
Abstract
The treatment of liver cirrhosis is currently being standardized and developed specifically to reduce activation of hepatic stellate cells (HSCs), inhibit fibrosis, increase degradation of matrix components, and reduce activated myofibroblasts. Cell therapy can be applied in the treatment of liver cirrhosis; however, the characteristic features of this therapy differ from those of other treatments because of the involvement of a living body origin and production of multiple cytokines, chemokines, matrix metalloproteinases (MMPs), and growth factors. Thus, cell therapies can potentially have multiple effects on the damaged liver, including alleviating liver cirrhosis and stimulating liver regeneration with affecting the host cells. Cell therapies initially involved autologous bone marrow cell infusion, and have recently developed to include the use of specific cells such as mesenchymal stem cells and macrophages. The associated molecular mechanisms, routes of administration, possibility of allogeneic cell therapy, and host conditions appropriate for cell therapies are now being extensively analyzed. In this review, we summarize the status and future prospects of cell therapy for liver cirrhosis.
Similar content being viewed by others
Introduction
Chronic liver injury caused by hepatitis virus infection, alcohol intake, nonalcoholic steatohepatitis (NASH), autoimmune hepatitis, and primary biliary cholangitis, among other factors, results in liver cirrhosis, leading to liver failure, with reduced liver function, portal hypertension, and increased risk of hepatocellular carcinoma (HCC). Owing to the severity of clinical symptoms, US, European [1–4], and Japanese clinical practice guidelines [5] have been developed for liver cirrhosis; these guidelines include standardized treatments such as nutrition therapy, antiviral therapy, management of portal hypertension, ascites, hepatorenal syndrome, hepatic encephalopathy, and portal thrombosis, and liver transplantation.
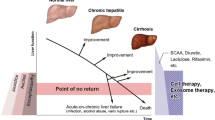
The liver is highly regenerative, and fibrosis and ductular reactions (DRs), which are assumed to include hepatic stem/progenitor cells (HPCs) and inflammatory cells, are closely associated and form the hepatic stem/progenitor cell niche [6–8]. A recent report revealed that matrix remodeling by matrix metalloproteinases (MMPs) is required for the proliferation of HPCs [9–11], indicating that resident and recruited cells in or surrounding the DR are involved in the alleviation of fibrosis and stimulation of the liver regenerative process. Although analysis of data following antiviral therapy has revealed that reducing the damage caused by hepatitis can improve liver fibrosis in the treatment for hepatitis B and C, there are also some populations in which liver fibrosis and function did not improve. Jang et al. reported that in patients with Child–Pugh class B and C disease, antiviral therapy for hepatitis B alone was not sufficient for prolonging survival in patients [12]. Additionally, Curry et al. reported that during antiviral therapy for hepatitis C, 58 % of patients showed no improvement in Child–Pugh score after sustained virological responses [13]. Furthermore, it is believed that there is a “point of no return” for patients with advanced cirrhosis; in these patients, liver transplantation is thought to be the only curative treatment [14]. However, liver transplantation has some problems, including donor shortage, severe complications, high cost, and the use of immunosuppressant agents [15]. Thus, a new strategy to improve liver fibrosis and promote liver regeneration is eagerly anticipated.
In this paper, we review the status and history of cell therapy, discuss the possible applications of mesenchymal stem cells (MSCs) and macrophages in such therapy, and present future prospects for cell therapies for liver cirrhosis.
Status of cell therapy in liver fibrosis treatment
Hepatic stellate cells (HSCs) are central players in liver fibrosis and the major precursors of activated myofibroblasts, which produce extracellular matrix (ECM) during liver fibrosis. Although the ECM is fundamentally important as a scaffold, excess ECM can cause complications such as liver dysfunction and portal hypertension; thus, the balance between ECM production and degradation must be controlled appropriately. During liver damage, quiescent HSCs, which contain vitamin A, are activated by the many cytokines present, including transforming growth factor (TGF)-β and platelet-derived growth factor (PDGF), and the activated myofibroblasts produce excess ECM. After liver injury, the number of activated myofibroblasts is reduced by apoptosis, senescence, or reversion to an inactivated phenotype. Excess ECM can be degraded by MMPs. Thus, understanding and controlling the molecular mechanisms involved in the activation of HSCs, production or degradation of ECM, and reduction of the number of activated myofibroblasts is important for developing novel therapies for liver fibrosis [16–21]. The current therapeutic approach for liver fibrosis is divided into five categories, as discussed below.
