
Abstract
Graphene/Fe3O4 nanocomposite was prepared for the immobilization of hemoglobin (Hb) to improve the electron transfer between Hb and glass carbon electrode (GCE). The characterization of nanocomposites was described by transmission electron microscopy, Fourier transform infrared, Raman spectroscopy, and X-ray photoelectron spectroscopy, respectively. The electrochemistry of Hb on the graphene/Fe3O4-based GCE was investigated by cyclic voltammetry and amperometric measurement. The modified electrode showed a wide linear range from 0.25 μmol/L to 1.7 mmol/L with a correlation coefficient of 0.9967. The detection limit of the H2O2 biosensor was estimated at 6.0 × 10−6 mol/L at a signal-to-noise ratio of 3.
Similar content being viewed by others

Introduction
Graphene (GE), with one-atom-thick planar sheets of sp2-bonded carbon atoms, has attracted much attention recently due to its high surface area, excellent electrical conductivity, good mechanical strength, and high thermal conductivity [1]. Based on these properties, GE has shown great potential applications in electronic devices [2], energy conversion and storage [3], and biosensors [4]. However, GE is limited to designing biosensors for its hydrophobicity and agglomerate in water [5, 6]. Therefore, the combination of GE with other functional materials may result in the composition integrating characterizations and functions of each component. Among these, poly(diallyldimethylammonium chloride) (PDDA), a linear positively charged polyelectrolyte, is an effective reducing agent and can prevent the aggregation of GE sheets through electrostatic repulsion [7, 8]. Fe3O4 with low toxicity, good biocompatibility, and strong adsorption ability may serve as an ideal matrix for investigating entrapped biomolecules. For these biomolecules, hemoglobin (Hb) is a heme protein containing four electroactive iron hemes, which can store and transport oxygen in red blood cells [9]. In contrast to most other proteins, Hb is highly stable and can construct H2O2 sensor as a result of its commercial availability and peroxidase activity [10]. However, it is difficult for an electron to transfer between Hb and the electrode, as the redox center of proteins is embedded in polypeptide chain structures. The objective of this work was to explore the ability of the GE/Fe3O4 nanocomposite in immobilizing Hb on the electrode surface and promoting direct electron transfer between Hb and electrodes.
In this paper, we prepared Fe3O4 and then added it to the reduction process, during which process the graphene oxide was reduced to GE by PDDA. The GE/Fe3O4 nanocomposite was modified onto a glass carbon electrode (GCE), where Hb was immobilized by drop casting. The amperometric H2O2 biosensor based on GE/Fe3O4/Hb GCE was fabricated for investigating the effect of the nanocomposite on the direct electrochemistry of Hb, including the linear range, detection limitation, solution pH, stability, and reproducibility towards H2O2.
Experimental
Reagents
Graphite powder (325 mesh) was obtained from Nanjing XF Nano Materials Tech Co., Ltd. PDDA (Mw, 2–3.5 × 105), Hb (Mw, 64,500), chitosan (Mw, 5–6 × 105, >85 % deacetylation), diethanolamine (DEA), and diethylene glycol (DEG) were purchased from Sigma. All other chemicals were of analytical reagent grade. The phosphate buffer solution (PBS, 0.1 mol/L) and the doubly distilled water were used in all the experiments unless otherwise stated.
Apparatus and measurements
Transmission electron microscopy (TEM) was carried out with Tecnai 12 operating at 200 kV accelerating voltage. Fourier transform infrared (FTIR) was recorded by Bruker Vector with the KBr method. Raman spectra were collected on Horiba-JY HR 800 equipped with a 514.5-nm laser source. X-ray photoelectron spectroscopy (XPS) was performed using Thermo Scientific ESCALAB 250 equipped with Mg Kα radiation (hν = 1,253.6 eV) at a power of 250 W. Cyclic voltammetry (CV) and amperometric measurement were carried out on CHI760D electrochemical workstation using three electrode systems. The working electrode was GCE (3 mm in diameter) and modified GCE. A saturated calomel electrode and a platinum wire served as reference and counter electrodes, respectively. All tested solutions were purged with N2 before experiments and during measurements. In the electrochemical testing and analysis, the abbreviated symbols are explained as follows: anodic and cathodic voltage (E p,a and E p,c), anodic and cathodic current (I p,a and I p,c), and correlation coefficient (R n).
