
Abstract
2-Deoxy-D-ribose was converted to α/β-mixtures of methyl 3-O-acetyl- and methyl 3-O-benzoyl-2-deoxy-5-(p-toluenesulfonyl)-D-ribofuranosides. These were reacted with boron trichloride to generate ribofuranosyl chlorides, which afforded precursors for tracers to image tumor hypoxia on substitution with salts of 2-nitroimidazole. The anomeric ratio of the nucleosides was delicately influenced by the reaction conditions.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
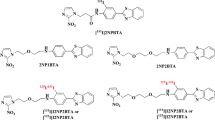
Tumor hypoxia has a negative prognosis predictive value for solid tumors, because it is associated with tumor aggressiveness, metastasis, and aberrant angiogenesis [1–3]. It reflects increased resistance to anticancer treatment by radio- and chemotherapy. Therefore, it is in the interest of cancer patients to identify and target hypoxic areas in solid tumors [4, 5]. Non-invasive in vivo quantification of hypoxic areas of solid tumors with radiolabeled tracers attracted much attention and was studied extensively in recent years [6, 7]. Fluorine-18 containing tracers derived from 2-nitroimidazole (azomycin) are the most important ones used for positron emission tomography (PET) to image hypoxia for diagnostic purposes. Under hypoxic conditions in cells, the 2-nitroimidazole moiety of the tracer is reduced stepwise by electron transfers via reactive intermediates [8, 9]. These attack low-molecular weight compounds, preferably glutathione, and to a lesser extent high molecular weight compounds, and the nitro group ends up as amino group. The modified compounds with the bound 18F, which is detected by PET, stay in the cells and are accumulated. Figure 1 is a compilation of those tracers, nucleosides derived from carbohydrates, such as various D-pentoses and D-hexoses, except compounds 1 and 2. The first azomycin-based tracer and, at the same time, the gold standard up to now for imaging tumor hypoxia are [18F]fluoromisonidazole (FMISO, 1) [7, 10]. A homologue thereof is [18F]fluoroerythronitroimidazole (2) [11]. From the [18F]fluoro nucleosides 3-8 derived from α-arabinose, tracer 3 [12, 13], from β-arabinose, tracer 4 [14], from β-xylose, tracer 5 [14], and from β-glucose, tracer 6 [15], only 3 gained prominence. Recently, we synthesized 2-nitroimadazole precursors derived from α- and β-2-deoxy-D-ribose and α- and β-D-allofuranose. The β-anomers were radiolabeled and deprotected to give tracers 7 [16] and 8 [17] so far and evaluated for imaging tumor hypoxia.

Known 2-nitroimidazole-based [18F]fluoro tracers
Results and discussion
The synthesis of the precursors for tracers α- and β-8 is given in Scheme 1 [16]. In brief, it started from 2-deoxy-D-ribose, which was converted via methyl glycosides 10 to fully protected methyl glycosides 11. Their mixture was treated with 8 M HCl/Et2O/CH2Cl2 at 0 °C to form a mixture of glycosyl chlorides which was reacted with the tetrabutylammonium salt of 2-nitroimidazole. The two nucleosides, α- and β-12, were separated by flash column chromatography and individually desilylated and finally tosylated to give the two desired precursors α- and β-14. This sequence was selected, because we thought that introduction of the tosyl group right from the beginning would not be tolerated by 8 M HCl in Et2O/CH2Cl2. However, if that worked, the synthesis of both precursors could be shortened. Furthermore, we wanted to replace the tedious preparation of 8 M HCl in Et2O by a commercially available and more reactive reagent, such as BCl3, for the conversion of the methyl glycosides into the glycosyl chlorides.

The improved synthesis is given in Scheme 2. Although the mixture of methyl glycosides α- and β-10 [18] was tosylated [19] at −25 °C for 3 days in 59% yield (α/β = 1.2/1), some ditosylate 16 was formed as well (11%, α/β = 1.4/1). Analytical samples of the anomers for characterization could not be obtained by column flash chromatography. However, they could be obtained in homogeneous form by deacetylation of (+)- and (−)-17 and allowed to assign the anomeric configuration as will be shown later. Acetylation of the mixture of tosylates α- and β-15 with Ac2O in dry pyridine delivered a mixture of acetates α- and β-17 in 92% (α/β = 1.2/1) yield. This mixture could be separated by flash column chromatography and Zemplen saponification of acetates (+)- and (−)-17 delivered homogenous samples of α- and β-15, respectively. The latter one is a literature known compound whose β-configuration has been determined by 2D NMR methods [20]. It allowed to assign α-configuration to (+)-15 and α and β to (+)- and (−)-17, respectively. As glycosides α- and β-17 were less reactive with HCl/Et2O in CH2Cl2, BCl3 in CH2Cl2 (1 M) was found to be an alternative to generate the glycosyl chlorides at 0 °C (general procedure A). Rapid aqueous work up at 0 °C allowed to isolate the labile chlorides, which were immediately reacted in two ways with 2-nitroimidazole. In the first case (general procedure B), the tetrabutylammonium salt of 2-nitroimidazole [21] was mixed with a solution of the 2-deoxy-D-ribofuranosyl chloride at −30 °C in CH2Cl2. The reaction mixture was allowed to warm slowly to 0 °C within 2 h and was then extractively worked up. Flash chromatography furnished known anomers α- and β-14 over two steps in 41 and 11% yield, respectively. When the reaction was started at −50 °C, the α/β-14 ratio was 5/1 (by NMR) and only the α-anomer was isolated in 53% yield. In the second case (general procedure C), the mixture of glycosyl chlorides was added to a mixture of 2-nitroimidazole/K2CO3/excess tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) as phase transfer catalyst [22] in CH3CN at 0 °C. Work up after 2 h and purification delivered 12% of nucleoside α-14 and 36% of β-14 starting from methyl glycosides. Satisfyingly, the two complementary procedures gave either preferably α- or β-anomer 14 [16].