Controlling the primary disease
The strategy of controlling primary liver disease is the most effective antifibrotic treatment. Damage to hepatocytes and cholangiocytes and the subsequent inflammatory response induce activated myofibroblasts. Antiviral therapy for hepatitis B and C virus infection and antioxidants such as vitamin E are the typical treatments for NASH [22, 23]. In this context, many strategies, including a small molecule antagonist of NADPH oxidase [24] and a carbohydrate molecule that inhibits galectin-3 [25], are currently being evaluated in animal and human trials.
Targeting the receptor–ligand interaction and intracellular signaling in stellate cells
Membrane and nuclear receptors expressed in HSCs and their intracellular signaling pathways are related to HSC activation and fibrosis. The renin-angiotensin system [26, 27], adipokines [28, 29], tyrosine kinase receptors [30, 31], and nuclear receptors such as peroxisome proliferator-activated receptor (PPAR)-γ [32] are representative targets.
Inhibiting fibrogenesis or increasing matrix degradation
TGF-β is the most potent stimulus for the synthesis of collagen and other matrix components; thus, the inhibition of this molecule is an attractive goal of therapy [33, 34]. gp46, the rat homolog of human heat shock protein 47, facilitates collagen secretion and has been implicated in the translational regulation of procollagen synthesis. An siRNA against gp46 delivered by vitamin A-coupled liposomes was evaluated in an animal model of liver fibrosis [35]. Collagen that is not fully cross-linked is more susceptible to degradation by endogenous MMPs and other enzymes; thus, a monoclonal antibody that inhibits the collagen cross-linking enzyme lysyl oxidase-like 2 was developed and has shown promising results in preclinical studies [36].
Reducing activated HSCs
Myofibroblast clearance is another target of therapy. Natural killer (NK) cell activation to kill activated myofibroblasts [37] and inhibition of nuclear factor kappa B (NF-κB) to enhance the expression of anti-apoptotic proteins [38] are included in this category.
Cell therapies
Cell therapies such as the use of bone marrow cells, mesenchymal cells, and macrophages are completely different from other therapeutic approaches [39, 40]. These cells are used in therapies because various factors, including cytokines, chemokines, growth factors, and MMPs, influence many targets and physiological functions (Fig. 1). Living donors and cultures are often necessary; thus, the quality of the cells is not always uniform. Furthermore, the cells themselves are frequently heterogeneous populations. Although the main target of these therapies is the regression of fibrosis, other effects such as reduced inflammation, enhanced angiogenesis, and increased liver regeneration can be achieved by these therapies, making them attractive approaches.