Preparation of GE/Fe3O4 nanocomposite
Firstly, Fe3O4 nanoparticles were synthesized by a solvothermal coprecipitation method [11, 12]. In brief, FeCl3·6H2O (10 mmol) and FeCl2·4H2O (5 mmol) were dispersed in DEG (50 mL) for 30 min at 90 °C. DEA (10 mL) was added to the above solution. Meanwhile, NaOH (40 mmol) and DEG (20 mL) were mixed to produce a stock solution and then introduced into the hot iron solution. The mixture was kept in a Teflon-lined autoclave for 8 h at 200 °C. After centrifugation and washing, Fe3O4 nanoparticles were obtained. Secondly, graphite oxide (GO) was prepared from graphite powder by a modified Hummers method [13]. Under ultrasonic irradiation, GO (100 mg) was dispersed in deionized water (100 mL) to yield a yellow–brown dispersion. The unexfoliated GO was removed by centrifugation. Subsequently, the homogeneous GO dispersion was mixed with PDDA solution (6 mL, 20 wt%) and Fe3O4 nanoparticles under vigorous stirring. When the suspension was maintained for 5 h in refluxing conditions, GE/Fe3O4 nanocomposite was obtained. If Fe3O4 was not added at the previous step, the PDDA-functionalized graphene (PDDA-G) was collected.
Configuration of modified electrode
The bare GCE was polished with 1.0, 0.3, and 0.05 μm alumina slurry, rinsed thoroughly with deionized water, and dried by N2 blowing. Each of GE/Fe3O4, PDDA-G, and Fe3O4 of 1.5 mg was added into 1 mL 0.5 % chitosan solution to form different suspending mixtures under ultrasonication for 2 h. The various suspensions of 5 μL were cast onto the surface of GCE or modified GCE and then dried at room temperature. The GE/Fe3O4/Hb-, PDDA-G/Hb-, and Fe3O4/Hb-modified electrodes were prepared by the simple drop-casting technique in a Hb solution of 5 mg/L.
Results and discussion
Characterization of GE/Fe3O4
The morphology of GE/Fe3O4 was observed by TEM. Figure 1a shows GO has been reduced to GE sheet by PDDA, clarifying the crumpled and wrinkled flake-like structure. Adding Fe3O4 to GO reducing reaction, Fe3O4 nanoparticles adhere uniformly to the surface of an individual GE sheet with little Fe3O4 agglomeration in Fig. 1b. As shown in Fig. 1c, the Fe3O4 particle diameter is roughly 7 nm, providing a catalytic reaction active center.

TEM image of a GE, b GE/Fe3O4, and c zoomed-in edge of b
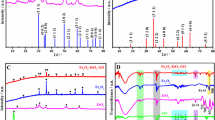
As shown in Fig. 2, FTIR was employed to confirm the chemistry structure of the GE/Fe3O4 nanocomposite and the functionalized and assembly process. For GE/Fe3O4, the peaks at 632 and 580 cm−1 are denoted to stretching vibrations of Fe–O, which suggests the presence of iron oxide. Compared with GO, the absorption intensities of GE/Fe3O4 at 960, 1,110, 1,395, and 1,733 cm−1 decrease or even disappear. These bands can be attributed to ether/ester (C–O–C), alkoxy (C–O), carboxy (C=O), and carbonyl/carboxy (C=O) groups, respectively. The results prove that the oxygen functional groups of GO are almost entirely removed from GE with PDDA acting as reducing reagent. Simultaneously, PDDA-G owns the characteristic bands of PDDA at 2,921 (CH n ), 1,630 (C=C) and 1,465 (C=C), 1,381 (N–O), and 862 cm−1 (C–N), which suggests the GE is successfully functionalized by PDDA.

