-rich conditions. Upon induction of autophagy, the ULK1 complex is activated by AMP-activated protein kinase (AMPK) phosphorylation and by autophosphorylation to phosphorylate Beclin1, which promotes the formation of the Vps34/PI3-kinase complex and hence generation of phosphatidylinositol-3-phosphate (PI(3)P). This recruits PI(3)P-binding proteins like DFCP1 and WIPIs to the membrane and promotes the formation of autophagosomes. The Atg12–Atg5–Atg16 complex is required for conjugating phosphatidylethanolamine (PE) to LC3 for its attachment to the autophagosomes and hence for elongation and closure of the isolation membrane. Once complete, the outer membrane of the autophagosome fuses with the lysosome, and the material is degraded in the autolysosome by acidic hydrolases. Ceramides (Cer) have been shown to reduce the abundance of nutrient transporters in the plasma membrane resulting in lowered uptake of nutrients, hence activation of AMPK, suppression of TORC1 activity, and activation of autophagy. Ceramides also promote dissociation of the Bcl2–Beclin1 complex and affect ER homeostasis and fusion between autophagosomes and lysosomes. Moreover, ceramides have also been shown to affect calpain-mediated cleavage of Atg5. Sphingoid long-chain base phosphates have furthermore been shown to induce autophagy
Without stimulation, autophagic activity is kept at a low basal level, but can be induced by extracellular cues such as stress, starvation, various pathologies, or by drug treatment. One very central gatekeeper of autophagy initiation is the target of rapamycin (TOR) complex, with the TOR kinase as a central catalytically active entity. The TOR kinase assembles into two structurally and functionally distinct complexes, referred to as TOR complex 1 (TORC1) and 2 (TORC2) [8]. If not inhibited by amino acid starvation, TORC1 inhibits the formation of the autophagosome at a very early step, namely the formation of the isolation membrane. By phosphorylation of Atg13, TORC1 prevents the formation of the Unc-51 like autophagy activating kinase 1 (ULK1) complex (Fig. 1). In times of amino acid starvation, TORC1 is inhibited [9] and ULK1 is free to phosphorylate Beclin-1, which in turn enhances the activity of VPS34, a class III phosphoinositide 3-kinase (PI3K) situated at the autophagosomal membrane. Hence, the formation of phosphatidylinositol-3-phosphate (PI(3)P) at the autophagosomal membrane is induced. This provides a platform for the gathering of autophagosomal actors and recruits PI(3)P-binding proteins like the Double FYVE-containing protein 1 (DFCP1) and WD-repeat protein interacting with phosphoinositides 1 and 2 (WIPI1 and WIPI2), which are both required for full induction of autophagy [10, 11].
In order for the isolation membrane to form and elongate, continuous recruitment and formation of cellular membrane structures are required. The endoplasmic reticulum (ER), Golgi, mitochondria, plasma membrane, lipid droplets, and ER-mitochondria contact sites have all been suggested to provide lipids for the formation of the autophagosomal isolation membrane [12–19]. The elongation of the growing pre-autophagosomal structure requires ubiquitination of Atg5 by the ubiquitin-like protein Atg12, which is activated by the E1-like enzyme Atg7 and the E2-like enzyme Atg10. The Atg12-Atg5 complex is subsequently linked to Atg16, forming a tetramer that dissociates from the mature autophagosome, which is required for the elongation process [20]. In a second ubiquitin-like reaction, in which the E1-like enzyme Atg7 and the E2-like enzyme Atg3 are involved, microtubule-associated protein 1A/1B-light chain 3 (LC3) is cleaved by Atg4 to form LC3-I, which is subsequently lipidated by the addition of phosphatidylethanolamine into LC3-II. LC3-II localizes both to the outer and inner autophagic membrane where it resides until degradation of the cargo and the inner autophagic membrane. Once the autophagosome is formed and matured, it can fuse with either an endosome to form an amphisome, which subsequently fuses with a lysosome, or directly with a lysosome to form an autolysosome. Although the precise molecular details underlying the fusion event still remain to be fully elucidated, it is known that the small GTP-binding protein named Rab7, the SNARE syntaxin Stx17, and components of the homotypic fusion and protein sorting (HOPS)-tethering complex serve as important factors in the autophagosome-lysosome fusion [21–23]. After the fusion and subsequent degradation of the autophagosomal cargo, the resulting material is transported back to the cytosol through lysosomal permeases, where it can be reused for energy production or as molecular building blocks [24].
Autophagy was originally considered to be a non-selective process, however during the past decade several studies have proven that autophagy can be highly selective. Indeed, proteins like nucleoporin p62 and Neighbor of BRCA1 gene 1 (NBR1) function as cargo receptors selectively collecting ubiquitinated substrates for autophagosomal degradation. p62 binds directly to LC3 and is degraded in parallel with the cargo, thus accumulating when degradation is inhibited. Recently, selective degradation of entire organelles such as lipid droplets, ER, mitochondria, and peroxisomes has been described [25], suggesting that autophagy can act specifically.
Whether autophagy is acting selectively or not, the regulation of this delicate process is highly critical. Within the past decade, it has become clear that sphingolipids serve as important regulators of this process at several levels.
Sphingolipid metabolism
Sphingolipids comprise a large family of lipids, which differs structurally from other lipid species by containing a sphingoid base as structural backbone. Sphingolipid metabolism constitutes an interconnected network where balancing of sphingolipid synthesis, turnover, and recycling are carefully regulated according to cell response and fate. This network revolves around ceramide as depicted in Fig. 2. De novo synthesis of sphingolipids is initiated at the cytosolic face of the ER membrane, where serine palmitoyl-transferase (SPT) catalyzes the condensation of serine and palmitoyl-CoA producing 3-ketodihydrosphinganine. Next, 3-ketodihydrosphinganine is reduced to produce the sphingoid base dihydrosphingosine (sphinganine), which along with sphingosine, comprises the backbone of sphingolipids. Besides sphinganine the yeast Saccharomyces cerevisiae (S. cerevisiae) also produces phytosphingosine as a sphingoid base. Ceramide synthases (CERS1-6) catalyze the N-acylation of sphingoid bases resulting in synthesis of ceramides. The CERSs display different chain length specificities [26], which adds to the complexity of sphingolipids. CERS1 prefers C18-CoAs, while CERS2 utilizes acyl-CoAs ranging from C20 to C26. CERS3 shows preference towards the ultra-long-chain acyl-CoAs (C26–36), whereas CERS4 has specificity for C18- and C20-CoAs. CERS5 and CERS6 both primarily incorporate C16-CoAs. Ceramides can be phosphorylated to ceramide-1-phosphate (C1P), modified by addition of phosphocholine to yield sphingomyelin, or be glycosylated to produce a vast number of different glycosylceramides. Ceramides can be resynthesized from sphingosine or regenerated by recycling of glycosylceramides and sphingomyelin, or by dephosphorylation of C1P [27]. Importantly, while de novo synthesis of ceramides by CERSs requires hours [28], generation of ceramides from the recycling pathways, e.g. degradation of sphingomyelin, occurs within minutes of activation [29]. Thus, immediate regulation of cellular processes by ceramides and other sphingolipids may be mediated by salvaging pathways, whereas de novo synthesis of sphingolipids may modulate long-term cellular processes.