We aimed to increase the yields of the nucleosides by replacing the acetyl protecting group by the more stable benzoyl group (Scheme 3). Therefore, the mixture of tosylates α- and β-15 was benzoylated and gave again a mixture of globally protected 2-deoxy-D-riboses α- and β-18, which could not be separated by flash column chromatography to obtain homogeneous analytical samples. Benzoylation of alcohols α- and β-15 with benzoyl chloride/pyridine affords the individual anomers of 18 for analytical purposes, although the mixture was used for the next step. It was converted to chlorides as before with BCl3 in CH2Cl2 according to general procedure A. Their isolation without purification was immediately followed by reaction with the tetrabutylammonium salt of 2-nitroimidazole, starting the reaction at −50 °C and allowing it to warm to ambient temperature. The mixture of the nucleosides α- and β-19 was isolated in 81% yield (α/β = 2/1) by flash chromatography. The individual anomers were obtained by a second flash chromatography. When general procedure C was used for the preparation of the nucleosides from the chlorides, the yield of α-19 was 14% and that of β-19 was 69%. As envisioned, the yields with the benzoyl protecting group were higher than with the acetyl version. The anomeric configuration of α- and β-19 (1H NMR; α-1-H′: d, J = 6.6 Hz; β-1-H′: dd, J = 7.5 and 5.6 Hz) was assigned in analogy to nucleosides α- and β-14 (1H NMR; α-1-H′: dd, J = 6.3, 0.8 Hz; β-1-H′: dd, J = 6.6, 6.1 Hz) and the literature known analogue [19] of β-14 with two 4-toluoyl protecting groups (1H NMR; β-1-H′: t, J = 6.5 Hz) instead of the acetyl and benzoyl group. The 1-H′ hydrogen atoms of the α-anomers resonate as doublets or as doublets of doublets with one coupling constant being very small. However, the 1-H′ hydrogen atoms of the β-anomers resonate as doublets of doublets or as triplet with two similar coupling constants.

Conclusions
The synthesis of known 2-nitroimidazole nucleosides derived from 2-deoxy-D-ribose used as precursors for tracers was shortened if tosylation is performed at the beginning instead of at the end of the reaction sequence. The yield was further improved using BCl3 for generation of 2-deoxy-D-ribofuranosyl chlorides and benzoyl instead of acetyl group as protecting group for OH at C-3.
Experimental
1H, 13C (J-modulated; not J-modulated spectra were recorded of 2-nitroimidazole derivatives) NMR spectra were recorded in CDCl3 on a Bruker AV III 400 (1H: 400.27 MHz, 13C: 100.65 MHz), AV 400 (1H: 400.13 MHz, 13C: 100.61 MHz), and AV III 600 (1H: 600.13 MHz, 13C: 150.90 MHz) spectrometer at 25 °C, respectively. Chemical shifts δ (ppm) were referenced to residual CHCl3 (δ H = 7.24 ppm) and CDCl3 (δ C = 77.00 ppm). IR spectra were recorded on a Bruker VERTEX 70 IR spectrometer as ATR spectra or of films on a silicon disc [23] on a Perkin Elmer 1600 FT-IR spectrometer. Optical rotations were measured at 20 °C on a Perkin Elmer 351 polarimeter in a 10 cm cell. Melting points were determined on a Reichert Thermovar instrument. Elemental analyses (C, H, N, S) were conducted using the Euro EA 3000 Elemental Analyser (for oxygen in combination with a high temperature pyrolysis furnace (1480 °C) and reduction with carbon) from Eurovector. Their results were found to be in good agreement (±0.3%) with the calculated values.
Flash (column) chromatography was performed with Merck silica gel 60 (230–400 mesh). TLC was carried out on 0.25 mm-thick Merck plates, silica gel 60 F254. Spots were visualized by UV and/or dipping the plate into a solution of 23.0 g (NH4)6Mo7O24·4H2O and 1.0 g Ce(SO4)2·4H2O in 500 cm3 10% aqueous H2SO4, followed by heating with a heat gun. Pyridine was dried by refluxing over powdered CaH2, then distilled and stored over molecular sieves (4 Å). Dichloromethane was dried by storage over molecular sieves (3 Å). All other chemicals and solvents were of the highest purity available and used as received.
