
Abstract
In this work, we were focused on the development of the electrochemical approach resulting in a stable boron doping of titania nanotubes. The doping procedure concerns anodic polarization of as-anodized titania in a H3BO3 solution acting as n boron precursor. The series of attempts were taken in order to elaborate the most beneficial doping conditions. The parameters of electrochemical doping allowing to obtain boron-doped titania characterized by the highest photoconversion efficiency are as follows: reaction voltage 1.8 V, process duration 0.5 h, and the concentration of boric acid 0.5 M. Spectroscopy techniques such as UV-vis, X-ray diffraction, photoluminescence emission, and X-ray photoelectron spectroscopy were used to characterize the absorbance capability and the crystalline phase, to confirm the presence of boron atoms and to study the nature of chemical compounds, respectively. The well-ordered structure of titania and resistance of its morphology toward electrochemical treatment in H3BO3 were confirmed by scanning electron microscopy images. However, cyclic voltammetry and electrochemical impedance spectroscopy studies showed the significant difference in conductivity and capacitance between doped and pristine titania. Moreover, the photocurrent densities of the B-doped sample were about seven times higher in comparison with those generated by the pure titania nanotube electrode.
Similar content being viewed by others


Introduction
In the last decades, the development of photochemical systems capable of harnessing solar energy to produce electricity or chemical fuels has attracted significant interest [1, 2]. The most of research strategies focus on novel types of semiconductors exhibiting photocatalytic activity [3]. In particular, titanium dioxide is regarded as a promising semiconducting material for photoelectrochemical applications due to its low cost, non-toxicity, and stability against photocorrosion [4–6]. However, the activity of titania nanotubes in processes carried out under illumination is limited by their wide bandgap (E bg = 3.2 eV) that corresponds to energy provided by ultraviolet light [7, 8]. For this reason, many approaches have been made to improve the activity of TiO2 that allows its efficient utilization under natural light source—sun. The vast majority of known strategies concerning material modification are based on metal [9–12] and non-metal doping [13–16], formation of organic-inorganic junctions [17], or surface sensitization by dye molecules [18]. To initiate the visible-light-driven activity of titania, doping with non-metal atoms (I, B, C, N, and S) has been considered as a one of the most effective approaches that results in stable material with improved photoactivity compared to the unmodified sample [19, 20].
Recently, doping of TiO2 with boron has been widely studied in terms of its effect on the surface and the bulk structure which changes the ability of tailoring the bandgap, suppresses electron-hole pairs from recombination, and improves electron conductivity [21–23]. Some papers have reported that B-doped TiO2 powders obtained by a modified sol-gel method showed a red-shifted absorption spectrum [24, 25], while other studies concern increase in the bandgap energy of TiO2 upon doping when H3BO3 was used as a boron precursor because of the decrease of the crystal size after incorporation of boron atoms [21]. According to Bettinelli et al. [21], boron used as a titania dopant could favor the transformation of anatase to rutile. On the other hand, Finazzi et al. [26] reported that B atoms in the anatase crystal could occupy different positions and promote changes in the electronic structure of boron-doped TiO2 that is in accordance to the results of density functional theory. In general, previous results on boron-doped titania nanoparticles suggest that preparation method highly impacts on material properties, but simultaneous decrease of bandgap energy value is not always observed.
In this work, we present a new boron doping of titania nanotube (TiO2 NT) approach realized during an electrochemical process carried out at constant voltage when as-anodized titanium plate is immersed in electrolyte containing boric acid as a boron precursor. This method could be regarded as a simple, cheap, and fast doping procedure that enables control of the photoactivity of obtained material. The series of attempts were undertaken in order to optimize the electrochemical doping conditions. The concentration of boric acid and process duration were the main factors that influenced the boron-doping effect on the registered photocurrent. Differences between pure and doped titania were emphasized using various spectroscopic techniques: UV-vis, X-ray diffraction, Fourier transformed infrared, photoluminescence emission spectroscopy, and X-ray photoelectron spectroscopy. The optimal experimental parameters based on the exhibited photocurrent density were determined. Furthermore, performed cyclic voltammetry and impedance spectroscopy measurements were used to characterize electrochemical activity of the most photoactive boron-doped titania.
Experimental
Material preparation
The substrate sample (Ti plate, Strem, 99.7 %) was ultrasonically cleaned in the following order: in acetone, ethanol, and water for 10 min in each solvent and finally dried in the air. Titania nanotubes were prepared via a two-step electrochemical anodization of Ti plate in solution containing fluoride ions that leads to formation of highly ordered tubular structure [10, 27]. The process was realized in a two-electrode configuration, where the titanium metal foil served as an anode and the platinum mesh as a cathode placed at a fixed separation of 2 cm. The first anodization took place in the electrolyte containing the following: ethylene glycol (EG), 0.27 M NH4F, and 1 vol% of deionized water. Anodization was performed at 40 V for 2 h, with the initial voltage rate of 0.1 V/s. Then, the titanium plate was immersed for 12 h in 0.5 % wt. solution of oxalic acid in order to remove the first rugged nanotube layer. The second anodization was performed on the as-cleaned titanium plate under the same conditions as during the first anodization. In order to remove any surface debris, the titanium plates covered with nanotubes were put into diluted HF (0.05 %) solution for 180 s.