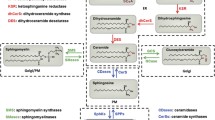
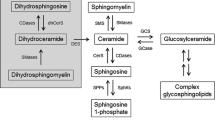
Overview of sphingolipid metabolism. Central in the sphingolipid metabolism is ceramide. Ceramide is de novo synthesized at the endoplasmatic reticulum (ER) with the condensation of serine and palmitoyl-CoA by serine palmitoyltransferase (SPT) being the first step. Further reduction and acylation by a ceramide synthase (CERS1-6) yields dihydrosphingosine (dHSph), which after desaturation results in the formation of ceramide. At the ER ceramide can be modified into galactosylceramide (GalCer), yet the majority of ceramide modification takes place at the Golgi in a manner depending on their further utilization. At the Golgi ceramide is used in the synthesis of sphingomyelin (SM) and glycosphingolipids in reactions catalyzed by sphingomyelin synthase 1 (SMS1) and glycosphingolipid synthases (GCSs), respectively. From the Golgi, SM and glycosphingolipids are transported to the plasma membrane (PM). Here SM can be turned into ceramide again by the actions of secretory and neutral sphingomyelinases (sSMase and nSMase, respectively). The ceramide can then be metabolized into ceramide-1-phosphate (C1P), sphingosine-1-phosphate (S1P), or be resynthesized back into SM. Complex sphingolipids residing in the PM can also be used as a pool for recycling of ceramide by entering the endolysosomal pathway. In this pathway acid SMase (aSMase) and glycosidases (GCase) produce ceramide, which in turn can be hydrolyzed into sphingosine and reused in the synthesis of ceramide or be degraded by phosphorylation into S1P followed by breakdown to hexadecenal and ethanolamine-1-phosphate (EA1P). In the Golgi, ceramide kinase (CERK) can phosphorylate ceramide thereby generating ceramide-1-phosphate (C1P). Other abbrevations: 3KSR 3-ketosphinganine reductase, CPP ceramide phosphatase, DES1 dihydroceramide desaturase 1, aCDase acid ceramidase, nCDase neutral ceramidase, SPHK sphingosine kinase, aSMase acid sphingomyelinase, SPL S1P lyase, S1PP sphingosine phosphate phosphatase
Compartmentalization of sphingolipids is controlled by subcellular transport
The diversity of biomembranes is based on the vast structural heterogeneity of lipid species and their subcellular and asymmetric distribution. Such heterogeneity in organelles and membrane leaflets is critical in modulating the localization of membrane-bound proteins and their functional properties. Sphingolipids in the plasma membrane are normally exclusively found in the outer leaflet of the bilayer, and form together with cholesterol specific membrane microdomains [30, 31]. Such microdomains are laterally segregated regions, which arise as a result of selective affinities between sphingolipids and membrane proteins and presumably act as signaling platforms [31–34].
The distribution of sphingolipids in cellular organelles is far from uniform. It has become evident that sphingolipids are compartmentalized according to their functions, and that subcellular transport aids this sorting. The sorting initiates at the ER from where ceramide must be transported to the Golgi apparatus for further modification. Non-vesicular transport of ceramide requires the ceramide transfer protein (CERT) [35]. CERT has a preference for ceramide species containing acyl chains shorter than C22, while CERT-mediated transport of C22 and C24:1 ceramides, and of dhCer is significantly less efficient [36, 37]. Together with the fact that ceramides transported by CERT to the Golgi preferentially are incorporated into sphingomyelin compared to glycosphingolipids [38], this implies that CERT contributes to the complexity and subcellular distributions of specific sphingolipids. The restricted specificity of CERT also suggests that alternative routes for intracellular trafficking of ceramide must exist. Although not delineated in molecular detail yet, such alternative routes may include vesicular transport or be mediated through specific inter-organelle contact sites [39, 40].
Recently, a novel lipid transfer protein was identified by virtue of its ability to specifically transfer C1P between membranes, thus named C1P transfer protein (CPTP) [41]. Despite poor sequence homology, CPTP shares a very similar structural fold with glycolipid transfer protein (GLTP). However, it does not bind glycosylated ceramides like galactosylceramide and lactosylceramide as GLTP does. CPTP is localized in the cytosol, but it is also associated with the trans-Golgi network, nucleus, and plasma membrane, and has been proposed to control C1P levels to maintain proper Golgi organization and inflammatory responses [41].
Although it does not transport ceramides, the evolutionary conserved GLTP has been shown to accelerate transfer of both diacylglycerols- and sphingoid-based glycolipids between lipid membranes [42]. GLTP enables intermembrane transfer of glucosylceramide, lactosylceramide, galactosylceramide, sulfatide, and the gangliosides GM1 and GM3, but not phosphatidylcholine, phosphatidylethanolamine, sphingomyelin, phosphatidylinositol, cholesterol, and cholesterol oleate [43, 44]. Thus, although its function is yet to be fully resolved, it has been proposed that GLTP mediates the transfer of glycosylceramides from the Golgi to the plasma membrane or functions as an intracellular sensor of glycosphingolipids [45].
To this end, another member of the GLTP superfamily, the four-phosphate adaptor protein 2 (FAPP2) has also been shown to mediate intermembrane lipid transfer between the Golgi and the plasma membrane, and to be crucial for synthesis of complex glycosphingolipids in the Golgi network [46, 47]. The fact that cellular processes are highly compartmentalized too, paves the way for the idea of a very tight and local control of sphingolipid metabolism in relation to regulation of cellular processes.
Sphingolipids and membrane fusion
Upon completion of the autophagic process, the autophagosome fuses with the lysosome to form the autolysosome, where sequestered organelles and proteins are degraded by acidic lysosomal hydrolases. The efficiency of this step depends on the cellular lipid composition, including the level of cholesterol and other lipids [48]. Even though sphingolipids have not directly been shown to affect fusion of autophagosomes with lysosomes, accumulating evidence suggests that this step also depends on sphingolipids. For example, it has recently been shown that myristate induces autophagy and autophagic flux in cardiomyocytes in a sphingolipid- and CERS5-dependent way, indicating that ceramide synthesis is involved in regulation of the autophagic flux [4]. Moreover, it has also been suggested that progression of autophagy in yeast depends on sphingolipid production. This is due to its role in formation of autophagosomes rather than conjugation of Atg12-Atg-5, lipidation of Atg8/LC3, maturation of vacuolar proteases, or the formation of the pre-autophagosomal structure [49].