Putative targets of cell therapies. Drugs targeting various processes ranging from prevention of tissue damage to ECM degradation may be useful in the treatment of liver fibrosis. Cell therapies that produce multiple factors can potentially affect multiple targets or processes
Autologous bone marrow cell injection therapy and related treatments
In 2003, autologous bone marrow cell infusion (ABMi) therapy was initiated at Yamaguchi University in patients with liver cirrhosis [41, 42]. For this therapy, 400 mL of autologous bone marrow cells is obtained under general anesthesia, and mononuclear cells are then infused through a peripheral vein after washing. Due to the need for general anesthesia, this therapy requires a total bilirubin level of 3 mg/dL or lower, platelet count of 50,000/μL or higher, and absence of hepatocellular carcinoma by magnetic resonance imaging (MRI) and computed tomography (CT). To date, this therapy has been performed at Yamaguchi University and collaborating institutions in more than 60 patients with cirrhosis of various etiologies [43, 44]. From the experience with this therapy, several novel points have been elucidated. Although direct evidence of alleviation of liver fibrosis, such as reduction of liver fibrosis by MMP-producing mononuclear cells in animal models, is difficult to obtain in humans, improved liver function has been observed, as indicated by increased serum albumin levels, decreased amount of ascites, and decreased Child–Pugh scores. The mechanisms through which liver function is improved may be related to increases in the number of proliferating cell nuclear antigen (PCNA)-positive hepatocytes and DRs after therapy. These results in animal models and human studies have confirmed the existence of bone marrow-derived cells that alleviate liver fibrosis and improve liver function. Similar and related studies have been performed worldwide using autologous cells with modified transplantation techniques. Some groups have used bone marrow mononuclear cells (BMNCs), whereas other groups have used G-CSF and CD34+ hematopoietic cells harvested from a peripheral vein, avoiding the need for invasive procedures. Furthermore, several routes of cell administration have been investigated, included peripheral veins, the hepatic artery, and the portal vein [45–54]. In a randomized control study, Spahr et al. reported that there were no significant differences in treatment outcomes when using autologous BMNCs harvested after G-CSF infusion through the hepatic artery, although several other randomized studies have shown favorable results, suggesting the usefulness of these therapies [50]. More recently, the Repeated AutoLogous Infusion of Stem cells In Cirrhosis (REALISTIC), a multicenter, phase II, open-label, randomized controlled trial of repeated autologous infusion of G-CSF-mobilized CD133+ bone marrow stem cells in patients with cirrhosis, was announced in the UK [55]. Although the effectiveness and potential applications of cell therapy have been reported, several problems need to be overcome. For example, bone marrow cells, which include hematopoietic, endothelial, and mesenchymal cells, and peripheral mononuclear cells are heterogeneous populations; thus, the specific cell population that is most important for alleviating liver fibrosis and improving liver function has not been identified. Further analysis is needed to determine which administration route is the most effective, whether autologous cells are necessary, and whether allogeneic cells can be employed for cell therapy against liver fibrosis. To address these unresolved problems, two promising cell populations, MSCs and macrophages, are now under extensive investigation. Below, we discuss recent progress in the study of MSCs and macrophages.
MSCs
The use of MSCs in cell therapy has recently received worldwide attention [56]. MSCs can be obtained from not only bone marrow but also medical wastes such as adipose tissue [57, 58], umbilical tissue [59–62], and dental pulp [63]. MSCs are positive for the common markers CD73, CD90, and CD105; however, they are negative for the endothelial marker CD31 and hematopoietic marker CD45 [56, 64]. The expansion of MSCs in culture is relatively easy, and under appropriate conditions, MSCs can differentiate into adipocytes, osteoblasts, and chondroblasts. MSCs are not a homogeneous population and are actually quite heterogeneous. For mouse bone marrow MSCs, there are at least four populations of cells: stem cell factor-positive cells, stromal cell-derived factor-1-positive cells, nestin-positive cells, and PDGF receptor (PDGFR)-α + sca-1 + CD45-Ter119- cells [65].
The effects of MSCs can be broadly divided into two mechanisms: (1) recruited MSCs differentiate into functional cells to replace damaged cells, permitting the treatment of bone and cartilage damage; and (2) in response to inflammatory cytokines, MSCs help prepare the microenvironment by producing immunoregulatory factors that modulate the progression of inflammation. MSCs also produce a large amount of cytokines and growth factors that stimulate angiogenesis, prevent apoptosis, block oxidation reactions, promote remodeling of the ECM, and induce differentiation of tissue stem cells. This mechanism can be applied for many diseases, including liver cirrhosis. Some studies have reported that the effects of MSCs are determined by host conditions, such as inflammation stage and the use of immunosuppressants [66].