As-anodized titania plates were used for further electrochemical doping. It was realized in a two-electrode arrangement, during which the previously prepared titania nanotubes served as an anode and the Pt mesh plate as a cathode immersed in solution of boric acid. The optimization of the electrochemical doping [28] approach was realized in different conditions: reaction times: 15, 30, and 60 min and H3BO3 concentrations: 0.1, 0.5, and 1 M. The doping process was performed under 1.8-V voltage between electrodes. Finally, the samples (pure and doped TiO2 NTs) were rinsed with deionized water, dried in the air, and thermally treated at 450 °C for 2 h with a heating rate of 2 °C min−1 to transform the amorphous TiO2 into a crystalline phase.
Characterization
The sample morphology (top-view and cross-sectional) was investigated by Schottky field emission scanning electron microscopy (FEI Quanta FEG 250) with ET secondary electron detector. The crystalline phase of obtained materials was identified by X-ray diffraction (XRD, Rigaku Miniflex 600) analysis. The measurements were performed with Cu Kα radiation at 2θ angles from 5° to 90° with scan speed of 0.02° min−1. Crystallite size was calculated from the half height width of the (101) diffraction peak of anatase using the Scherrer equation. The reflectance spectra of titania nanotubes were measured with a dual-beam UV-vis spectrophotometer (Lambda 35, PerkinElmer) equipped with a diffuse reflectance accessory. Bandgap energy values were determined as the intercept of the tangent of the plot of transformation of the Kubelka-Munk function (KM0.5 E ph 0.5) vs. photon energy. The Fourier transform infrared spectroscopy (FTIR) measurements were performed using a Nicolet iS50 spectrometer equipped with the Specac Quest single-reflection diamond attenuated total reflectance (ATR) accessory. Spectral analysis was controlled by the OMNIC software package. The FTIR spectra were recorded in the range of 400–4000 cm−1. The photoluminescence spectroscopy (PL) measurements were carried out using laboratory setup consisting of 0.3 m Czerny-Turner spectrograph (SR303i, Andor) equipped with an ICCD camera (DH740, Andor). Samples of pure and boron-doped TiO2 were excited with UV LED (center wavelength 365 nm, FWHM 9 nm, output power 350 mW). PL experiments were carried out in order to determine the quenching efficiency of charge carrier in semiconductor. Escalab 250Xi by Thermo Fisher Scientific was used to perform high-resolution X-ray photoelectron spectroscopy (XPS) measurements to determine the chemical binding properties of the surface, utilizing monochromatic Al Kα source with charge neutralization implemented by means of a flood gun. High-resolution spectra were recorded at energy step size of 0.1 eV at pass energy of 10 eV. In order to normalize spectroscopic measurements, X-axis (binding energy, E bin) from XPS spectrum was calibrated for peak characteristics of neutral carbon 1s (E bin = 284.6 eV). Data analysis was performed using the Avantage software provided by the manufacturer.
Electrochemical characterization
Electrochemical experiments (cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS)) were performed using an Autolab PGStat302N potentionstat-galvanostat system (Methrom, Autolab) in the standard three-electrode arrangement, where titanium foil covered with 6+ nanotubes served as a working electrode (geometric surface area of 0.5 cm2), Ag/AgCl/0.1 M KCl was used as a reference electrode, and platinum mesh acted as a counter electrode. Electrodes were tested in the contact with 0.5-M K2SO4 solution at 23 °C. CV was performed to characterize materials in contact with the deaerated aqueous electrolyte (pH = 7). EIS measurements were conducted at the frequency range from 20 kHz to 0.1 Hz, covering 75 points and with 10-mV amplitude of the AC signal. In order to perform the Mott-Schottky analysis, impedance spectra were recorded at different potentials in the range from +0.6 to −0.5 V vs. Ag/AgCl/0.1 M KCl. Before each spectra registration, the potential was held for 60 s to achieve a steady-state condition.
Photoactivity characterization
Measurements carried out under illumination were performed in the photoelectrochemical cell equipped with a quartz window and the cooling jacket keeping constant temperature at 23 °C (±1 °C) provided by thermostat (Julabo F-12) control. A high-pressure 150-W xenon lamp (Osram XBO 150) equipped with AM1.5 filter was used as a light source. The light intensity was adjusted to 100 mW cm−2 (controlled by power meter, Ophir).
Results and discussion
Morphology and structure studies
In Fig. 1, the top-view and cross-sectional images of pure and boron-doped titania nanotubes are presented. Since the morphologies for the entire set of B-doped TiO2 NTs arrays prepared in different conditions do not change significantly, the representative sample obtained under 1.8 V, for 30 min and in 0.5-M concentrated H3BO3 solution, is shown. The homogeneous layers are composed of highly ordered and aligned nanotubes. When comparing Fig. 1a, b SEM images, any noticeable difference was observed in the architecture between the doped and undoped materials. It suggests that electrochemical doping realized in boric acid solution did not cause any changes in the morphology of the TiO2 NT sample. The layer thickness was determined on the basis of a cross-sectional image. For pure and doped material, the length of nanotubes equals 2.5 μm. The nanotubes were characterized by an average internal diameter of 100 nm and a wall thickness of 25 nm.