It has previously been shown that the production of C1P from sphingomyelin, by the joint action of sphingomyelinase and ceramide kinase, promotes Ca2+-dependent liposomal fusion, which enhances the vesicle fusion. Interestingly, C1P levels increase during phagocytosis, indicating that C1P may promote fusion between phagosomes and lysosomes [50], and in turn signifying that C1P regulates autophagosome-to-lysosome fusion as well. Given the role of CPTP in controlling intracellular C1P levels [41], CPTP may also control such fusion events. Moreover, a recent study suggests a role for sphingosine kinase 1 (SPHK1) and its product, S1P, in the endo-and exocytotic membrane trafficking pathways [51], which are tightly linked to the autophagic pathway [52, 53].
The heterogeneity in organelles and membrane leaflets is, as previously mentioned, critical in modulating the localization of membrane-bound proteins and their functional properties. Changes in membrane lipid species can alter the membranes biophysical properties, which in turn affect biological properties. Ceramide can induce rigidization of membranes and hence may also affect the curvature of the membrane [54]. Accordingly, acid sphingomyelinase-induced synthesis of ceramide increases the packing of the lipids and is associated with enhanced order in membranes [55, 56]. It has been suggested that the hydrolysis of sphingomyelin to ceramide within the inner leaflet of the plasma membrane causes an outward curvature of the membrane important for exocytosis [57]. This function of membranous ceramide production may also apply to fusion of autophagosomes with lysosomes. By analogy, homotypic vacuole fusion, which shares a number of components with the fusion between autophagosomes and lysosomes, has been suggested to be affected by ceramide and other lipids in yeast [58, 59]. Moreover, sphingolipids have been shown to support fusion of enveloped animal viruses, such as Semliki Forest virus, with lipid bilayers [60, 61], arguing that sphingolipids are important in membrane fusion events.
In line with these studies, it has been found that an increase in sphingosine and a concomitant decrease in S1P are of pathological importance in the early events of the lysosomal storage disorder, Niemann-Pick type C1. The increase in sphingosine disturbs lysosomal Ca2+ homeostasis, subsequently blocking the late endosome-to-lysosome transport [62]. Moreover, accumulation of sphingomyelin is known to have a destabilizing effect on lysosomes [63] and to result in leakage of the lysosomal proteases to the cytosol [64]. A very recent study suggests that the increased levels of sphingomyelin observed in Niemann Pick disease type A, cause lysosomal dysfunction due to lysosomal membrane permeabilization [65]. This underlines that sphingolipids also function at another step of the lysosomal degradation pathway.
Collectively, these studies reveal an additional function of sphingolipids, besides their role as messenger molecules, in the control of the autophagic and lysosomal degradation pathways.
The pollice verso of sphingolipids
The regulation of the delicate balance between proliferation and cell death is another important aspect where sphingolipids act as second messengers. Specifically, S1P and ceramide have proved important in the regulation of cell fate [66, 67], however, their effect on cell fate are very different [68, 69]. Both acting through autophagy, S1P is believed to promote cell survival and proliferation, whereas ceramide has been found to induce growth arrest and cell death [70]. These opposing roles of two so easily inter-convertible biomolecules have led to the manifestation of the sphingolipid rheostat [2], which describes the intimate balance between the intracellular levels of ceramide and S1P and its importance in regulating cell fate (Fig. 3). Furthermore, this sphingolipid rheostat is controlling cell fate, at least partly, by modulating autophagy [67]. As the conversion of ceramide to S1P only requires two steps [69, 71], the regulation of the enzymes balancing the concentrations and localizations of ceramide and S1P, is rather significant to cell fate.

Sphingolipid-mediated regulation of cell death and survival. Regulation of the subcellular synthesis and localization of sphingolipids is crucial for their pro-apoptotic or pro-survival roles. Sphingosine kinase 2 (SPHK2)-dependent synthesis of sphingosine-1-phosphate (S1P) at the ER has been shown to promote apoptosis, while translocation of SPHK1 from the cytosol to the plasma membrane promotes synthesis of S1P at the plasma membrane and induces cell survival
In favor of cell survival, S1P has been observed as a critical player through its induction of autophagy [66]. S1P levels are balanced by their synthesis from sphingosine catalyzed by SPHKs, their dephosphorylation catalyzed by S1P phosphatases and phosphohydratases as well as their irreversible degradation catalyzed by S1P lyase (Fig. 2). Sphingosine, in turn, can be generated by hydrolysis of ceramide catalyzed by the ceramidase and removed by synthesis of ceramide by the action of the CERS. In agreement with this, knockdown of the ER-residing S1P phosphatase, the enzyme responsible for the degradation of S1P, augments S1P levels and induces autophagy [72].
Two distinct isoforms of SPHK have been characterized: SPHK1 and -2 sharing five conserved domains; however, their tissue distribution and developmental expression are distinct [73, 74]. SPHK1, which is cytosolic, is activated by several external stimuli to specifically signal growth and survival [69]. It has been recognized as a nutrient-sensitive regulator of autophagy [68], and starvation-induced autophagy is dependent on SPHK1 activity, a mechanism, which is conserved from yeast to mammals [68, 75]. Through its production of S1P, SPHK1 is therefore considered pro-survival, and accordingly SPHK1 has been shown to induce DNA transcription and cell proliferation [76].
It is not clear whether S1P signaling acts through or parallel to the mammalian TOR (mTOR) pathway in regulating autophagy [68, 72]. In a study by Lavieu et al. phosphorylation of two known downstream effectors of mTOR was found to be induced as response to overexpression of SPHK1 [68]. However, down-regulation of S1P phosphorylase-1, and thus the degradation of S1P, does not affect phosphorylation of mTOR nor its downstream effectors [72, 77]. Surprisingly, several studies have described S1P as an inhibitor of autophagy through activation of the mTOR pathway via specific S1P receptors in the plasma membrane [67, 78–80]. However, it is important to note that S1P signaling through cell surface receptors might differ from signaling within the cell.