Although studies have analyzed the behaviors of MSCs after administration, and MSCs have been shown to migrate to the injured site, MSC behaviors in humans have not been fully elucidated. Some studies have reported that MSCs disappear within a few weeks and remain in the target tissue for only a short time [64]. Recent studies have reported that only culture-conditioned media or exosomes induce a treatment effect, suggesting that the trophic effect is the most important effect of MSCs [67–69]. As MSCs can be obtained relatively easily and have multiple functions, more than 300 clinical trials are ongoing, most of which are in phase I or II, in the bone/cartilage, heart, neuron, immune/autoimmune, diabetes/kidney, lung, liver, and gastrointestinal fields [56]. These studies aim to elucidate the effectiveness of MSCs in the treatment of various diseases. Another important characteristic of MSCs is that they generally have low immunogenicity; thus, injection of autologous or allogeneic MSCs has been employed in clinical studies, including some studies of liver cirrhosis. For example, Lalu et al. performed a meta-analysis of the safety of MSCs in clinical trials and showed that autologous and allogeneic MSC therapies were related to transient fever but not to infusion toxicity, organ system complications, infection, death, or malignancies [64]. Thus allogeneic MSC therapy has the potential to expand MSC treatment to many patients.
In the field of liver cirrhosis, as shown in Table 1, many autologous [70–83] and four allogeneic [59–62] MSC therapies involving a variety of tissue origins and administration routes have been reported. Although details regarding the mechanisms of therapeutic effects in humans remain unclear, most studies have reported favorable effects with improvement in MELD scores, Child–Pugh scores, and some serum markers such as albumin and bilirubin. As shown in Table 1, allogeneic umbilical tissue-derived MSCs have also improved MELD scores and serum levels of albumin and bilirubin. Moreover, as shown in Table 2, human MSCs have been safely administered in rodents, also exhibiting some therapeutic effects, thereby supporting the low immunogenicity of MSCs [84–88].
One concern regarding the administration of MSCs in patients with liver cirrhosis is the potential for stimulation of tumor formation. In a study by Peng et al., MSCs from 120 mL of bone marrow were administered through the hepatic artery into more than 50 patients, and no severe adverse events or significant differences in tumor formation were detected compared with those in the control group [75]. Cell administration routes through the peripheral vein, portal vein, and hepatic artery (intra-arterial) have been reported. Differences regarding the administration route will be elucidated as studies accumulate. More recently, Suk et al. reported a phase II trial of autologous bone marrow-derived MSC (BM–MSC) therapy for alcoholic liver cirrhosis. The authors showed that although tracing the administered MSCs was difficult, intra-arterial autologous BM–MSC injection was able to alleviate liver fibrosis and improve Child–Pugh scores [83].
Macrophages
Recent studies have examined the role of macrophages in the liver [39, 89]. Functionally distinct subpopulations of macrophages exist in the damaged liver, and these macrophages play critical roles in both scar progression and regression. Duffield et al. examined the dual role of macrophages. To show whether macrophages have a dual role during liver injury, a CD11b-DTR transgenic mouse was generated in which macrophages could be depleted after the administration of diphtheria toxin. This mouse line was used in a carbon tetrachloride (CCl4) model of liver cirrhosis. Macrophages were depleted either during the injury phase or during the recovery phase. Macrophage depletion during the injury phase, when liver fibrosis was advancing, resulted in reduced scarring and fewer myofibroblasts. However, macrophage depletion during the recovery phase resulted in a reduction in matrix degradation [90]. These results clearly show the dual role of macrophages during liver injury. Ramachandran et al. examined the specific phenotypes of macrophages during the injury and resolution phases in mice. During the pro-fibrotic injury phase, macrophages associated with liver tissue scars were predominantly CD11BhiF4/80intLy-6Chi, whereas after cessation of the injury and during the resolution of the scars, the scar-associated macrophages were predominantly CD11BhiF4/80intLy-6Clo. Interestingly, this phenotypic switch appears to occur in response to phagocytosis and is associated with the upregulation of a key mediator of scar resolution, MMPs [91].