Top-view and cross-sectional SEM image of a pure and b B-TiO2 NTs. The doping conditions are as follows: reaction voltage 1.8 V, reaction time 0.5 h, and electrolyte concentration 0.5 M
According to a geometric model [29] that considers a regular network of identical and equally spaced nanotubes, the real surface area of the layer composed of nanotube titania was estimated using the following equation:
where R 1 and R 2, respectively, are the internal radium and the sum of R 1 and half thickness of the wall of the nanotubes, y is the thickness of the slit between the tubes, h is the average length of nanotube, and S geo states for a geometric surface area. Because the distance between each tube is almost imperceptible, the dimension of y was not taken into account for calculations. As the value of the geometric surface, 1 cm2 was used for simplicity. Based on Eq. (1), the real surface area of the layer composed of nanotubes was estimated to be 120 cm2 both for pure and doped materials. Knowing that any difference in tube dimensions and overall real surface area between pristine and modified titania is not observed, only the presence of dopant atoms can have an impact on their structural properties and photoactivity [30, 31].
The plot of Kubelka-Munk function vs. photon energy and the absorbance curves are presented in Fig. 2 and Fig. 2 inset, respectively. It can be clearly observed that the absorption edge position of B-doped TiO2 moves toward longer wavelengths. It is confirmed that electrochemical doping results in TiO2 with the obvious red shifted photoactivity and results in the bandgap narrowing [32]. The values of E bg determined on the basis of the Tauc plot are 2.91 and 3.09 eV for B-doped and pure TiO2 NTs, respectively. Among other non-metals used as titania dopands, boron atoms can be introduced into the TiO2 lattice and the p orbital of B is mixed with O2p orbitals, leading to a shift in the optical response to the visible range [33]. This is due to the fact that the ion radius of B(III) (0.023 nm) is much smaller than that of Ti(IV) (0.064 nm). Therefore, boron oxides existing at the surface of the material can easily incorporate into the crystalline framework of titania [21, 34].

The absorbance spectra and the Kubelka-Munk function for pure and B-doped titania. The doping conditions are as follows: reaction voltage 1.8 V, reaction time 0.5 h, and electrolyte concentration 0.5 M
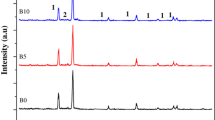
Figure 3a shows XRD spectra registered for boron-doped and pure titania layer. All detected crystallite phases were indexed as characteristic peaks of anatase (A) (JCPDS No. 21-1272) and the titanium (Ti) phase (JCPDS No. 44-1294) [21] acting as TiO2 NT support. It should be noted that any peak typical for rutile was not observed. Furthermore, the lack of signal attributed to dopant atoms could result from their low content in the B-TiO2 material and cannot be detected by XRD [35]. However, as it is shown in Fig. 3b, the addition of B atoms leads to changes in shape of A(101) peak. The increase of its intensity is related to the crystallinity improvement. According to Li et al. [22], such phenomena observed for B-doped TiO2 was due to the reduction of the grain boundaries and amorphous regions acting as charge carrier recombination centers. Basing on the Scherrer equation, crystallite size was calculated and equals 17.3 nm for pure and 20.4 nm for boron-doped titania. The increase of crystallite size can be ascribed to the incorporation of dopant atoms into the structure, e.g., substitution of the lattice oxygen/titanium atoms or introduction of boron at the interstitial sites [36].

a XRD patterns of pure and boron-doped titania and b the magnification of A(101) peak extracted from XRD patterns. The doping conditions are as follows: reaction voltage 1.8 V, reaction time 0.5 h, and electrolyte concentration 0.5 M.
Figure 4 shows FTIR spectra of pure and B-TiO2 samples. Both titania samples exhibit the main band between 400 and 700 cm−1, which are attributed to Ti–O stretching and Ti–O–Ti bridging stretching modes [37]. The peak found around 1000 cm−1 could be assigned to the stretching vibration of Ti–O and Ti–O–Ti [22]. The bands observed at about 3400 and 1600 cm−1 are assigned to the stretching of hydroxyl groups and the bending vibration of water molecules adsorbed on the material surface [38]. In the case of B-TiO2 sample, the intensity of the FTIR spectra is higher than that of pristine titania. It is related to higher surface energy and larger amount of hydroxyl groups at the B-doped comparing to pristine titania [31].