Mixture of methyl 2-deoxy-5-O-(p-toluenesulfonyl)-α- and methyl 2-deoxy-5-O-(p-toluenesulfonyl)-β-D-ribofuranoside (α- and β-15, C13H18O6S) and mixture of methyl 3,5-bis(p-toluenesulfonyl)-α- and methyl 3,5-bis(p-toluenesulfonyl)-β-D-ribofuranoside (α- and β-16, C20H24O8S2)
Dry pyridine (2.20 cm3, 27.24 mmol) was added to a mixture of 1.345 g methyl glycosides α- and β-10 (9.08 mmol) [18] in 17 cm3 dry CH2Cl2 under Ar. The stirred reaction mixture was cooled to 0 °C and 1.868 g p-toluenesulfonyl chloride (9.08 mmol) was added. The flask was stored at −25 °C for 3 days and afterwards 1 cm3 water was added. After stirring for 15 min, the reaction mixture was concentrated under reduced pressure and 10 cm3 EtOAc was added. The organic phase was washed with 10 cm3 2 M HCl, 10 cm3 water, and 10 cm3 NaHCO3, then dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc = 1/1; R f = 0.34 for monotosylates, R f = 0.75 for ditosylates) giving 1.631 g mixture of monotosylates α- and β-15 (59%; α/β = 1.22/1) and 0.458 g mixture of ditosylates α- and β-16 (11%), both as colorless oils. The data of the individual anomers are given later. Mixture of ditosylates α- and β-16: [α] 20D = + 24.3 cm2 g−1 (c = 1.55, acetone); IR (ATR, NMR sample in CDCl3): \(\bar{\nu }\) = 1359, 1190, 1174, 1096, 976 cm−1.
1H NMR (400.27 MHz, CDCl3): α/β = 1.4/1.0; contained 5% by weight of toluene; α-16: δ = 7.76–7.71 (m, 4H, HAr), 7.36–7.30 (m, 4H, HAr), 4.92 (bd, J = 5.2 Hz, 1H, 1-H), 4.77 (ddd, J = 8.3, 3.4, 2.0 Hz, 1H, 3-H), 4.29 (q, J = 3.4 Hz, 1H, 4-H), 4.09 (d, J = 3.4 Hz, 2H, 5-H), 3.27 (s, 3H, OCH3), 2.43 (s, 6H, CH tol3 ), 2.13 (ddd, J = 14.8, 8.3, 5.2 Hz, 1H, 2-H), 1.96 (ddd, J = 14.8, 2.0. 0.8 Hz, 1H, 2-H) ppm; β-16: δ = 7.76–7.71 (m, 4H, HAr), 7.36–7.30 (m, 4H, HAr), 5.00 (dd, J = 4.9, 2.6 Hz, 1H, 1-H), 4.88 (ddd, J = 9.0, 5.9 Hz, 1H, 3-H), 4.18 (td, J = 5.9, 3.5 Hz, 1H, 4-H), 3.92 (AB part of ABX system, J AB = 10.4 Hz, J AX = J BX = 5.9 Hz, 2H, 5-H), 3.18 (s, 3H, OCH3), 2.43 (s, 6H, CH tol3 ), 2.20–2.15 (m, 2H, 2-H) ppm; 13C NMR (100.65 MHz, CDCl3) of mixture: δ = 145.4 (Cq, β), 145.2(Cq, α), 145.0 (Cq, α), 145.0 (Cq, β), 133.0 (Cq, α), 132.8 (Cq, β), 132.5 (Cq, β), 132.5 (Cq, α), 130.1 (2 CH, β), 130.0 (2 CH, α), 129.8 (4 CH, α and β), 127.9 (4 CH), 127.8 (2 CH), 127.8 (2 CH), 105.1 (C-1, β), 104.7 (C-1, α), 80.5 (C-4, β), 80.4 (C-4, α), 80.1 (C-3, β), 79.1 (C-3, α), 68.8 (C-5, β), 68.4 (C-5, α), 55.1 (CH3O, β), 64.99 (OCH3, α), 39.0 (C-2, β), 38.9 (C-2, α), 21.6 (2 CH3), 21.55 (2 CH3) ppm.
Mixture of (+)- and (−)-methyl 3-O-acetyl-2-deoxy-5-O-(p-toluenesulfonyl)-D-ribofuranoside ((+)- and (−)-17, C15H20O7S)
To 1.631 g mixture of monotosylates α- and β-5 (5.39 mmol), 1.02 cm3 Ac2O (10.78 mmol) and 1.67 cm3 dry pyridine (21.56 mmol) in 13 cm3 dry CH2Cl2 were added under Ar. The reaction mixture was heated at 40 °C until the starting material was consumed (about 4 h). After addition of 4 cm3 water, stirring was continued for 15 min. The organic phase was separated and washed with 15 cm3 2 M HCl and 15 cm3 saturated aqueous solution of NaHCO3, dried (MgSO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc = 1/1, R f = 0.82, 0.75 for anomers) to yield 1.704 g mixture of anomers (92%, α/β = 1.2/1.0) as colorless oil. Part of the mixture was flash chromatographed to get analytical samples of anomers (+)- and (−)-17 (less polar isomer: R f = 0.20, more polar isomer: R f = 0.11 for hexanes/EtOAc = 3/1) as colorless oils.