The class III PI3K inhibitor, 3-methyladenine, which is known to prevent the formation of autophagosomes, does not affect S1P induced autophagy, whereas silencing of Atg5, a protein required at a later step in the autophagic process, does indeed inhibit S1P-induced autophagy [72]. This might either suggest that S1P is controlling the autophagic state through an entirely different signaling pathway, or that S1P is able to interact directly with the class III PI3K, preventing its inhibition by 3-methyladenine. In turn, the effect of S1P on cell survival is believed to be conducted through an inositol-independent Ca2+ mobilization from intracellular stores [81], activation of the mitogen activated protein pathway through the extracellular signal-regulated kinase ERK [82], and by suppression of the apoptotic effect of ceramide via inhibition of the stress-activated protein kinase c-Jun N-terminal protein kinase (JNK) [2].
Surprisingly, the other isoform of the kinase responsible for S1P production, SPHK2, is pro-apoptotic and has been found to localize in both the nucleus and cytosol, where it prevents DNA synthesis and cell proliferation, thus counteracting the positive effect of SPHK1 on cell division [83, 84]. Upon starvation SPHK2 is redirected to the ER, which is essential for its pro-apoptotic function [85]. Due to this re-localization, it has been suggested that SPHK2’s pro-apoptotic role is due to indirect production of ceramide from external sources of sphingoid bases by the combined actions of SPHK2 and lipid phosphohydrolases in the ER [69, 85]. Accordingly, down-regulation of SPHK2 reduces the conversion of sphingosine to ceramide via the recycling pathway, whereas down-regulation of SPHK1 increases it [85]. Interestingly, targeting of the pro-survival SPHK1 to the ER makes it pro-apoptotic [85], collectively suggesting that differently distributed intracellular pools of S1P might participate in different metabolic and signaling pathways. The death-inducing mitochondrial protein BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) is involved in ceramide-induced cell death and can displace Beclin 1 from the Beclin 1/Bcl-2 complex due to its Bcl-2 homology-3 (BH3) domain [86, 87]. The fact that SPHK2 also contains a BH3 domain that enables it to displace Beclin 1, supports the pro-apoptotic effect of SPHK2 [88]. In line with this, mutation of the BH3 domain causes suppression of the SPHK2 induced apoptosis [89]. Accordingly, it has been suggested that SPHK2 functions independently of its catalytic activity by production of S1P, but rather through its ability to interact with Bcl-2 [88].
Ceramide orchestrates programmed cell death through two different pathways referred to as type I and type II programmed cell death. Type I programmed cell death, apoptosis, is induced by increased ceramide levels [69]. The mechanisms for ceramide-induced apoptosis are numerous, including activation of caspase-9 through inactivation of protein kinase B (PKB), activation of Bad through Ras and protein phosphatase 2A (PP2A), dephosphorylation of retinoblastoma gene product by the activation of protein phosphatase 1, as well as activation of protein kinase Cζ and ceramide activated protein kinase [90–95].
Ceramide has not only been associated with apoptotic cell death, but also with another type of cell death distinct from apoptosis [96–98]. Whereas autophagy is normally associated with protection of the cell, an alternative strategy has been known for a decade, where cells consume their own interior resulting in a programmed cell death occurring independently of the apoptotic cell death [99]. This autophagic cell death is referred to as type II cell death [100] and is caspase-independent and characterized by a large number of autophagic vacuoles, early degradation of organelles, and preservation of cytoskeletal elements [100, 101], and by being independent of apoptosis and can be rescued by autophagic inhibition [1]. Furthermore, type II cell death can occur in the absence of the pro-apoptotic members of the Bcl-2 family [102]. It is activated by ceramide via induction of autophagy through lowering of the mitochondrial membrane potential, and by activation of the transcription of BNIP3 [98].
Whereas ceramide-induced autophagy and cell death act by activation of PP2A, thus inhibiting the pro-survival PKB [103], and subsequently augment Beclin 1 accumulation [1, 66], S1P promotes cell survival in a PKB-independent manner [103]. Since the anti-apoptotic protein Bcl-2 inhibits the function of Beclin 1 in the early stages of autophagy [104], the dissociation of the Beclin-1/Bcl-2 complex is required for induction of autophagy. Yet, ceramide has proved a potent inducer of this dissociation [105]. The release of Beclin 1 from the complex can be mediated by phosphorylation of Bcl-2 by the stress-activated JNK and is stimulated by supplementation of short-chain ceramides or by enhancing de novo synthesis of ceramide [105]. Accordingly, Beclin 1 mutation, which disables the protein to bind Bcl-2, induces autophagy resulting in cell death [104].
In contrast to the cell death-inducing effects of ceramide described above, Demarchi et al. found that C2-ceramide triggers an NF-кB dependent survival pathway, and that the induction of pro-survival genes is dependent on the protease calpain [106]. Calpain is induced upon starvation [107], inhibits apoptosis [108], and has been found to be required for autophagy as well as for the pro-survival effect of C2 ceramide [106, 109]. These opposing functions of ceramide have recently been coined the autophagy paradox [1], and have been suggested to function as a brake of the ceramide-induced cell death [106]. However, these observations may also be due to the artificial effects caused by the use of biologically irrelevant ceramides used in these studies.
In yeast TORC2 positively controls the synthesis of phytoceramide and dhCer [110]. Since ceramide, as described above, controls the induction of type II programmed cell death through activation of autophagy, it might seem rather conflicting that TORC2, which is otherwise known to support growth, activates the synthesis of ceramide. However, it is likely that phytoceramide and dhCers exert effects different from ceramides. Indeed, the action of ceramide and its analogs on cell death has been shown to be specific among the different lipid species [111]. The steady-state levels of sphingoid long-chain-bases and their phosphorylated derivatives in yeast were also shown to decrease in mutants lacking an ortholog of the mammalian TORC2 component Rictor [110]. This observation suggests that TORC2 might be involved in the production of S1P as well, which has been shown to reverse ceramide-mediated apoptosis [2]. In fact, the TORC2-dependent ceramide and S1P production might work as a feedback loop since both lipids are activators of autophagy, which is negatively regulated by TOR [68]. Since S1P is synthesized from recycling of ceramide [28], ceramide synthesis is required for production of this pro-survival second messenger. Interestingly, increasing levels of dhCer in response to inhibition of sphingolipid delta(4)-desaturase DES1 have been shown to delay the G1/S transition of the cell cycle in an autophagy-dependent manner, suggesting a pro-survival role for dhCer [112].