FTIR spectra of pure and boron-doped TiO2 nanotubes
The presence of boron and its chemical nature in TiO2 NTs layers were studied using the XPS. The high-resolution XPS spectra of the boron region were registered only in the case of doped titania. The XPS spectra obtained for pure and boron-doped TiO2 were shown and discussed in our previous article [39]. According to these results, signals attributed to titanium, oxygen, and carbon were found in both TiO2 NT samples. The positions of maxima of fitted spectra obtained for investigated materials are listed in Table 1. The XPS spectrum of B1s region registered at the titania surface exhibits two singlet peaks. One of them with the binding energy of 190.71 eV is assigned to boron directly linked to titanium atoms [40], whereas the second signal with maximum found at 192.64 eV results from presence of Ti–O–B or B–O–B arrangement [41]. According to literature [27], boron atoms could occupy interstitial sites in anatase crystals and thereby take part in the balance of residual charge. Such location of boron ions is justified by almost three times smaller radius of B(III) comparing to Ti(IV) [17]. Taking into account only oxygen linked to boron and titanium, the atomic ratio of Ti/O/B found at the material surface equals: 1:2.39:0.73. However, XPS investigation performed after subsequent etching of titania layer shows decrease in boron content down to 2.74 % and the signal attributed to the B–O arrangement disappears (see Fig. 5). The difference in boron distribution at the external layer and inside the material could result from adsorption of boric acid on the titania surface in a form of monodentate arrangement: –HOB(OH)O(H)-Ti [42]. Therefore, because of significant amount of surface boron species, the increase in hydroxyl groups amount observed on FTIR spectra comparing to pristine titania provides the neutrality of B-TiO2 surface [43].

High-resolution XPS spectra recorded at the boron region for B-TiO2 sample
In the case of XPS spectra registered for titanium and oxygen, the shift in binding energy value both for Ti2p and O1s is observed. That may be caused by the introduction of boron ions into the titania lattice. Furthermore, in the case of B-TiO2, additional oxygen signal was found that is related to the oxygen linked to boron.
The photoluminescence spectra (PL) of undoped and boron-doped samples excited at a wavelength of 365 nm at room temperature are presented in Fig. 6. PL is commonly used for examination of the rate of recombination, the electronic structure of nanomaterials, and the transfer behavior of the photoexcited electron-hole pairs in semiconductors. The luminescence intensity is strongly related with the radiative recombination of photogenerated electrons and holes, so the higher the intensity of PL, the greater the likelihood of occurrence of electron-hole recombination processes [44]. For both materials, the registered PL spectra are characterized by a broad band in the range of 480–600 nm with a maximum at 530 nm that is attributed to activity of surface states [45]. The doped titania nanotubes exhibit lower intensity of photoluminescence than pure TiO2. Thus, it can be expected that boron atoms efficiently participate in a charge carrier separation and significantly inhibit radiative recombination processes [46]. Furthermore, according to Begum et al. [47], boron atoms can be located in the interstitial positions of the TiO2 lattice leading to the partial reduction of Ti(IV) to Ti(III), which could act as an electron trap.

Photoluminescence spectra of pure and B-doped sample. The doping conditions are as follows: reaction voltage is 1.8 V, reaction time is 0.5 h, and electrolyte concentration is 0.5 M
Electrochemical properties
To evaluate the electrochemical properties of the obtained samples, CV and electrochemical impedance spectra were registered in 0.5 M K2SO4. In Fig. 7, typical CV curves of the B-TiO2 and TiO2 electrodes collected at the scan rate of 20 mV s−1 were shown. As it could be seen, the area limited by the CV curve for boron-doped titania is much higher than that for pure material, which indicates higher electrical double-layer capacitance of B-TiO2 comparing to undoped titania. Similar relations between double-layer capacitance of boron-containing and boron-free titania nanotubes were reported by Li et al. and were attributed to the presence of Ti(III) traces leading to increase of material conductivity [48].

Cyclic voltammetry curves recorded for doped and pure TiO2 immersed in 0.5 M K2SO4, v = 50 mV s−1
Apart from higher capacitive current, on the reverse scan of CV registered for B-TiO2, at about −0.24 V vs. Ag/AgCl/0.1 M KCl, additional cathodic peak arised (see B inset). As it was reported for other non-metal-doped titania nanotubes, it could be attributed to the reduction of Ti(IV) sites in TiO2 that causes change in the electronic structure of titania. It is related to the formation of additional energy levels within the bandgap, and as the result, both electrical conductivity and optical properties of material are different than that of pristine TiO2. Further cathodic polarization of electrode leads to initiation of the hydrogen evolution process. Additionally, the difference in CV run between boron-doped and pristine titania is observed in anodic potential range. In the case of B-TiO2, the current increases more steeply comparing to unmodified sample, showing that also anodic oxygen evolution reaction is facilitated upon boron doping [49].
EIS measurements were conducted on pure and boron-doped titania in the wide potential range. In accordance to proposed approach for interpretation of EIS spectra registered for TiO2 nanotube arrays [8], Mott-Schottky analysis was performed. Thus, the plot shown in Fig. 8 was prepared on the basis of relation given as follows:

The Mott-Schottky plot for pristine and boron-doped titania nanotubes
where C is the areal capacity of the space charge layer, q—elementary charge, ε 0 —vacuum permittivity, ε—dielectric constant, N D concentration of donors, E—applied potential bias, E fb —flatband potential, k—Boltzmann’s constant, and T—absolute temperature. The non-linear relation in the range from +0.8 to −0.5 V vs. Ag/AgCl/0.1 M KCl is due to intrinsic electronic state [50] and could result from charging of bulk or surface states [51] or irregular inhomogeneous distribution of O2− ions [52]. Both doped and pristine titania exhibit positive slope that is characteristic for n-type semiconductor, and from the intercept with E-axis, flatband potential could be determined [53]. For pure titania, E fb equals −0.04 V vs. Ag/AgCl/0.1 M KCl, whereas in the case of B-TiO2, E fb value is anodically shifted to −0.14 V vs. Ag/AgCl/0.1 M KCl. Negative shift in E fb can be attributed to the improved electron concentration and trap distribution [54] caused by boron doping.