FTIR spectra of GO, PDDA-G, and GE/Fe3O4
Raman spectroscopy was used to probe the disorder of carbon structures during the change from graphite to GO and GE. Figure 3 shows the typical D band (∼1,350 cm−1) and G band (∼1,580 cm−1) of graphite. The D band is assigned to the vibrations of sp3 carbon atoms of disordered graphite. The G band is related to the coplanar vibration of sp2 carbon atoms in a 2D hexagonal lattice. Generally, the intensity ratio of the D to G band (I D/I G) is a useful indicator to evaluate the ordered and disordered crystal structures of carbon. The I D/I G value of 0.28 for graphite is very small, reflecting the low defect density of graphite. Then, the value increases to 1.56 for GO, indicating the presence of some defects for GO in the oxidative process. The I D/I G value of 2.34 for GE/Fe3O4 suggests that the PDDA assistant reducing reaction decreases sp2 graphitic domains and increases edge planes and disordered structures in GE sheets.

Raman spectra of a GE/Fe3O4, b GO, and c graphite
The surface chemical states of GO, PDDA-G, and GE/Fe3O4 were discussed by XPS. Figure 4a shows the N1s peak was visible in PDDA-G, which implied the strong interaction between PDDA and GE. If Fe3O4 was added to the procedure of PDDA-functionalized GE, Fe3O4 became the part of the homogeneous PDDA-functionalized GE/Fe3O4 composite. This process was confirmed by the Fe2p peak in GE/Fe3O4. For the C1s spectrum of Fig. 4b and c, the calculated oxygen content reduced from 28 to 17 %, indicating GO being effectively reduced to GE. Furthermore, the binding energy peaks at 285.9, 711.8, and 725.1 eV in Fig. 4d and e, corresponding to the N1s, Fe 2p3/2, and Fe 2p1/2, respectively. The disappearance of the charge transfer satellite of Fe 2p3/2 around 720 eV reveals the formation of mixed oxides of Fe(II) and Fe(III) such as Fe3O4.

XPS spectra of GO, PDDA-G, and GE/Fe3O4: a survey scan, high-resolution scan in the b C1s region of GO, c C1s, and d N1s regions of PDDA-G and e Fe2p region of GE/Fe3O4
Direct electrochemistry of Hb
The direct electrochemistry of Hb-modified electrodes was recorded by CV. The specific peak was observed at PDDA-G/Hb/GCE and Fe3O4/Hb/GCE in Fig. 5a, indicating the electro-inactiveness of PDDA-G/Hb and Fe3O4/Hb. However, a pair of well-defined and quasi-reversible redox peaks GE/Fe3O4/Hb/GCE was observed at −0.394 and −0.255 V with a peak-to-peak separation value (\( \varDelta {E_{\mathrm{p}}}=\left| {{E_{\mathrm{p}\text{,}\mathrm{a}}}-\left. {{E_{\mathrm{p}\text{,}\mathrm{c}}}} \right|} \right. \)) of about 139 mV, revealing the direct electron transfer reaction. The formal potential (\( {E^{0' }}=\frac{{{E_{\mathrm{p}\text{,}\mathrm{a}}}+{E_{\mathrm{p}\text{,}\mathrm{c}}}}}{2} \)) is −0.324 V in PBS, which indicates that the redox reactions are associated with the reduction/oxidation of the HbFe(III)/Fe(II) couple [14]. The cathodic and anodic peak currents were located at −0.09367 and −0.1002 mA, respectively. The peak current ratio is about 1.0, indicating that the direct electron transfer process is a stable and reversible electron transfer reaction. Figure 5b shows the scan rate effects on both the peak voltage and current. As the scan rate increases from 20 to 320 mV/s, both I p,a and I p,c can be described by the linear regression equations as \( {I_{\mathrm{p}\text{,}\mathrm{a}}}=0.00134\upsilon -0.00618 \) (R n = 0.9994) and \( {I_{\mathrm{p}\text{,}\mathrm{c}}}=-0.0014\upsilon +0.0117 \) (R n = 0.9991) in Fig. 5c. The linear changing suggests that the redox of Hb on the GE/Fe3O4/GCE is a quasi-reversible surface-controlled electrochemical process. Similarly, both E p,a and E p,c shift slightly with increasing scan rate, resulting in the increment of the peak-to-peak separation. The electrochemical parameters were calculated according to Laviron equations [15]:
where α is the charge transfer coefficient, υ is the scan rate, n is the electron transfer number, k s is the apparent charge transfer rate constant, R is the gas constant, and F is Faraday’s constant. Figure 5d shows the linear relationship of E p and logυ(υ/mV s−1) with the linear regression equations as \( {E_{\mathrm{p}\text{,}\mathrm{a}}}=0.1359\log \upsilon -0.5384 \) (R n = 0.9966) and \( {E_{\mathrm{p}\text{,}\mathrm{c}}}=-0.09629\log \upsilon -0.2143 \) (R n = 0.9840). Obviously, the slopes of the linear segments are equal to −2.3RT/αnF and 2.3RT/(1 − α)nF for the E p,c and E p,a curves, respectively. The evaluated value of α and n is 0.58 and 1.05. According to the Eq. (3), k s can be estimated to be 0.3 s−1. The estimated k s value is larger than that obtained for Hb immobilized on polymer-MWCNT-modified [9].

CV plots of a (I) GE/Fe3O4/Hb-, (II) PDDA-G/Hb-, and (III) Fe3O4/Hb-modified GCE at the scan rate of 100 mV/s, b GE/Fe3O4/Hb-modified GCE with different scan rates (20, 50, 70, 90, 150, 180, 210, 240, 270, 300, and 320 mV/s), c the linear dependence of anodic and cathodic peak currents on scan rate (20 to 320 mV/s), d variation of E p versus the logarithm of scan rates
Effect of pH on the direct electrochemistry
The direct electrochemistry of Hb immobilized on GE/Fe3O4 showed a strong dependence on solution pH in Fig. 6a. The stable and well-defined CV plots were obtained, and E 0′ showed a negative shift with the increase of buffer solution pH. The linear relationship of E 0′ and the pH value was obtained from the linear regression equation as \( {E^{0' }}=-0.0396\mathrm{pH}-0.0391 \) (R n = 0.9921). The slope of −39.6 mV/pH is smaller than the expected theoretical value of 56 mV/pH at 20 °C for a single proton coupled to the reversible one-electron transfer [16]. It may be due to the influence of the protonization of ligands to the heme iron and amino acids around the heme [13]. Thus, the electron transfer between Hb and the electrode can be presented by:

a CV plots of the Hb immobilized on GE/Fe3O4/GCE in 0.2 mol/L PBS at different pH values with scan rate of 100 mV/s, b effect of pH on E 0′
Sensitive detection for H2O2
Considering the sensitivity and stability, the typical steady-state amperometric response of the biosensor is investigated by successively increasing the H2O2 concentration under the optimized conditions. Figure 7a shows a rapid current response with continuous addition of H2O2 to the testing solution. The maximum steady-state current could be achieved within 5 s, indicating a fast H2O2 diffusion process in the modified electrode due to the porous structure of the GE/Fe3O4 film. Figure 7b shows the calibration curve between amperometric current and H2O2 concentration. The regression equations were expressed as a linear equation \( {I_{\mathrm{p}}}=-0.3837c-1.5954 \) (R n = 0.9967). The detection limit of the biosensor was estimated at 6.0 × 10−6 mol/L at a signal-to-noise ratio of 3. The linear ranger is broader than that of the Hb/MCMS/GCE [9]. The result indicates that the magnetic microspheres in the experiment displayed better catalytic ability than the microspheres using the magnetic core of carbon-coated iron nanoparticles.