The potential role of monocytes and mature macrophages in cell therapy was examined in a mouse model of chronic liver fibrosis. In this analysis, mouse bone marrow macrophage precursor cells and differentiated bone marrow-derived macrophages (BMMs) generated after 7 days in culture with colony-stimulating factor (CSF)-conditioned medium were injected into the portal vein in mice, in which liver damage was induced through repeated injections of CCl4. The BMMs were grown in low adherence conditions to allow them to reach full maturity and enable easy removal from the culture containers. This methodology resulted in macrophages that did not conform to either the traditional classically (M1) or alternatively activated (M2) macrophage phenotype. However, the BMMs expressed anti-inflammatory (IL-10), antifibrotic (MMP-13), pro-regenerative (tumor necrosis factor-like weak inducer of apoptosis [TWEAK]), and chemotactic (monocyte chemotactic protein [MCP]-1, macrophage inflammatory protein [MIP]-1α, and MIP-2) mediators. Injection of 1 × 106 wild-type BMMs into the portal vein of recipient mice subjected to CCl4-induced liver fibrosis resulted in a significant reduction in fibrosis, whereas injection of BMM precursor cells did not alleviate fibrosis. The underlying mechanisms mediating the beneficial effects of mature BMMs were further analyzed. Although engraftment of injected BMMs was transient, the cells released chemokines that attracted host inflammatory cells to the scar areas in the liver (including recipient macrophages), and that presumably amplified their effects, accounting for the significant changes observed with small numbers of injected macrophages. The animals that received injections of the cultured macrophages had lower numbers of hepatic-activated myofibroblasts and higher numbers of hepatic MMP-expressing cells. Increases in the levels of pro-regenerative growth factors and anti-inflammatory cytokines were also observed in the liver. All of these effects induced by BMM injection likely contributed to the regression of liver fibrosis and stimulated regeneration [10].
In addition to reduced fibrosis, increased serum albumin levels have been observed in BMM recipient mice, revealing that BMMs facilitated the reduction of liver fibrosis and improved liver regeneration. Bird et al. reported that the in vitro induction of bone marrow cells (BMCs) to macrophages resulted in a 20-fold upregulation of TWEAK mRNA, and that macrophage-secreted TWEAK stimulated the DR via its receptor Fn14 [92]. Furthermore, Boulter et al. found that Notch and Wnt signaling determines the behavior and fate of HPCs, including their differentiation. In mice, during the activation of HPCs in biliary diseases, HPCs express the receptors Notch1 and Notch2, and these are activated through interaction with myofibroblast-derived Jagged1. This high Notch state appears to delineate the default pathway for the differentiation of HPCs into cholangiocytes and is inhibited during the regeneration of hepatocytes by the ubiquitin ligase Numb. In HPCs during hepatocyte differentiation, Numb transcription is activated by Wnt/β-catenin signaling. Wnt-mediated, Numb-dependent repression of Notch acts as a node at which the Notch and Wnt pathways can interact to enable lineage specification. During this process, macrophages secrete the Wnt ligand and thereby induce the differentiation of HPCs into hepatocytes. When macrophages phagocytize cell debris, they strongly upregulate the production of Wnt3A, which then helps to promote the differentiation of HPCs towards a hepatocyte fate [93, 94]. Moore et al. showed that human murine monocyte-derived macrophages alleviated liver fibrosis and function in NOD CB17 Prkdc/SCID mice; in addition, they reported no significant difference in the mean number of CD14+ monocytes isolated from a peripheral vein by apheresis or in the mean yield of cultured mature macrophages between patients with cirrhosis and healthy controls. These mature human macrophages caused upregulation of a number of genes, including MMP-9, CCL2, IL-10, and TWEAK, suggesting that macrophages can be differentiated from apheresis-derived CD14+ monocytes isolated from patients with cirrhosis and developed into “regenerative” macrophages as control macrophages [95]. Based on these results, although few clinical trials using macrophages have been performed, macrophage therapy may be a promising approach.