Regulation of nutrient uptake by sphingolipids
Sphingolipids not only constitute significant structural entities in the plasma membrane, they also regulate the activity and abundance of nutrient transporters, thus indirectly affecting autophagy. Erdinger and co-workers showed that addition of C2-ceramides diminishes the surface abundance of the amino acid transferase, 4F2, resulting in impaired amino acid uptake, induced autophagy, and decreased viability [3]. Supplementation of methyl pyruvate, a membrane-permeant derivative of pyruvate, reversed C2-ceramide-induced cell death and autophagy independent of the surface abundance of 4F2, arguing that C2-ceramides cause starvation-induced cell death [3]. Similarly, Rosales et al. recently demonstrated that sphingolipid-based drugs down-regulate nutrient transporters, thereby inducing autophagy and killing cancer cells by nutrient deprivation [113]. Interestingly, inhibition of TORC1 in S. cerevisiae activates the nitrogen permease reactivator 1 kinase, which phosphorylates and hence relieves the inactivating effects of Orm1 and Orm2 on the SPT, resulting in increased de novo synthesis of complex sphingolipids. In turn, plasma membrane localization and activity of the general amino acid permease Gap1 is stimulated [114]. To this end, inhibition of SPT and complex sphingolipid synthesis have been shown to inhibit autophagy [49]. Moreover, when sphingolipid levels are low, the two phosphoinositide PI4,5P(2) binding proteins Slm1 and Slm2 recruit the kinases Ypk1 and Ypk2 to TORC2 at the plasma membrane, where they are phosphorylated and activated by TORC2 and the kinases Pkh1 and Pkh2 [115, 116]. Ypk1 and Ypk2 subsequently phosphorylate Orm1 and Orm2, which relieves their inhibition of SPT, thereby stimulating the synthesis of long-chain bases and sphingolipids [116, 117]. Interestingly, the ORMDL gene family encoding mammalian homologs of Orm1 and Orm2 has also been found to repress SPT activity and sphingolipid synthesis in mammalian cells in a phosphorylation-dependent manner [118], indicating that this regulatory mechanism is evolutionary conserved. These observations imply that nutrient uptake, TORC activities, and sphingolipid synthesis are coordinately regulated. Zimmerman et al. recently found that TORC1- and GSK3-dependent phosphorylation of Elo2 in S. cerevisiae promotes very-long chain fatty acid synthesis, while impaired phosphorylation results in a profound decrease in ceramide levels and a concomitant increase in the level of phosphorylated long-chain bases. The increase in phosphorylated long-chain bases resulted in constitutive induction of autophagy, which negatively affected cell viability, which again could be prevented by inactivation of the sphingoid long-chain base kinase Lcb4 [119].
Collectively, these observations suggest that import of nutrients via specific transporters and permeases is sensitive to changes in the sphingolipid level in the plasma membrane, which in turn is carefully controlled by networks of kinases and phosphatases in an auto-regulatory loop.
Lifespan and sphingolipids
It is widely accepted that autophagy and lifespan are tightly linked [120–123]. Sphingolipids may therefore also affect organismal and cellular lifespan by modulating autophagy. Liu et al. have recently shown that both chemical- and genetic-inhibition of SPT activity increase lifespan in S. cerevisiae through reduced TORC1 activity and enhanced autophagy [124, 125], arguing that sphingolipids modulate lifespan. They also observed that lifespan extension induced by both calorie restriction and inhibition of S6 kinase was further augmented by myriocin in a dose-dependent manner [125], suggesting that impaired de novo sphingolipid synthesis induces longevity in parallel to caloric restriction and S6 kinase inhibition. Consistently, inhibition of TORC1 by rapamycin further enhanced myriocin-induced longevity, underlining that inhibition of sphingolipid synthesis synergistically with impaired TORC1 activity can extend life span [124, 125]. In line with this, inhibition of SPT1 activity in Caernorhabditis elegans (C. elegans) slowed the development rate and extended longevity [126]. Mosbech et al. recently found that impaired sphingolipid synthesis in C. elegans, caused by functional loss of the two ceramide synthases HYL-1 and LAGR-1, extended longevity in an autophagy-dependent manner [127]. Interestingly, loss of HYL-2 and LAGR-1 function had the opposite effect on lifespan, arguing that unique sphingolipid species and/or tissue-specific synthesis of sphingolipids are important in determining organismal longevity [127]. Moreover, C. elegans lacking ceramide glucosyl transferases arrests at the first larval stage which can be rescued by expression of ceramide glucosyl transferases in the most anterior- and posterior intestinal cells, implying that cell-specific synthesis of glycosphingolipids are indispensable for growth and survival [128]. In summary, lifespan and development are under the control of sphingolipid metabolism, possibly through regulation of autophagy.
Concluding remarks
Sphingolipids comprise a diverse group of lipid species, which are highly interchangeable, and therefore constitute an ideal second messenger. The exact mechanisms governing sphingolipid-mediated regulation of autophagy and other cellular processes still remain enigmatic; however, their roles in autophagy appear to be mediated at different stages of the autophagic process. Sphingolipids constitute a major part of the plasma membrane and by virtue of their biophysical properties and interactions with membrane proteins, sphingolipids and proteins cluster to form microdomains with unique regulatory functions. Alterations in membrane sphingolipids can modulate the level and activity of nutrient transporters [3, 113, 129, 130] and therefore impair nutrient uptake and indirectly induce autophagy. Sphingolipids may also affect autophagy by regulating the assembly of the autophagic machinery, and affect fusion between autophagosomes and lysosomes, either by modulating the membrane properties, abundance of fusogenic SNARE proteins, or acidification of the lysosomes [48, 58, 59, 131]. However, since specific sphingolipids like S1P modulate particular cellular processes, while other closely related species may exert opposite effects [54, 132, 133], the enzymes responsible for their interconversions serve central roles in regulation of cellular metabolism and cell fate. It is therefore also critical that the localization of sphingolipids in specific cellular compartments is tightly controlled. Thus, intracellular transport of sphingolipids by transfer proteins like CERT, CPTP, FAPP2, and GLTP, is crucial for their regulatory properties.
Considering the regulatory roles of sphingolipids in autophagy, it is interesting that autophagy regulates sphingolipid levels including ceramide levels [134] and mobilization as well as storage of glycerolipids in lipid droplets [19, 135, 136]. Collectively, this shows that autophagy and lipid metabolism are coordinately controlled, and underlines the importance of sphingolipids in metabolic regulation. However, some of the reported effects of sphingolipids are based on short-chain ceramides, which may have completely different effects from ceramides produced in vivo. Therefore, rather than using such non-natural ceramides, the roles of sphingolipids in autophagy should be examined further via loss-of-function and overexpression studies in genetically tractable model systems. Such studies will broaden our understanding of how sphingolipids affect specific stages of autophagy.