Assuming that difference between E fb and the bottom of the conduction band (CB) is negligible, the position of CB could be identified with flatband potential value [13]. Knowing that bandgap energy for B-TiO2 equals 2.91 V, the position of valence band (VB) according to the relation E CB + E bg ≈ E VB could be estimated to be 2.77 V vs. Ag/AgCl/0.1 M KCl, whereas for pure titania, E VB ≈ 3.05 V vs. Ag/AgCl/0.1 M KCl. Thus, anodic shift of the CB in the case of B-TiO2 is accompanied by an anodic shift of the VB edge. This should improve the oxidation potential of the holes generated upon sample illumination [55].
Apart from change in E fb position, the difference in slopes in Mott-Schottky plots is observed and indicates a disparity of donor densities in pure and boron-doped titania. The carrier density N D could be calculated from the following equation:
when the following values are taken into account: e = 1.6 × 10−19, ε 0 = 8.86 × 10−14 F/cm, and ε = 31 for anatase [56]. The calculated donor densities of TiO2 NTs and B-TiO2 NTs equal to 1.98 × 1020 and 1.82 × 1021 cm−3, respectively. Despite the fact that the Mott-Schottky plot does not allow to estimate the absolute value of N D [57], it is shown that upon electrochemical doping, donor density is ten times higher comparing to the unmodified sample. Such change was observed for other modified titania samples and is commonly attributed to the introduction of substantial oxygen vacancies that are regarded as electron donors in titania [58, 59] and implied faster carrier transfer in doped TiO2 comparing to pure titania [60].
Photoelectroactivity
The photocurrent densities as a function of potential of undoped and B-doped TiO2 nanotube (exhibiting the highest photocurrent) electrodes exposed to radiation from a solar simulator equipped with an AM1.5 filter are presented in Fig. 9. The dark current is very low and can be negligible; thus, measured current under UV-vis illumination could be regarded as a pure photocurrent. In the case of doped material, the significant increase in photocurrent compared to pristine titania nanotubes is observed in the wide potential range from −0.6 up to +0.9 V vs. Ag/AgCl/0.1 M KCl. This increase in resulting from electrode illumination is significantly higher than reported by others [15, 22, 61] , indicating that proposed experimental approach could be regarded as an attractive alternative for other doping methods. The improved photoelectrochemical properties could be assigned to the boron doping which lead to bandgap narrowing, conductivity improvement, and inhibition of charge carrier recombination processes.

Variation of photocurrent density under UV-vis illumination versus potential for pure and boron-doped titania in 0.5 M K2SO4 (v = 20 mV s−1)
In Fig. 10a, b, the transient photocurrent response is given for the pure and the set of boron-doped TiO2 samples prepared under different conditions. The specific parameters of each electrochemical doping procedure (concentration of boric acid, reaction time) are listed in Table 2 together with the photocurrent density registered after 10 min of measurement duration and enhancement factor (EF). EF is defined as a ratio between photocurrent registered for boron-doped sample and unmodified titania. Such approach used during optimization procedure allows to determine experimental conditions resulting in the most photoactive B-TiO2 nanotube arrays. Independently from doping conditions (sample: A—119.7, B—199.3. C—123.3, D—199.3, E—310.7, and F—232.8 μA cm−2), all titania samples doped with boron exhibit much higher photocurrents than non-doped titania (41.7 μA cm−2). Furthermore, the lack of steady current diminution suggests that the material is characterized by resistance toward photocorrosion processes. The highest steady-state photocurrent was observed for B-TiO2 and reached the value of almost 311 μA cm−2. The increase of generated photocurrent is influenced by the presence of dopant and can be caused by the decrease of the recombination rate and charge transfer resistance and also enhanced light absorption in the visible range as reported for other types of non-metal-doped nanotube arrays [62]. Summarizing, on the basis of the EF value, the optimal B-doping conditions were determined as follows: reaction time 0.5 h and electrolyte concentration 0.5 M.

Photocurrent response of pure titania and boron-doped nanotube array samples (A–F)
As can see in Fig. 11a, even during 120 min of illumination, boron-doped material generates stable photocurrent; thus, obtained material seems to be photocorrosion resistant. Additionally, the CV curves of B-TiO2 electrode were recorded in 0.5-M K2SO4 solution before and after long-term illumination (see Fig. 10b). Therefore, electrochemical measurements also confirm high stability of doped TiO2 nanotube layer in wide potential range.