Conclusions
Cell therapies for liver cirrhosis were first attempted with ABMi and have since evolved to include the use of specific cell populations such as MSCs and macrophages (Fig. 2). Although both MSCs and macrophages have been reported to alleviate liver fibrosis and improve liver regeneration, these cell types seem to have distinct characteristics, and it is unclear which population yields better results (Table 3). Furthermore, because MSCs have low immunogenicity, allogeneic treatment with MSCs derived from adipose tissue, umbilical tissue, and dental pulp may provide a means of expanding cell therapy for liver cirrhosis. Thus, additional clinical studies on allogeneic MSCs for the treatment of liver cirrhosis are needed. However, given that MSCs have been reported to influence the host disease state, e.g., inducing inflammation in certain contexts, the indications for MSC treatment should be evaluated carefully, and the most suitable conditions will need to be determined. For macrophages, although recently reported procedures involved the induction of “regenerative” macrophages from monocytes ex vivo after apheresis, the in vivo induction of these “regenerative” macrophages from monocytes may be an interesting approach. The cellular kinetics and relationship with host cells remain unclear, and elucidating the details of these factors will clarify the related mechanisms. Recently, injected MSCs or macrophages have been reported to direct host macrophages and inflammatory cells through the production of growth factors and immune-modulating factors such as prostaglandin (PG) E2, PGE1α, IL-6, IL-8, indoleamine 2,3-dioxygenase, nitric oxide, TGF-β, and hepatocyte growth factor. Thus, cocktails of MSCs and macrophages may be more effective. Further studies on the interactions among MSCs, macrophages, and host cells are needed to obtain valuable information for overcoming the so-called point of no return.

History of cell therapy for liver cirrhosis. Cell therapies for liver cirrhosis were first applied using ABMi and have evolved to include the use of specific cell populations, including MSCs and macrophages. MSC and macrophage therapies are still being investigated and improved
References
Runyon BA. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology. 2013;57:1651–3.
O’Shea RS, Dasarathy S, McCullough AJ, et al. Alcoholic liver disease. Hepatology. 2010;51:307–28.
American Association for the Study of Liver Diseases, European Association for the Study of the Liver. Hepatic encephalopathy in chronic liver disease practice guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases. J Hepatol. 2014;2014(61):642–59.
European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397–417.
Fukui H, Saito H, Ueno Y, et al. Evidence-based clinical practice guidelines for liver cirrhosis 2015. J Gastroenterol. 2016;51:629–50.
Williams MJ, Clouston AD, Forbes SJ. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014;146:349–56.
Lu WY, Bird TG, Boulter L, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol. 2015;17:971–83.
Tsuchiya A, Lu WY, Weinhold B, et al. Polysialic acid/neural cell adhesion molecule modulates the formation of ductular reactions in liver injury. Hepatology. 2014;60:1727–40.
Kallis YN, Robson AJ, Fallowfield JA, et al. Remodelling of extracellular matrix is a requirement for the hepatic progenitor cell response. Gut. 2011;60:525–33.
Thomas JA, Pope C, Wojtacha D, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53:2003–15.
Sakaida I, Terai S, Yamamoto N, et al. Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology. 2004;40:1304–11.
Jang JW, Choi JY, Kim YS, et al. Long-term effect of antiviral therapy on disease course after decompensation in patients with hepatitis B virus-related cirrhosis. Hepatology. 2015;61:1809–20.
Curry MP, O’Leary JG, Bzowej N, et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med. 2015;373:2618–28.
Forbes SJ, Gupta S, Dhawan A. Cell therapy for liver disease: From liver transplantation to cell factory. J Hepatol. 2015;62:S157–69.
Merion RM, Schaubel DE, Dykstra DM, et al. The survival benefit of liver transplantation. Am J Transplant. 2005;5:307–13.
Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64:830–41.
Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat Sci. 2015;22:512–8.
Noureddin M, Anstee QM, Loomba R. Review article: emerging anti-fibrotic therapies in the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2016;43:1109–23.
Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–901.
Rockey DC. Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin Gastroenterol Hepatol. 2013;11(224–31):e1–5.
Berardis S, Dwisthi Sattwika P, Najimi M, et al. Use of mesenchymal stem cells to treat liver fibrosis: current situation and future prospects. World J Gastroenterol. 2015;21:742–58.