Abbreviations
- Atg:
-
Autophagy-related
- BH3:
-
Bcl-2 homology-3
- BNIP3:
-
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- C. elegans :
-
Caernorhabditis elegans
- C1P:
-
Ceramide-1-phosphate
- CERT:
-
Ceramide transfer protein
- CERS:
-
Ceramide synthase
- CPTP:
-
Ceramide-1-phosphate transfer protein
- dhCer:
-
Dihydroceramide
- DFCP1:
-
Double FYVE-containing protein 1
- ER:
-
Endoplasmic reticulum
- FAPP2:
-
Four-phosphate adaptor protein 2
- GLTP:
-
Glycolipid transfer protein
- JNK:
-
c-Jun N-terminal protein kinase
- LC3:
-
Microtubule-associated protein 1 light chain 3
- mTOR:
-
Mammalian target of rapamycin
- NBR1:
-
Neighbor of BRCA1 gene 1
- PI3 K:
-
Phosphoinositide 3-kinase
- PI(3)P:
-
Phosphatidylinositol-3-phosphate
- PKB:
-
Protein kinase B
- PP2A:
-
Protein phosphatase 2A
- S. cerevisiae :
-
Saccharomyces cerevisiae
- SPT:
-
Serine palmitoyltransferase
- SPHK:
-
Sphingosine kinase
- S1P:
-
Sphingosine-1-phosphate
- TOR:
-
Target of rapamycin
- TORC:
-
TOR complex
- ULK1:
-
Unc-51 like autophagy activating kinase 1
- WIPI:
-
WD-repeat protein interacting with phosphoinosides
References
Jiang W, Ogretmen B (2014) Autophagy paradox and ceramide. Biochim Biophys Acta 1841(5):783–792
Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381(6585):800–803
Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL (2008) Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci USA 105(45):17402–17407
Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA (2012) Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest 122(11):3919–3930
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y (2003) A unified nomenclature for yeast autophagy-related genes. Dev Cell 5(4):539–545
Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH (1994) Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett 349(2):275–280
Melendez A, Neufeld TP (2008) The cell biology of autophagy in metazoans: a developing story. Development 135(14):2347–2360
Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL (2008) The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4(7):851–865
Liao XH, Majithia A, Huang X, Kimmel AR (2008) Growth control via TOR kinase signaling, an intracellular sensor of amino acid and energy availability, with crosstalk potential to proline metabolism. Amino Acids 35(4):761–770
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15(7):741–750
Obara K, Ohsumi Y (2011) PtdIns 3-kinase orchestrates autophagosome formation in yeast. J Lipids. doi:10.1155/2011/498768
Tooze SA, Yoshimori T (2010) The origin of the autophagosomal membrane. Nat Cell Biol 12(9):831–835
Cuervo AM (2010) The plasma membrane brings autophagosomes to life. Nat Cell Biol 12(8):735–737
Geng J, Klionsky DJ (2010) The Golgi as a potential membrane source for autophagy. Autophagy 6(7):950–951
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495(7441):389–393
Ge L, Melville D, Zhang M, Schekman R (2013) The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2:e00947
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4):656–667
Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC (2014) ATG16L1 meets ATG9 in recycling endosomes: additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy 10(1):182–184
Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V (2014) neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol 24(6):609–620
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90(4):1383–1435
Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117(Pt 20):4837–4848
Itakura E, Kishi-Itakura C, Mizushima N (2012) The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151(6):1256–1269
Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N (2014) The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell 25(8):1327–1337
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22(2):124–131
Lamark T, Kirkin V, Dikic I, Johansen T (2009) NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8(13):1986–1990
Stiban J, Tidhar R, Futerman AH (2010) Ceramide synthases: roles in cell physiology and signaling. Adv Exp Med Biol 688:60–71
Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23
Huwiler A, Zangemeister-Wittke U (2007) Targeting the conversion of ceramide to sphingosine 1-phosphate as a novel strategy for cancer therapy. Critical reviews in oncology/hematology 63(2):150–159
Kolesnick RN, Kronke M (1998) Regulation of ceramide production and apoptosis. Annu Rev Physiol 60:643–665
Gupta G, Surolia A (2010) Glycosphingolipids in microdomain formation and their spatial organization. FEBS Lett 584(9):1634–1641
Dart C (2010) Lipid microdomains and the regulation of ion channel function. J Physiol 588(Pt 17):3169–3178
Inder KL, Davis M, Hill MM (2013) Ripples in the pond–using a systems approach to decipher the cellular functions of membrane microdomains. Mol Biosyst 9(3):330–338
Parton RG, del Pozo MA (2013) Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14(2):98–112
Ernst AM, Brugger B (2014) Sphingolipids as modulators of membrane proteins. Biochim Biophys Acta 1841(5):665–670
Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M (2003) Molecular machinery for non-vesicular trafficking of ceramide. Nature 426(6968):803–809
Kumagai K, Kawano M, Shinkai-Ouchi F, Nishijima M, Hanada K (2007) Interorganelle trafficking of ceramide is regulated by phosphorylation-dependent cooperativity between the PH and START domains of CERT. J Biol Chem 282(24):17758–17766
Kudo N, Kumagai K, Tomishige N, Yamaji T, Wakatsuki S, Nishijima M, Hanada K, Kato R (2008) Structural basis for specific lipid recognition by CERT responsible for nonvesicular trafficking of ceramide. Proc Natl Acad Sci USA 105(2):488–493
Hanada K, Kumagai K, Tomishige N, Kawano M (2007) CERT and intracellular trafficking of ceramide. Biochim Biophys Acta 1771(6):644–653
Perry RJ, Ridgway ND (2005) Molecular mechanisms and regulation of ceramide transport. Biochim Biophys Acta 1734(3):220–234
Holthuis JC, Levine TP (2005) Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol 6(3):209–220
Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, Molotkovsky JG, Malinina L, Hinchcliffe EH, Chalfant CE, Brown RE, Patel DJ (2013) Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 500(7463):463–467
Tuuf J, Mattjus P (2014) Membranes and mammalian glycolipid transferring proteins. Chem Phys Lipids 178:27–37
Yamada K, Abe A, Sasaki T (1985) Specificity of the glycolipid transfer protein from pig brain. J Biol Chem 260(8):4615–4621
Brown RE, Stephenson FA, Markello T, Barenholz Y, Thompson TE (1985) Properties of a specific glycolipid transfer protein from bovine brain. Chem Phys Lipids 38(1–2):79–93
Malakhova ML, Malinina L, Pike HM, Kanack AT, Patel DJ, Brown RE (2005) Point mutational analysis of the liganding site in human glycolipid transfer protein. Functionality of the complex. J Biol Chem 280(28):26312–26320
Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA (2004) FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 6(5):393–404