a The long-term chronoamperometry test (E = 0.5 V vs. Ag/AgCl (0.1 M KCl)) recorded during illumination of B-TiO2 in 0.5 M K2SO4 electrolyte. b The cyclic voltammetry curves of material electrode recorded in 0.5 M K2SO4 before and after long-term illumination, scan rate 50 mV s−1. The doping conditions are as follows: reaction voltage is 1.8 V, reaction time is 0.5 h, and electrolyte concentration is 0.5 M.
Conclusions
In this work, the optimization procedure in order to achieve highly photoactive and stable titania nanotube doped with boron was proposed. It has been found that electrolyte concentration, reaction voltage, and time are the most crucial factors affecting the photoelectrochemical response of the doped material. According to the inspection performed via scanning electron microscopy, the electrochemical doping in electrolyte containing H3BO3 as boron precursor does not affect the morphology of the as-anodized titania nanotubes. The existence of boron atoms both at the surface and inside the layer confirmed by the energy-dispersive X-ray spectroscopy allows to treat electrochemical doping as efficient technique for boron incorporation. Titania doped by boron atoms possesses lower bandgap energy than pure titania, but the material is still characterized by an anatase crystalline phase. On the basis of the EF, the optimal conditions were determined as follows: reaction time 0.5 h and boric acid concentration 0.5 M. The test performed under sample irradiation showed that B doping radically improves the photoelectrochemical activity and B-TiO2 exhibits 7.5 times higher photocurrent than unmodified sample. Summarizing, boron-doped TiO2 NTs could be regarded as a promising photoelectrode material for photovoltaic cells, water splitting, or electrocatalytic processes.
References
Balzani V, Credi A, Venturi M (2008) Photochemical conversion of solar energy. ChemSusChem 1:26–58
Momeni MM, Ghayeb Y, Mohammadi F (2015) Solar water splitting for hydrogen production with Fe2O3 nanotubes prepared by anodizing method: effect of anodizing time on performance of Fe2O3 nanotube arrays. J Mater Sci Mater Electron 26:685–692
Memming R (2001) Semiconductor Electrochemistry. Weinheim, Wiley–VCH
Fujishima A, Rao TN, Tryk DA (2000) Titanium dioxide photocatalysis. J Photochem Photobiol C 1:1–21
Carp O, Huisman CL, Reller A (2004) Photoinduced reactivity of titanium dioxide. Prog Solid State Chem 32:33–177
Zhong P, Liao Y, Que W, Jia Q, Lei T (2014) Enhanced electron collection in photoanode based on ultrafine TiO2 nanotubes by a rapid anodization process. J Solid State Electrochem 18:2087–2098
Beranek R, Macak JM, Gärtner M, Meyer K, Schmuki P (2009) Enhanced visible light photocurrent generation at surface-modified TiO2 nanotubes. Electrochim Acta 54:2640–2646
Momeni MM, Ghayeb Y (2015) Visible light-driven photoelectrochemical water splitting on ZnO–TiO2 heterogeneous nanotube photoanodes. J Solid State Electrochem 45:557–566
Momeni MM, Ghayeb Y (2015) Fabrication, characterization and photoelectrochemical behavior of Fe–TiO2 nanotubes composite photoanodes for solar water splitting. J Solid State Electrochem 751:43–48
Gong J, Pu W, Yang C, Zhang J (2013) Novel one-step preparation of tungsten loaded TiO2 nanotube arrays with enhanced photoelectrocatalytic activity for pollutant degradation and hydrogen production. Catal Commun 36:89–93
Momeni MM, Ghayeb Y, Ghonchegi Z (2015) Fabrication and characterization of copper doped TiO2 nanotube arrays by in situ electrochemical method as efficient visible-light photocatalyst. Ceram Int 41:8735–8741
Momeni MM, Ghayeb Y (2015) Photoelectrochemical water splitting on chromium-doped titanium dioxide nanotube photoanodes prepared by single-step anodizing. J Alloys Compd 637:393–400
Siuzdak K, Szkoda M, Sawczak M, Lisowska-Oleksiak A (2015) Novel nitrogen precursors for electrochemically driven doping of titania nanotubes exhibiting enhanced photoactivity. New J Chem 39:2741–2751
Siuzdak K, Szkoda M, Sawczak M, Lisowska-Oleksiak A, Karczewski J, Ryl J (2015) Enhanced photoelectrochemical and photocatalytic performance of iodine-doped titania nanotube arrays. RSC Adv 5:50379–50391
Lu N, Zhao H, Li J, Quan X, Chen S (2008) Characterization of boron-doped TiO2 nanotube arrays prepared by electrochemical method and its visible light activity. Sep Purif Technol 62:668–673