(−)-17: R f = 0.20 (hexanes/EtOAc = 3/1); colorless crystals, m.p.: 50–51 °C (i-Pr2O/hexanes); [α] 20D = −40.7 cm2 g−1 (c = 1.01, acetone); 1H NMR (600.13 MHz, CDCl3): δ = 7.81–7.78 (m, 2H, Htos), 7.34–7.30 (m, 2H, Htos), 5.08 (ddd, J = 7.3, 5.3, 2.3 Hz, 1H, 3-H), 5.04 (dd, J = 5.4, 2.0 Hz, 1H, 1-H), 4.19–2.13 (m, 2H, 4-H and 5-H), 4.04 (dd, 9.7, 6.6 Hz, 1H, 5-H), 3.21 (s, 3H, OCH3), 2.42 (s, 3H, CH tos3 ), 2.31 (ddd, J = 14.0, 7.3, 2.0 Hz, 1H, 2-H), 2.09 (d, J = 14.0, 5.3 Hz, 1H, 2-H), 2.01 (s, 3H, CH3CO) ppm; 13C NMR (150.90 MHz, CDCl3): δ = 171.0 (C=O), 144.9 (Cqtos), 132.8 (Cqtos), 129.8 (2 CH), 127.9 (2 CH), 105.2 (C-1), 80.9 (C-4), 74.0 (C-3), 69.5 (C-5), 55.2 (OCH3), 38.8 (C-2), 21.6 (CH tos3 ), 21.0 (CH3) ppm; and IR (ATR): \(\bar{\nu }\) = 2925, 1737, 1360, 1235, 1175, 1047, 973, 955 cm−1.
(+)-17: R f = 0.11 (hexanes/EtOAc = 3/1), oil; [α] 20D = +93.6 (c = 1.05, acetone); 1H NMR (600.13 MHz, CDCl3): δ = 7.79–7.75 (m, 2H, Htos), 7.35–7.30 (m, 2H, Htos), 4.98 (dd, J = 5.3, 0.7 Hz, 1H, 1-H), 4.94 (ddd, J = 8.3, 3.5, 1.9 Hz, 1H, 3-H), 4.21 (AB part of ABX system, J AB = 10.6 Hz, J 4,5 = 3.5 and 3.2 Hz, 2H, 5-H), 4.15 (~q, J = ~3.5 Hz, 1H, 4-H), 3.31 (s, 3H, OCH3), 2.42 (s, 3H, CH3 tol), 2.28 (ddd, J = 14.5, 8.3, 5.3 Hz, 1H, 2-H), 2.02 (s, 3H, CH3CO), 1.95 (ddd, J = 14.5, 1.9, 0.7 Hz, 1H, 2-H) ppm; 13C NMR (150.90 MHz, CDCl3): δ = 171.0 (C=O), 144.9 (Cqtos), 132.8 (Cqtos), 129.8 (2 CH), 127.9 (2 CH), 105.2 (C-1), 80.9 (C-4), 74.0 (C-3), 69.5 (C-5), 55.2 (OCH3), 38.8 (C-2), 21.6 (CH tos3 ), 21.0 (CH3) ppm; and IR (ATR): \(\bar{\nu }\) = 2836, 1736, 1364, 1240, 1177, 1070, 1020, 978 cm−1.
Methyl 2-deoxy-5-O-(p-toluenesulfonyl)-α-D-ribofuranoside (α-15, C13H18O6S)
A solution of 0.291 g acetate (+)-17 (0.84 mmol, [α] 20D = +93.6 (c = 1.05, acetone)), 4.25 cm3 dry MeOH, and 0.43 cm3 NaOMe/MeOH (0.425 mmol, 1 M) was stirred for 30 min at 0 °C (TLC). Dry ice was added to neutralize base. The reaction mixture was concentrated under reduced pressure. Water (10 cm3) and 5 cm3 CH2Cl2 were added. The organic phase was separated and the aqueous one extracted with CH2Cl2 (2 × 5 cm3). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc = 1/1, R f = 0.47) to yield 0.199 g alcohol α-15 (77%) as colorless oil. [α] 20D = + 95.09° g cm2 (c = 1.12, acetone); 1H NMR (400.13 MHz, CDCl3): δ = 7.78–7.73 (m, 2H, Htos), 7.35–7.30 (m, 2H, Htos), 5.01 (d, J = 4.4 Hz, 1H, 1-H), 4.18 (≈td, J = 4.1, 1.8 Hz, 1H, 4-H), 4.10 (bd, J = 5.9 Hz, 1H, 3H), 4.04 (AB part of ABX system, J AB = 10.7 Hz, J = 4.3, 3.8 Hz, 2H, 5-H), 3.32 (s, 3H, CH3O), 2.75 (bs, 1H, OH), 2.43 (s, 3H, CH3), 2.05 (ddd, J = 13.9, 5.9, 4.4 Hz, 1H, 2-H), 1.96 (dd, J = 13.9, 0.8 Hz, 1H, 2-H) ppm; 13C NMR (100.61 MHz, CDCl3): δ = 145.0 (Cq, CSO3), 132.7 (Cqtos), 129.90 (2 HCtos), 127.9 (2 HCtos), 105.7 (C-1), 84.6 (C-4), 72.8 (C-3), 69.4 (C-5), 55.0 (OCH3), 41.0 (C-2), 21.6 (CH tos3 ) ppm; and IR (Si): \(\bar{\nu }\) = 3445, 2923, 1354, 1173, 1081, 961, 908 cm−1.