D’Angelo G, Polishchuk E, Di Tullio G, Santoro M, Di Campli A, Godi A, West G, Bielawski J, Chuang CC, van der Spoel AC, Platt FM, Hannun YA, Polishchuk R, Mattjus P, De Matteis MA (2007) Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449(7158):62–67
Koga H, Kaushik S, Cuervo AM (2010) Altered lipid content inhibits autophagic vesicular fusion. FASEB J 24(8):3052–3065
Yamagata M, Obara K, Kihara A (2011) Sphingolipid synthesis is involved in autophagy in Saccharomyces cerevisiae. Biochem Biophys Res Commun 410(4):786–791
Hinkovska-Galcheva VT, Boxer LA, Mansfield PJ, Harsh D, Blackwood A, Shayman JA (1998) The formation of ceramide-1-phosphate during neutrophil phagocytosis and its role in liposome fusion. J Biol Chem 273(50):33203–33209
Shen H, Giordano F, Wu Y, Chan J, Zhu C, Milosevic I, Wu X, Yao K, Chen B, Baumgart T, Sieburth D, Camilli PD (2014) Coupling between endocytosis and sphingosine kinase 1 recruitment. Nat Cell Biol 510:552–555
Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA (2012) TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol 197(5):659–675
Szatmari Z, Kis V, Lippai M, Hegedus K, Farago T, Lorincz P, Tanaka T, Juhasz G, Sass M (2014) Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol Biol Cell 25(4):522–531
Goni FM, Alonso A (2009) Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim Biophys Acta 1788(1):169–177
Ira Johnston LJ (2008) Sphingomyelinase generation of ceramide promotes clustering of nanoscale domains in supported bilayer membranes. Biochim Biophys Acta 1778(1):185–197
Holopainen JM, Subramanian M, Kinnunen PK (1998) Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membrane. Biochemistry 37(50):17562–17570
Draeger A, Babiychuk EB (2013) Ceramide in plasma membrane repair. Handb Exp Pharmacol 216:341–353
Faergeman NJ, Feddersen S, Christiansen JK, Larsen MK, Schneiter R, Ungermann C, Mutenda K, Roepstorff P, Knudsen J (2004) Acyl-CoA-binding protein, Acb1p, is required for normal vacuole function and ceramide synthesis in Saccharomyces cerevisiae. Biochem J 380(Pt 3):907–918
Wickner W (2010) Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu Rev Cell Dev Biol 26:115–136
Samsonov AV, Chatterjee PK, Razinkov VI, Eng CH, Kielian M, Cohen FS (2002) Effects of membrane potential and sphingolipid structures on fusion of Semliki Forest virus. J Virol 76(24):12691–12702
Nieva JL, Bron R, Corver J, Wilschut J (1994) Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO J 13(12):2797–2804
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14(11):1247–1255
Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jaattela M (2010) Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 463(7280):549–553
Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lonborg A, Vindelov SD, Hanahan D, Arenz C, Ejsing CS, Kirkegaard T, Rohde M, Nylandsted J, Jaattela M (2013) Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 24(3):379–393
Gabande-Rodriguez E, Boya P, Labrador V, Dotti CG, Ledesma MD (2014) High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ 21(6):864–875
Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P (2004) Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem 279(18):18384–18391
Taniguchi M, Kitatani K, Kondo T, Hashimoto-Nishimura M, Asano S, Hayashi A, Mitsutake S, Igarashi Y, Umehara H, Takeya H, Kigawa J, Okazaki T (2012) Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem 287(47):39898–39910
Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P (2006) Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem 281(13):8518–8527
Le Stunff H, Galve-Roperh I, Peterson C, Milstien S, Spiegel S (2002) Sphingosine-1-phosphate phosphohydrolase in regulation of sphingolipid metabolism and apoptosis. J Cell Biol 158(6):1039–1049
Ogretmen B, Hannun YA (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4(8):604–616
Spiegel S, Milstien S (2000) Sphingosine-1-phosphate: signaling inside and out. FEBS Lett 476(1–2):55–57
Lepine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S (2011) Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ 18(2):350–361
Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S (1998) Molecular cloning and functional characterization of murine sphingosine kinase. J Biol Chem 273(37):23722–23728
Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem 275(26):19513–19520
Lanterman MM, Saba JD (1998) Characterization of sphingosine kinase (SK) activity in Saccharomyces cerevisiae and isolation of SK-deficient mutants. Biochem J 332(Pt 2):525–531
Olivera A, Kohama T, Edsall L, Nava V, Cuvillier O, Poulton S, Spiegel S (1999) Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol 147(3):545–558
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403(6765):98–103
Potteck H, Nieuwenhuis B, Luth A, van der Giet M, Kleuser B (2010) Phosphorylation of the immunomodulator FTY720 inhibits programmed cell death of fibroblasts via the S1P3 receptor subtype and Bcl-2 activation. Cell Physiol Biochem 26(1):67–78
Maeurer C, Holland S, Pierre S, Potstada W, Scholich K (2009) Sphingosine-1-phosphate induced mTOR-activation is mediated by the E3-ubiquitin ligase PAM. Cell Signal 21(2):293–300
Kluk MJ, Hla T (2001) Role of the sphingosine 1-phosphate receptor EDG-1 in vascular smooth muscle cell proliferation and migration. Circ Res 89(6):496–502
Mattie M, Brooker G, Spiegel S (1994) Sphingosine-1-phosphate, a putative second messenger, mobilizes calcium from internal stores via an inositol trisphosphate-independent pathway. J Biol Chem 269(5):3181–3188
Wu J, Spiegel S, Sturgill TW (1995) Sphingosine 1-phosphate rapidly activates the mitogen-activated protein kinase pathway by a G protein-dependent mechanism. J Biol Chem 270(19):11484–11488
Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem 278(47):46832–46839
Ding G, Sonoda H, Yu H, Kajimoto T, Goparaju SK, Jahangeer S, Okada T, Nakamura S (2007) Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J Biol Chem 282(37):27493–27502
Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH Jr, Milstien S, Spiegel S (2005) SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem 280(44):37118–37129
Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G (2007) BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 3(4):374–376
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581
Sheng R, Zhang TT, Felice VD, Qin T, Qin ZH, Smith CD, Sapp E, Difiglia M, Waeber C (2014) Preconditioning stimuli induce autophagy via sphingosine kinase 2 in mouse cortical neurons. J Biol Chem 289(30):20845–20857
Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S (2003) Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem 278(41):40330–40336
Basu S, Bayoumy S, Zhang Y, Lozano J, Kolesnick R (1998) BAD enables ceramide to signal apoptosis via Ras and Raf-1. J Biol Chem 273(46):30419–30426
Huwiler A, Brunner J, Hummel R, Vervoordeldonk M, Stabel S, van den Bosch H, Pfeilschifter J (1996) Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc Natl Acad Sci USA 93(14):6959–6963