Serviddio G, Bellanti F, Vendemiale G. Free radical biology for medicine: learning from nonalcoholic fatty liver disease. Free Radic Biol Med. 2013;65:952–68.
Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85.
Paik YH, Iwaisako K, Seki E, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91phox mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730–41.
Traber PG, Chou H, Zomer E, et al. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS One. 2013;8:e75361.
Granzow M, Schierwagen R, Klein S, et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology. 2014;60:334–48.
Rozenfeld R, Gupta A, Gagnidze K, et al. AT1R-CB1R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 2011;30:2350–63.
Ueno T, Nakamura A, Nakayama H, et al. Adiponectin suppresses endoplasmic reticulum stress in nonalcoholic steatohepatitis. Exp Ther Med. 2011;2:1035–40.
Moreno M, Chaves JF, Sancho-Bru P, et al. Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology. 2010;51:974–85.
Hong F, Chou H, Fiel MI, et al. Antifibrotic activity of sorafenib in experimental hepatic fibrosis: refinement of inhibitory targets, dosing, and window of efficacy in vivo. Dig Dis Sci. 2013;58:257–64.
Choi SS, Sicklick JK, Ma Q, et al. Sustained activation of Rac 1 in hepatic stellate cells promotes liver injury and fibrosis in mice. Hepatology. 2006;44:1267–77.
Staels B, Rubenstrunk A, Noel B, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013;58:1941–52.
Puthawala K, Hadjiangelis N, Jacoby SC, et al. Inhibition of integrin alpha (v) beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177:82–90.
Henderson NC, Arnold TD, Katamura Y, et al. Targeting of alpha v integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–24.
Sato Y, Murase K, Kato J, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26:431–42.
Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1009–17.
Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–28.
Chakraborty JB, Mann DA. NF-kB signalling: Embracing complexity to achieve translation. J Hepatol. 2010;52:285–91.
Forbes SJ, Rosenthal N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat Med. 2014;20:857–69.
Than NN, Tomlinson CL, Haldar D, et al. Clinical effectiveness of cell therapies in patients with chronic liver disease and acute-on-chronic liver failure: a systematic review protocol. Syst Rev. 2016;5:100.
Terai S, Ishikawa T, Omori K, et al. Improved liver function in patients with liver cirrhosis after autologous bone marrow cell infusion therapy. Stem Cells. 2006;24:2292–8.
Terai S, Takami T, Yamamoto N, et al. Status and prospects of liver cirrhosis treatment by using bone marrow-derived cells and mesenchymal cells. Tissue Eng Part B Rev. 2014;20:206–10.
Kim JK, Park YN, Kim JS, et al. Autologous bone marrow infusion activates the progenitor cell compartment in patients with advanced liver cirrhosis. Cell Transplant. 2010;19:1237–46.
Saito T, Okumoto K, Haga H, et al. Potential therapeutic application of intravenous autologous bone marrow infusion in patients with alcoholic liver cirrhosis. Stem Cells Dev. 2011;20:1503–10.
Gordon MY, Levicar N, Pai M, et al. Characterization and clinical application of human CD34+ stem/progenitor cell populations mobilized into the blood by granulocyte colony-stimulating factor. Stem Cells. 2006;24:1822–30.
Levicar N, Pai M, Habib NA, et al. Long-term clinical results of autologous infusion of mobilized adult bone marrow derived CD34+ cells in patients with chronic liver disease. Cell Prolif. 2008;41(Suppl 1):115–25.
Khan AA, Parveen N, Mahaboob VS, et al. Safety and efficacy of autologous bone marrow stem cell transplantation through hepatic artery for the treatment of chronic liver failure: a preliminary study. Transplant Proc. 2008;40:1140–4.
Pai M, Zacharoulis D, Milicevic MN, et al. Autologous infusion of expanded mobilized adult bone marrow-derived CD34+ cells into patients with alcoholic liver cirrhosis. Am J Gastroenterol. 2008;103:1952–8.