Momeni MM, Ghayeb Y, Ghonchegi Z (2015) Visible light activity of sulfur-doped TiO2 nanostructure photoelectrodes prepared by single-step electrochemical anodizing process. J Solid State Electrochem 19:1359–1366
Spanggaard H, Krebs FC (2004) A brief history of the development of organic and polymeric photovoltaics. Sol Energy Mater Sol Cells 83:125–146
Shankar K, Mor GK, Prakasam HE, Varghese OK, Grimes CA (2007) Self-assembled hybrid polymer-TiO2 nanotube array heterojunction solar cells. Langmuir 23:12445–12449
Lu N, Quan X, Li JY, Chen S, Yu HT, Chen GH (2007) Fabrication of boron-doped TiO2 nanotube array electrode and investigation of its photoelectrochemical capability. J Phys Chem C 111:11836–11842
Lisowska-Oleksiak A, Szybowska K, Jasulaitienè V (2010) Preparation and characterisation of visible light responsive iodine doped TiO2 electrodes. Electrochim Acta 55:5881–5885
Chen D, Yang D, Wang Q, Jiang Z (2006) Effects of boron doping on photocatalytic activity and microstructure of titanium dioxide nanoparticles. Ind Eng Chem Res 45:4110–4116
Li J, Lu N, Quan X, Chen S, Zhao H (2008) Facile method for fabricating boron-doped TiO2 nanotube array with enhanced photoelectrocatalytic properties. Ind Eng Chem Res 47:3804–3808
Subramanian A, Wang HW (2012) Effects of boron doping in TiO2 nanotubes and the performance of dye-sensitized solar cells. Appl Surf Sci 258:6479–6484
Zhao W, Ma WH, Chen CC, Zhao JC, Shuai ZG (2004) Efficient degradation of toxic organic pollutants with Ni2O3/TiO2−xBx under visible irradiation. J Am Chem Soc 126:4782–4783
In S, Orlov A, Berg R, Garcia F, Pedrosa-Jimenez S, Tikhov MS, Wright DS, Lambert RM (2007) Effective visible light-activated B-doped and B N-codoped TiO2 photocatalysts. J Am Chem Soc 129:13790–13791
Bettinelli M, Dallacasa V, Falcomer D, Fornasiero P, Gombac V, Montini T, Roman‘o L, Speghini A (2007) Photocatalytic activity of TiO2 doped with boron and vanadium. J Hazard Mater 146:529–534
Finazzi E, Valentin CD, Pacchioni G (2009) Boron-doped anatase TiO2: pure and hybrid DFT calculations. J Phys Chem C 113:220–228
Momeni MM, Ghayeb Y, Davarzadeh M (2015) Single-step electrochemical anodization for synthesis of hierarchical WO3–TiO2 nanotube arrays on titanium foil as a good photoanode for water splitting with visible light. J Electroanal Chem 739:149–155
Szkoda M, Siuzdak K, Lisowska-Oleksiak A (2015) Optimization of electrochemical doping approach resulting in highly photoactive iodine-doped titania nanotubes. J Solid State Electrochem. doi:10.1007/s10008-015-3081-7
Pu P, Cachet H, Ngaboyamahina E, Sutter EMM (2013) Relation between morphology and conductivity in TiO2 nanotube arrays: an electrochemical impedance spectrometric investigation. J Solid State Electrochem 17:817–828
Bessegato GG, Cardoso JC, Zanoni MVB (2015) Enhanced photoelectrocatalytic degradation of an acid dye with boron-doped TiO2 nanotube anodes. Catal Today 240:100–106
Zhuang H, Lin C, Lai Y, Sun L, Li J (2007) Some critical structure factors of titanium oxide nanotube array in its photocatalytic activity. Environ Sci Technol 41:4735–4740
Deng L, Chen Y, Yao M, Wang S, Zhu B, Huang W, Zhang S (2010) Synthesis, characterization of B-doped TiO2 nanotubes with high photocatalytic activity. J Sol-Gel Sci Technol 53:535–541
Dozzi MV, Selli E (2013) Doping TiO2 with p-block elements: effects on photocatalytic activity. J Photochem Photobiol C: Photochem Rev 14:13–28
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A 32:751–767
Yu JG, Dai GP, Cheng B (2010) Effect of crystallization methods on morphology and photocatalytic activity of anodized TiO2 nanotube array films. J Phys Chem C 114:19378–19385
Cheng X, Liu H, Chen Q, Li J, Wang P (2013) Construction of N, S codoped TiO2 NCs decorated TiO2 nano-tube array photoelectrode and its enhanced visible light photocatalytic mechanism. Electrochim Acta 103:134–142
Jeong JH, Jung DW, Shin EW, Oh ES (2014) Boron-doped TiO2 anode materials for high-rate lithium ion batteries. J Alloys Compd 604:226–232
Yu JG, Yu HG, Cheng B, Zhao WJ, Yu JC, Ho WK (2003) The effect of calcination temperature on the surface microstructure and photocatalytic activity of TiO2 thin films prepared by liquid phase deposition. J Phys Chem B 107:13871–13879
Nam SH, Cho SJ, Boo JH (2012) Growth behavior of titanium dioxide thin films at different precursor temperatures. Nanoscale Res Lett 7:1–6
Kamal MSK, Mohamed IZJ (1997) Synthesis of high surface area titania powders via basic hydrolysis of titanium (IV) isopropoxide. Powder Technol 92:233–239