Ruvolo PP, Deng X, Ito T, Carr BK, May WS (1999) Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem 274(29):20296–20300
Ruvolo PP, Deng X, Carr BK, May WS (1998) A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem 273(39):25436–25442
Dbaibo GS, Pushkareva MY, Jayadev S, Schwarz JK, Horowitz JM, Obeid LM, Hannun YA (1995) Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc Natl Acad Sci USA 92(5):1347–1351
Pettus BJ, Chalfant CE, Hannun YA (2002) Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta 1585(2–3):114–125
Mullen TD, Obeid LM (2012) Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med Chem 12(4):340–363
Woodcock J (2006) Sphingosine and ceramide signalling in apoptosis. IUBMB Life 58(8):462–466
Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S (2004) Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 64(12):4286–4293
Edinger AL, Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16(6):663–669
Levine B, Yuan J (2005) Autophagy in cell death: an innocent convict? J Clin Investig 115(10):2679–2688
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304(5676):1500–1502
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6(12):1221–1228
Van Brocklyn JR, Williams JB (2012) The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: oxidative stress and the seesaw of cell survival and death. Comp Biochem Physiol B 163(1):26–36
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122(6):927–939
Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P (2008) Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 90(2):313–323
Demarchi F, Bertoli C, Greer PA, Schneider C (2005) Ceramide triggers an NF-kappaB-dependent survival pathway through calpain. Cell Death Differ 12(5):512–522
Gomez-Vicente V, Donovan M, Cotter TG (2005) Multiple death pathways in retina-derived 661 W cells following growth factor deprivation: crosstalk between caspases and calpains. Cell Death Differ 12(7):796–804
Chua BT, Guo K, Li P (2000) Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem 275(7):5131–5135
Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, Schneider C (2006) Calpain is required for macroautophagy in mammalian cells. J Cell Biol 175(4):595–605
Aronova S, Wedaman K, Aronov PA, Fontes K, Ramos K, Hammock BD, Powers T (2008) Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab 7(2):148–158
Bielawska A, Crane HM, Liotta D, Obeid LM, Hannun YA (1993) Selectivity of ceramide-mediated biology. Lack of activity of erythro-dihydroceramide. J Biol Chem 268(35):26226–26232
Gagliostro V, Casas J, Caretti A, Abad JL, Tagliavacca L, Ghidoni R, Fabrias G, Signorelli P (2012) Dihydroceramide delays cell cycle G1/S transition via activation of ER stress and induction of autophagy. Int J Biochem Cell Biol 44(12):2135–2143
Romero Rosales K, Singh G, Wu K, Chen J, Janes MR, Lilly MB, Peralta ER, Siskind LJ, Bennett MJ, Fruman DA, Edinger AL (2011) Sphingolipid-based drugs selectively kill cancer cells by down-regulating nutrient transporter proteins. Biochem J 439(2):299–311
Shimobayashi M, Oppliger W, Moes S, Jeno P, Hall MN (2013) TORC1-regulated protein kinase Npr1 phosphorylates Orm to stimulate complex sphingolipid synthesis. Mol Biol Cell 24(6):870–881
Niles BJ, Mogri H, Hill A, Vlahakis A, Powers T (2012) Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc Natl Acad Sci USA 109(5):1536–1541
Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R (2012) Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol 14(5):542–547
Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J (2011) Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 108(48):19222–19227
Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463(7284):1048–1053
Zimmermann C, Santos A, Gable K, Epstein S, Gururaj C, Chymkowitch P, Pultz D, Rodkaer SV, Clay L, Bjoras M, Barral Y, Chang A, Faergeman NJ, Dunn TM, Riezman H, Enserink JM (2013) TORC1 inhibits GSK3-mediated Elo2 phosphorylation to regulate very long chain fatty acid synthesis and autophagy. Cell Rep 5(4):1036–1046
Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C (2008) A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4(2):e24
Morselli E, Galluzzi L, Kepp O, Criollo A, Maiuri MC, Tavernarakis N, Madeo F, Kroemer G (2009) Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging 1(12):961–970
Jia K, Levine B (2007) Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3(6):597–599
Greer EL, Brunet A (2009) Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 8(2):113–127
Liu J, Huang X, Withers BR, Blalock E, Liu K, Dickson RC (2013) Reducing sphingolipid synthesis orchestrates global changes to extend yeast lifespan. Aging Cell 12(5):833–841
Huang X, Liu J, Dickson RC (2012) Down-regulating sphingolipid synthesis increases yeast lifespan. PLoS Genet 8(2):e1002493
Cutler RG, Thompson KW, Camandola S, Mack KT, Mattson MP (2014) Sphingolipid metabolism regulates development and lifespan in Caenorhabditis elegans. Mech Ageing Dev 143–144:9–18
Mosbech MB, Kruse R, Harvald EB, Olsen AS, Gallego SF, Hannibal-Bach HK, Ejsing CS, Faergeman NJ (2013) Functional loss of two ceramide synthases elicits autophagy-dependent lifespan extension in C. elegans. PLoS One 8(7):e70087
Marza E, Simonsen KT, Faergeman NJ, Lesa GM (2009) Expression of ceramide glucosyltransferases, which are essential for glycosphingolipid synthesis, is only required in a small subset of C. elegans cells. J Cell Sci 122(Pt 6):822–833
Hearn JD, Lester RL, Dickson RC (2003) The uracil transporter Fur4p associates with lipid rafts. J Biol Chem 278(6):3679–3686
Lauwers E, Grossmann G, Andre B (2007) Evidence for coupled biogenesis of yeast Gap1 permease and sphingolipids: essential role in transport activity and normal control by ubiquitination. Mol Biol Cell 18(8):3068–3080
Finnigan GC, Ryan M, Stevens TH (2011) A genome-wide enhancer screen implicates sphingolipid composition in vacuolar ATPase function in Saccharomyces cerevisiae. Genetics 187(3):771–783
Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B (2010) Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J 24(1):296–308
Nybond S, Bjorkqvist YJ, Ramstedt B, Slotte JP (2005) Acyl chain length affects ceramide action on sterol/sphingomyelin-rich domains. Biochim Biophys Acta 1718(1–2):61–66
Alexaki A, Gupta SD, Majumder S, Kono M, Tuymetova G, Harmon JM, Dunn TM, Proia RL (2014) Autophagy regulates sphingolipid levels in the liver. J Lipid Res 55(12):2521–2531
Liu K, Czaja MJ (2012) Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 20:3–11
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458(7242):1131–1135
Acknowledgments
This work was supported by The Danish Research Councils and by The Lundbeck Foundation. We gratefully appreciate Dr. Dennis Pultz, Dr. Steven V. Rødkær and Esben Schøler Nielsen for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Harvald, E.B., Olsen, A.S.B. & Færgeman, N.J. Autophagy in the light of sphingolipid metabolism. Apoptosis 20, 658–670 (2015). https://doi.org/10.1007/s10495-015-1108-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-015-1108-2