Methyl 2-deoxy-5-O-(p-toluenesulfonyl)-β-D-ribofuranoside (β-15, C13H18O6S)
A mixture of 0.066 g acetate (−)-17 (0.19 mmol, less polar acetate, [α] 20D = −40.7 cm2 g−1 (c = 1.01, acetone)), 2 cm3 dry MeOH, and 0.064 cm3 MeONa/MeOH (0.064 mmol, 0.33 equiv, 1 M) was stirred at -30 °C. The ester was consumed after 5 h (TLC). Work up as for α-15 yielded 0.047 g alcohol β-15 (82%) as colorless oil. [α] 20D = −40.4 g cm2 (c = 1.08, acetone); 1H NMR (400.13 MHz, CDCl3): δ = 7.81–7.74 (m, 2H, Htos), 7.36–7.31 (m, 2H, Htos), 5.01 (dd, J = 5.2, 1.8 Hz, 1H, 1-H), 4.41 (bs, 1H, 3-H), 4.07–3.99 (m, 3H, 5-H, 4-H), 3.20 (s, 3H, OCH3), 2.43 (s, 3H, CH tos3 ), 2.19 (ddd, J = 13.4, 6.9, 1.8 Hz, 1H, 2-H), 2.10 (bs, 1H, OH), 2.03 (ddd, J = 13.4, 6.2, 5.2 Hz, 1H, 2-H) ppm; 13C NMR (100.61 MHz, CDCl3): δ = 145.1 (Cq, CSO3), 132.7 (Cq, Ctos), 129.9 (2C, HCtos), 128.0 (2C, HCtos), 105.3 (C-1), 82.9 (C-4), 72.65 (C-3), 70.14 (C-5), 50.02 (CH3O), 41.32 (C-2), 21.64 (CH3 tol) ppm.
Preparation of anomeric 3-O-acetyl-2-deoxy-5-O-(p-toluenesulfonyl)-D-ribofuranosyl chlorides (general procedure A) and their conversion to 1-(3′-O-acetyl-2′-deoxy-5′-O-(p-toluenesulfonyl)-α- and 1-(3′-O-acetyl-2′-deoxy-5′-O-(p-toluenesulfonyl)-β-D-ribofuranosyl)-2-nitroimidazole (α- and β-14 )
General procedure A: To a solution of 0.507 g methyl glycosides, α- and β-17 (1.47 mmol) in 4.5 cm3 dry Et2O at 0 °C under Ar 3.68 cm3 BCl3 (3.68 mmol, 2.5 equiv, 1 M in CH2Cl2) was added. The reaction mixture was stirred for 2 h (TLC: hexanes/EtOAc = 1/1; virtually no starting material was present; new strong spot with R f = 0.34) at 0 °C. CH2Cl2 (12 cm3, 0 °C) was added and the mixture was washed with 4 cm3 cold brine (−18 °C), which was then extracted with 5 cm3 cold CH2Cl2 (0 °C). The combined organic phases were washed with 5 cm3 cold aqueous solution of NaHCO3 (0 °C), dried (Na2SO4) at 0 °C, and concentrated first to 5–10 cm3 on a rotavapor without warming with the water bath and then the remaining solvent was removed on the vacuum pump (1 mbar) within a few min without warming. The clear somewhat coloured solution was used immediately for the next step after withdrawing a sample for 1H NMR spectroscopy; ratio of chlorides: α/β = 3.6/1.0.
1H NMR of anomeric 2-deoxy-D-ribofuranosyl chlorides (400.27 MHz, CDCl3): δ = 6.21 ppm (d, J = 5.3 Hz, 1-H of α-chloride), 1-H of β-chloride overlapping with 1-H of α-chloride, integration was referenced to resonance at 4.46 ppm (q, J = 2.9 Hz, 4-H).
Reaction of anomeric 2-deoxy-D-ribofuranosyl chlorides with tetrabutylammonium salt of 2-nitroimidazole (general procedure B)
A solution of the above 2-deoxy-D-ribofuranosyl chlorides derived from α- and β-17 in 3.5 cm3 dry CH2Cl2 (0 °C) was added to a solution of the 0.450 g tetrabutylammonium salt of 2-nitroimidazole (1.32 mmol, 0.9 equiv. relative to methyl glycosides) [21] in dry 4 cm3 CH2Cl2 at −30 °C under Ar. Stirring was continued for 2 h, while the cooling bath was allowed to reach 0 °C. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in 15 cm3 EtOAc and washed with water (2 × 5 cm3). The organic phase was dried (MgSO4) and concentrated under reduced pressure. The residue (α/β = 2/1 by 1H NMR) was flash chromatographed (hexanes/EtOAc = 1/1, α: R f = 0.29; β: R f = 0.49) to yield 0.060 g β-14 (11%) and 0.231 g α-14 (41%), both spectroscopically (1H, 13C NMR) identical to the ones described in Ref. [14].
Similarly, 0.536 g mixture of anomeric methyl glycosides (1.56 mmol) were converted via chlorides to nucleosides (reaction was started at −50 °C); ratio of α/β = 5/1 by 1H NMR in crude product. Flash chromatography furnished 0.318 g α-14 (53%).
Reaction of 3-O-acetyl-glycosyl chlorides with 2-nitroimidazole/K 2 CO 3 /tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) (general procedure C)
A mixture of 0.118 g 2-nitroimidazole (1.04 mmol, 0.8 equiv.), 0.225 g K2CO3 (1.63 mmol), 10 mm3 TDA-1 [23], and 20 cm3 dry CH3CN was stirred for 10 min at RT under Ar and then cooled to 0 °C. The chlorides prepared from 0.449 g methyl glycosides α- and β-17 (1.30 mmol) by the above given general procedure A were dissolved in dry CH3CN at 0 °C and added. Stirring was continued for 2 h at 0 °C and then the reaction mixture was filtered through Celite (washing with CH2Cl2). The filtrate was concentrated under reduced pressure and 20 cm3 EtOAc was added to the residue. The mixture was washed with water (2 × 10 cm3), dried (MgSO4), and concentrated under reduced pressure. The residue (α/β = 1/3, by 1H NMR) was purified by flash chromatography (hexanes/EtOAc = 1/1) to yield 54 mg α-14 (12%) and 160 mg β-14 (36%).