a Amperometric responses of H2O2 upon successive addition of 20 μL H2O2 in N2-saturated PBS, b the peak current versus H2O2 concentration
Repeatability and stability of the H2O2 biosensor
Considering the fabrication repeatability of the biosensor based on the GE/Fe3O4/Hb GCE, five modified electrodes were prepared and measured independently. The relative standard deviation of peak currents was 2.3 % for measuring 25 μmol/L H2O2 in PBS, revealing an excellent reproducibility. After the electrode was stored for 2 weeks at 4 °C in a humid environment, it could retain 95 % of its original response, suggesting acceptable storage stability.
Conclusion
In this work, the GE/Fe3O4 nanocomposite consisting of PDDA-functionalized GE covered with high-density Fe3O4 nanoparticles was prepared by a simple and cost-effective procedure. Then, the direct electrochemistry of Hb was investigated in detail by various electrochemical methods. The GE/Fe3O4/GCE showed a fast response on H2O2, a wide linear range, and a low detection limit towards H2O2. In addition, the fabricated H2O2 biosensor exhibited excellent reproducibility and stability. Based on these, a low-cost, highly efficient, and direct electrochemical method for H2O2 determination was developed.
References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Science 306:666–669
Biswas C, Lee Y (2011) Adv Funct Mater 21:3806–3826
Hou J, Shao Y, Ellis MW, Moore RB, Yi B (2011) Phys Chem Chem Phys 13:15384–15042
Shao Y, Wang J, Wu H, Liu J, Aksay IA, Lin Y (2010) Electroanal 22:1027–1036
He Y, Shen Q, Zheng J, Wang M, Liu B (2011) Electrochim Acta 56:2471–2476
Lu D, Zhang Y, Wang L, Lin SX, Wang CM, Chen XF (2012) Talanta 88:181–186
Zhang S, Shao Y, Liao H, Engelhard M, Yin G, Lin Y (2011) ACS Nano 5:1785–1791
Liu X, Zhu H, Yang XR (2011) Talanta 87:243–248
Tan XC, Zhang JL, Tan SW, Zhao DD, Huang ZW, Mi Y, Huang ZY (2009) Sensors 9:6185–6199
Yu CM, Zhou XH, Gu HY (2010) Electrochim Acta 55:8738–8743
Caruntu D, Caruntu G, Chen Y, O’Connor CJ, Goloverda G, Kolesnichenko VL (2004) Chem Mater 16:5527–5534
Chen M, Kim Y, Li C, Cho S (2008) J Phys Chem C 112:6710–6716
Li D, Müller M, Gilje S, Kaner R, Wallace GG (2008) Nat Nanotechol 3:101–105
Liu H, Wan Y, Zou G (2006) Anal Bioanal Chem 385:1470–1476
Laviron E (1979) J Electroanal Chem 101:19–28
Sun W, Li X, Liu S, Jiao K (2009) Bull Korean Chem Soc 30:582–588
Acknowledgments
We gratefully acknowledge the financial support of the 973 Program (2013CB932902), National Natural Science Foundation of China (21173042), National Natural Science Foundation of Jiangsu (BK201123694) and Hebei (B2012402006), Jiangsu Key Laboratory of Environmental Material and Environmental Engineering (JHCG201012), and Foundation of Southeast of University (3212001102 and 3212002205).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, Y., Zhang, H., Yao, D. et al. Direct electrochemistry of hemoglobin on graphene/Fe3O4 nanocomposite-modified glass carbon electrode and its sensitive detection for hydrogen peroxide. J Solid State Electrochem 17, 881–887 (2013). https://doi.org/10.1007/s10008-012-1939-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-012-1939-5