Szkoda M, Siuzdak K, Lisowska-Oleksiak A, Karczewski J, Ryl J (2015) Facile preparation of extremely photoactive boron-doped TiO2 nanotube arrays. Electrochem Commun 60:212–215
Wei C, Rang X, Liang J, Tan S (2007) Growth behavior of titanium dioxide thin films at different precursor temperatures. J Environ Sci 19:90–96
Li L, Yang Y, Liu X, Fam R, Shi Y, Li S, Zhang L, Fan X, Tang P, Xu R, Zhang W, Wang Y, Ma L (2013) Growth behavior of titanium dioxide thin films at different precursor temperatures. Appl Surf Sci 265:36–40
Raghunath P, Lin MC (2008) Adsorption configurations and reactions of boric acid on a TiO2 anatase (101) surface. J Phys Chem C 112:8276–8287
Feng N, Zheng A, Wang Q, Ren P, Gao X, Liu SB, Shen Z, Chen T, Deng F (2011) Boron environments in B-doped and (B, N)-codoped TiO2 photocatalysts: a combined solid-state NMR and theoretical calculation study. J Phys Chem C 115:2709–2719
Siuzdak K, Szkoda M, Lisowska-Oleksiak A, Grochowska K, Karczewski J, Ryl J (2015) Thin layer of ordered boron-doped TiO2 nanotubes fabricated in a novel type of electrolyte and characterized by remarkably improved photoactivity. Appl Surf Sci 357:942–950
Fross L, Schubnell M (1993) Temperature dependence of the luminescence of TiO2 powder. Appl Phys B: Photophys Laser Chem 56:363–366
Jung KY, Park SB, Matsuoka M, Anpo M (2003) In-situ investigations of the photoluminescence properties of SiO2/TiO2 binary and Boron-SiO2/TiO2 ternary oxides prepared by the sol-gel method and their photocatalytic reactivity for the oxidative decomposition of trichloroethylene. Int J Photogr 5:31–36
Begum NS, Ahmed HMF, Hussain OM (2008) Characterization and photocatalytic activity of boron-doped TiO2 thin films prepared by liquid phase deposition technique. Bull Mater Sci 31:741–745
Li H, Xing J, Xia Z, Chen J (2014) Preparation of extremely smooth and boron-fluorine co-doped TiO2 nanotube arrays with enhanced photoelectrochemical and photocatalytic performance. Electrochim Acta 139:331–336
Kim S, Yoo H, Rhee O, Choi J (2015) Doping of Pt into anodic TiO2 nanotubes for Water Oxidation: Underpotential shock method in Cl- solution. J Phys Chem C 119:21497–21503
Munoz AG, Chen Q, Schmuki P (2007) Interfacial properties of self-organized TiO2 nanotubes studied by impedance spectroscopy. J Solid State Electrochem 11:1077–1084
Bonham DB, Orazem ME (1992) a mathematical model for the influence of deep‐level electronic states on photoelectrochemical impedance spectroscopy II. Assessment of characterization methods based on mott‐schottky theory. J Electrochem Soc 139:127–131
Wang Z, Zhao J, Wang X, Zhou J (2015) Causes for the Formation of Titania Nanotubes During Anodization. IEEE Trans Nanotechnol 14:113–117
Gelderman K, Lee DSW (2007) Flat-band potential of semiconductor: using the Mott-Schottky equation. J Chem Educ 84:685–689
Gu F, Huang W, Wang S, Cheng X, Hu Y, Lee PS (2014) Open-circuit voltage improvements in tantalum-doped TiO2 nanocrystals. Phys Chem Chem Phys 16:25679–25683
Macyk W, Burgeth G, Kisch H (2003) Photoelectrochemical properties of platinum (IV) chloride surface modified TiO2. Potochem Photobiol Sci 2:322–328
Tang H, Prasad K, Sanjines R P, Levy F (1994) Electrical and optical properties of TiO2 anatase thin films. J Appl Phys 75:2042–2047
Chen B, Beach JA, Maurya D, Moore RB, Priya S (2014) Fabrication of black hierarchical TiO2 nanostructures with enhanced photocatalytic activity. RSC Adv 4:29443–29449
Cui H, Zhao W, Yang C, Yin H, Lin T, Shan Y, Xie Y, Gu H, Hang F (2014) Black TiO2 nanotube arrays or high-efficiency photoelectrochemical water-splitting. J Mater Chem A 2:8612–8616
Lu X, Ang GW, Zhai T, Yu M, Gan J, Tong Y, Li Y (2012) Hydrogenated TiO2 nanotube arrays for supercapacitors. Nano Lett 12:1690–1696
Acknowledgments
This work received financial support from the Polish National Science Centre: Grant No. 2012/07/D/ST5/02269. The authors are grateful to K. Karczewski (Gdańsk University of Technology) and K. Grochowska (The Szewalski Institute of Fluid Flow Machinery) for technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szkoda, M., Lisowska-Oleksiak, A. & Siuzdak, K. Optimization of boron-doping process of titania nanotubes via electrochemical method toward enhanced photoactivity. J Solid State Electrochem 20, 1765–1774 (2016). https://doi.org/10.1007/s10008-016-3185-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3185-8