Mixture of methyl 3-O-benzoyl-2-deoxy-5-O-(p-toluenesulfonyl)-α- and methyl 3-O-benzoyl-2-deoxy-5-O-(p-toluenesulfonyl)-β-D-ribofuranoside (α- and β-18, C20H22O7S)
To 0.800 g, mixture of anomeric monotosylates 15 (2.65 mmol) and 0.64 cm3 dry pyridine (7.95 mmol) in dry CH2Cl2 (6.3 cm3) under Ar was added 0.64 cm3 benzoyl chloride. The reaction mixture was stirred at RT for 18 h. After addition of 0.5 cm3 water, stirring was continued for 15 min. The mixture was concentrated under reduced pressure and 15 cm3 EtOAc and 5 cm3 water were added. The organic phase was separated and washed with 5 cm3 2 M HCl, 5 cm3 water, and 5 cm3 saturated aqueous solution of NaHCO3, dried (MgSO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc = 2/1, R f = 0.76) to yield 0.972 g mixture of anomeric benzoates 18 (90%; α/β = 1.2/1.0 by 1H NMR) as a colorless oil possibly containing some benzoic acid; [α] 20D = + 41.0 g cm2 (c = 1.35, acetone). The individual anomers of 18 for characterization were prepared by esterification of homogeneous anomers α- and β-15 with PhC(O)Cl/pyridine.
Methyl 3-O-benzoyl-2-deoxy-5-O-(p-toluenesulfonyl)-α- and methyl 3-O-benzoyl-2-deoxy-5-O-(p-toluenesulfonyl)-β-D-ribofuranoside (α- and β-18, C20H22O7S)
Benzoyl chloride (0.143 g, 1.02 mmol, 0.118 cm3) and 0.120 g dry pyridine (1.52 mmol, 0.122 cm3) were added to 0.153 g alcohol α-15 (0.51 mmol) dissolved in 1.5 cm3 dry CH2Cl2 and the solution was stirred for 20 h at RT. Water (0.5 cm3) was added and the reaction mixture was stirred for 15 min. The mixture was concentrated under reduced pressure, 10 cm3 water was added, and it was extracted with ethyl acetate (3 × 5 cm3), dried with Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography (hexanes/ethyl acetate = 1/1, R f = 0.55) to yield 0.183 g benzoate α-18 (88%) as colorless oil. Similarly, 0.096 g alcohol β-15 (0.32 mmol) was converted to 0.104 g benzoate β-18 (81%).
α-18: [α] 20D = + 98.23 (c = 1.015, acetone); 1H NMR (400.13 MHz, CDCl3): δ = 8.01–7.96 (m, 2H, HPh), 7.81–7.76 (m, 2H, Htos), 7.58–7.52 (m, 1H, HPh), 7.45–7.38 (m, 2H, HPh), 7.34–7.28 (m, 2H, Htos), 5.22–5.16 (m, 1H, 3-H), 5.06 (d, J = 5.2 Hz, 1H, 1-H), 4.35–4.27 (m, 3H, 4-H, 5-H), 3.34 (s, 3H, CH3O), 2.41 (s, 3H, CH tos3 ), 2.40 (ddd, J = 14.5, 8.1, 5.2 Hz, 1H, 2-H), 2.11 (dd, J = 14.5, 1.5 Hz, 1H, 2-H) ppm; 13C NMR (100.61 MHz, CDCl3): δ = 166.41 (CO), 144.92 (CSO3), 133.27 (HCPh), 132.86 (CH3 C tos), 129.86 (2C, HCtos), 129.73 (2C, HCPh), 129.58 (CCO), 128.39 (2C, HCar), 127.99 (2C, HCar), 105.26 (C-1), 80.945 (C-4), 74.49 (C-3), 69.56 (C-5), 55.14 (CH3O), 38.90 (C-2), 21.62 (CH tos3 ) ppm; and IR (Si): \(\bar{\nu }\) = 3016, 2970, 2946, 1738, 1725, 1365, 1229, 1217 cm−1.
β-18: [α] 20D = −16.75 (c = 0.83, acetone); 1H NMR (400.13 MHz, CDCl3): δ = 7.98–7.93 (m, 2H, HPh), 7.83–7.77 (m, 2H, Htos), 7.59–7.53 (m, 1H, HPh), 7.46–7.38 (m, 2H, HPh), 7.33–7.27 (m, 2H, Htos), 5.32 (ddd, J = 7.5, 5.4, 3.3 Hz, 1H, 3-H), 5.13 (dd, J = 5.4, 2.0, Hz, 1H, 1-H), 4.32 (ddd, J = 7.1, 5.1, 3.3 Hz, 1H, 4-H), 4.26 (dd, J = 10.1, 5.1 Hz, 1H, 5-H), 4.14 (dd, J = 10.1, 7.1 Hz, 1H, 5-H), 3.26 (s, 3H, OCH3), 2.43 (ddd, J = 14.2, 7.3, 2.0 Hz, 1H, 2-H), 2.39 (s, 3H, CH tos3 ), 2.25 (td, J = 14.2, 5.4 Hz, 1H, 2-H) ppm; 13C NMR (100.61 MHz, CDCl3): δ = 165.95 (CO), 144.90 (CSO3), 133.38 (HCPh), 132.84 (CH3 C tos), 129.86 (2C, HCtos), 129.64 (2C, HCPh), 129.35 (CCO), 128.44 (2C, HCPh), 128.02 (2C, HCtos), 105.70 (C-1), 81.39 (C-4), 75.10 (C-3), 70.35 (C-5), 55.19 (CH3O), 39.07 (C-2), 21.60 (CH tos3 ) ppm; and IR (Si): \(\bar{\nu }\) = 2924, 1721, 1365, 1274, 1178, 1110 cm−1.
Preparation of mixture of anomeric 3-O-benzoyl-2-deoxy-5-O-(p-toluenesulfonyl)-D-ribofuranosyl chlorides and their conversion to 1-(3′-O-benzoyl-2′-deoxy-5′-O-(p-toluenesulfonyl)-α- and 3′-O-benzoyl-2′-deoxy-5′-O-(p-toluenesulfonyl)-β-D-ribofuranosyl)-2-nitroimidazole (α- and β-19, C22H21N3O8S)
A mixture of 0.609 g methyl glycosides α- and β-19 (1.50 mmol) was converted to 3-O-benzoyl-glycosyl chlorides (TLC: hexanes/EtOAc = 1/1, R f = 0.58) by the procedure used for methyl glycosides α- and β-17 (general procedure A). The crude product was used immediately for the next step. 1H NMR spectrum of crude 3-benzoyl 2-deoxy-D-ribofuranosyl chlorides (400.27 MHz, CDCl3): δ = 6.31 (d, J = 5.0 Hz, 1-H of α-chloride), 6.28 (dd, J = 5.5, 1.6 Hz, 1-H of β-chloride), integration referenced to resonance at 4.60 ppm (q, J = 2.6 Hz, 4-H); α/β = 1/0.13; fairly pure.
The above mixture of chlorides was converted to a mixture of α- and β-19 using the procedure (general procedure B) given for the corresponding 3-O-acetyl-glycosyl chlorides. Tetrabutylammonium salt of 2-nitroimidazole (0.481 g, 1.36 mmol) was used; the reaction was started at −50 °C; and the reaction mixture was allowed to warm to RT in 18 h. The crude product (α/β = 2/1, by 1H NMR) was flash chromatographed (hexanes/EtOAc = 2/1, α-19: R f = 0.25; β-19: R f = 0.21) to yield 0.536 g mixture (81%, α/β = 2/1) of α- and β-19. The anomers were separated by flash chromatography (CH2Cl2/EtOAc = 20/1; α: R f = 0.42; β: R f = 0.36) using a long column to yield homogenous anomers and mixture of anomers.
α-19: Oil, which decomposed at room temperature within a few days, but it was more stable at 4 °C. When α-19 was crystallized from C2H4Cl2/i-Pr2O by slowly cooling from RT to −18 °C, a white powder was obtained, which contained after drying at 0.5 mbar/RT for 10 h 0.05 mol% of i-Pr2O; m.p.: 63–65 °C (powder became glassy); this powder was ideal for storage at 4 °C and handling. [α] 20D = −9.78 g cm2 (c = 1.15, acetone); 1H NMR (400.13 MHz, CDCl3): δ = 7.85–7.80 (m, 2H, Har), 7.66–7.62 (m, 2H, Har), 7.57–7.52 (m, 1H, Har), 7.41–7.33 (m, 4H, Har), 7.32 (d, J = 1.0 Hz, 1H, Him), 7.13 (d, J = 1.0 Hz, 1H, Him), 6.62 (d, J = 6.6 Hz, 1H, 1′-H), 5.43 (d, J = 6.6, 0.7 Hz, 1H, 3′-H), 4.74 (td, J = 3.0, 1.0 Hz, 1H, 4′-H), 4.37 (AB part of ABX system, J AB = 11.4 Hz, J AX = J BX = 3.0 Hz, 2H, 5′-H), 3.05 (td, J = 15.5, 6.6 Hz, 1H, 2′-H), 2.48 (d, J = 15.5 Hz, 1H, 2′-H), 2.45 (s, 3H, CH3) ppm; 13C NMR (100.61 MHz, CDCl3): δ = 165.7 (CO), 145.6 (Cqtos), 143.7 (Cqim), 133.9 (HC), 132.43 (Cq), 130.1 (2 HCtos), 129.5 (2 HCar), 128.6 (2 HCar), 128.4 (Cqar), 128.2 (HCim), 127.9 (2 HCar), 122.2 (HCim), 91.4 (C-1′), 86.1 (C-4′), 74.6 (C-3′), 69.0 (C-5′), 41.1 (C-2′), 21.7 (CH tos3 ) ppm; and IR (ATR, NMR sample): \(\bar{\nu }\) = 2971, 1709, 1535, 1476, 1355, 1270, 1240, 1175, 1092, 1075 cm−1.
β-19: [α] 20D = −16.89 g cm2 (c = 1.06, acetone); m.p.: 90 °C (decomp., CH2ClCH2Cl/i-Pr2O, solution not heated above 50 °C); 1H NMR (400.13 MHz, CDCl3): δ = 8.03–7.96 (m, 2H, Har), 7.82–7.75 (m, 2H, Har), 7.63–7.57 (m, 2H, Har), 7.60 (d, J = 1.0 Hz, 1H, Him), 7.50–7.43 (m, 2H, Har), 7.38–7.32 (m, 2H, Har), 7.17 (d, J = 1.0 Hz, 1H, Him), 6.78 (dd, J = 7.6, 5.6 Hz, 1H, 1′-H), 5.42 (td, J = 6.6, 2.3 Hz, 1H, 3′-H), 4.48–4.37 (m, 3H, 5′-H and 4′-H), 2.99 (ddd, J = 14.3, 5.6, 2.3 Hz, 1H, 2′-H), 2.43 (s, 3H, CH3), 2.41 (ddd, J = 14.3, 7.6. 6.6 Hz, 1H, 2′-H) ppm; 13C NMR (100.61 MHz, CDCl3): δ = 165.9 (CO), 145.7 (C tosq ), 144.0 (Cqim), 133.9 (HCPh), 132.2 (Cqtos), 130.2 (2C, HCtos), 129.7 (2C, HCtos), 128.96 (HCim), 128.7 (CqPh), 128.6 (2CPh), 127.9 (2CPh), 121.8 (Cim), 88.8 (C-1′), 83.2 (C-4′), 74.4 (C-3′), 68.5 (C-5′), 40.1 (C-2′), 21.7 (CH tos3 ) ppm; and IR (ATR, NMR sample): \(\bar{\nu }\) = 1713, 1544, 1352, 1279, 1174, 1096 cm−1.
Preparation of 3-O-benzoyl-2-deoxy-5-O-tosyl-D-ribofuranosyl chlorides and their conversion to α- and β-19 by general procedure C
A mixture of 0.495 g methyl glycosides α- and β-18 (1.22 mmol) was transformed via glycosyl chlorides (general procedure A) into nucleosides α- and β-19 by general procedure C. The crude product (α/β = 1/5, by 1H NMR) was flash chromatographed (hexanes/EtOAc = 2/1) using a long column to yield 0.066 g nucleoside α-19 (14%) and 0.327 g β-19 (69%).
References
Fyles AW, Milosevic M, Wong R, Kavanagh MC, Pintilie M, Sun A, Chapman W, Levin W, Manchui L, Keane TJ, Hill RP (1998) Radiother Oncol l48:149
Hanahan D, Weinberg RA (2011) Cell 144:646
Eales KI, Hollinshead KE, Tennant DA (2016) Oncogenesis 5:e190
Vaupel P, Mayer A (2007) Cancer Metastasis Rev 26:225
Dhani N, Fyles A, Hedley D, Milosevic M (2015) Semin Nucl Med 45:110
Kelada OJ, Carlson DJ (2014) Radiat Res 181:335
Kumar P, Bacchu V, Wiebe LJ (2015) Semin Nucl Med 45:122
Nunn A, Linder K, Strauss HW (1995) Eur J Nucl Med 22:265
Masaki Y, Shimizu Y, Yoshioka T, Tanaka Y, Nishijima K-i, Zhao S, Higashino K, Sakamoto S, Numata Y, Yamaguchi Y, Tamaki N, Kuge Y (2015) Nature Sci Rep 5:16802
Rasey JS, Koh WJ, Evans ML, Peterson LM, Lewellen TK, Graham MM, Krohn KA (1996) Int J Radiat Oncol Biol Phys 36:417
Lehtio K, Oikonen V, Gronroos T, Eskola O, Kalliokoski K, Bergman J, Solin O, Grenman R, Nuutila P, Minn H (2001) J Nucl Med 42:1643
Piert M, Machulla H-J, Picchio M, Reischl G, Ziegler S, Kumar P, Wester H-J, Beck R, McEwan AJB (2005) J Nucl Med 46:106
Halmos GB, Bruine de Bruin L, Langendijk JA, van der Laan BF, Pruim J, Steenbakkers RJ (2014) Clin Nucl Med 39:44
Kumar P, Emami S, Kresolek Z, Yang J, McEwan AJB, Wiebe LI (2009) Med Chem 5:118
Patt M, Sorger D, Scheunemann M, Stöcklin G (2002) Appl Radiat Isot 57:705
Schweifer A, Maier F, Ehrlichmann W, Laparter D, Kneilling M, Pichler BJ, Hammerschmidt F, Reischl G (2016) Mol Med Biol 43:759
Wanek T, Kreis K, Križková P, Schweifer A, Denk C, Stanek J, Mairinger S, Filip T, Sauberer M, Edelhofer P, Traxl A, Muchitsch VE, Mereiter K, Hammerschmidt F, Cass CE, Damaraju VL, Langer O, Kuntner C (2016) Bioorg Med Chem 24:5326
Bath CC (1968) In: Zorbach WW, Tipson RS (eds) Synthetic procedures in nucleic acid chemistry. Wiley, New York, p 521
Wang D, Nugent WA (2007) J Org Chem 72:7307
Schmidt L, Pedersen EB, Nielsen C (1994) Acta Chem Scand 48:215
Searcey M, Pye PL, Lee JB (1989) Synth Commun 19:1309
Rao P, Benner SA (2001) J Org Chem 66:5012
Mikenda W (1992) Vib Spectrosc 3:327
Acknowledgements
Open access funding provided by University of Vienna. The authors thank S. Felsinger for recording NMR spectra and J. Theiner for combustion analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Križková, P., Wieczorek, A. & Hammerschmidt, F. Efficient preparation of 2-nitroimidazole nucleosides as precursors for hypoxia PET tracers. Monatsh Chem 148, 83–90 (2017). https://doi.org/10.1007/s00706-016-1874-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-016-1874-8