Han Y, Yan L, Han G, et al. Controlled trials in hepatitis B virus-related decompensate liver cirrhosis: peripheral blood monocyte transplant versus granulocyte-colony-stimulating factor mobilization therapy. Cytotherapy. 2008;10:390–6.
Spahr L, Lambert JF, Rubbia-Brandt L, et al. Granulocyte-colony stimulating factor induces proliferation of hepatic progenitors in alcoholic steatohepatitis: a randomized trial. Hepatology. 2008;48:221–9.
Salama H, Zekri AR, Bahnassy AA, et al. Autologous CD34+ and CD133+ stem cells transplantation in patients with end stage liver disease. World J Gastroenterol. 2010;16:5297–305.
Salama H, Zekri AR, Zern M, et al. Autologous hematopoietic stem cell transplantation in 48 patients with end-stage chronic liver diseases. Cell Transplant. 2010;19:1475–86.
Garg V, Garg H, Khan A, et al. Granulocyte colony–stimulating factor mobilizes CD34+ cells and improves survival of patients with acute-on-chronic liver failure. Gastroenterology. 2012;142(505–512):e1.
Kedarisetty CK, Anand L, Bhardwaj A, et al. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology. 2015;148(1362–70):e7.
King A, Barton D, Beard HA, et al. REpeated AutoLogous Infusions of STem cells In Cirrhosis (REALISTIC): a multicentre, phase II, open-label, randomised controlled trial of repeated autologous infusions of granulocyte colony-stimulating factor (GCSF) mobilised CD133+ bone marrow stem cells in patients with cirrhosis. A study protocol for a randomised controlled trial. BMJ Open. 2015;5:e007700.
Trounson A, McDonald C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell. 2015;17:11–22.
Banas A, Teratani T, Yamamoto Y, et al. Adipose tissue-derived mesenchymal stem cells as a source of human hepatocytes. Hepatology. 2007;46:219–28.
Seki A, Sakai Y, Komura T, et al. Adipose tissue-derived stem cells as a regenerative therapy for a mouse steatohepatitis-induced cirrhosis model. Hepatology. 2013;58:1133–42.
Shi M, Zhang Z, Xu R, et al. Human mesenchymal stem cell transfusion is safe and improves liver function in acute-on-chronic liver failure patients. Stem Cells Transl Med. 2012;1:725–31.
Zhang Z, Lin H, Shi M, et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J Gastroenterol Hepatol. 2012;27(Suppl 2):112–20.
Wang L, Li J, Liu H, et al. Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 2013;28(Suppl 1):85–92.
Wang L, Han Q, Chen H, et al. Allogeneic bone marrow mesenchymal stem cell transplantation in patients with UDCA-resistant primary biliary cirrhosis. Stem Cells Dev. 2014;23:2482–9.
Sharpe PT. Dental mesenchymal stem cells. Development. 2016;143:2273–80.
Lalu MM, McIntyre L, Pugliese C, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS One. 2012;7:e47559.
Omatsu Y, Seike M, Sugiyama T, et al. Foxc 1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature. 2014;508:536–40.
Wang Y, Chen X, Cao W, et al. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014;15:1009–16.
Katsuda T, Kosaka N, Takeshita F, et al. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics. 2013;13:1637–53.
Monsel A, Zhu YG, Gudapati V, et al. Mesenchymal stem cell derived secretome and extracellular vesicles for acute lung injury and other inflammatory lung diseases. Expert Opin Biol Ther. 2016;16:859–71.
Park CM, Kim MJ, Kim SM, et al. Umbilical cord mesenchymal stem cell-conditioned media prevent muscle atrophy by suppressing muscle atrophy-related proteins and ROS generation. In Vitro Cell Dev Biol Anim. 2016;52:68–76.
Mohamadnejad M, Namiri M, Bagheri M, et al. Phase 1 human trial of autologous bone marrow-hematopoietic stem cell transplantation in patients with decompensated cirrhosis. World J Gastroenterol. 2007;13:3359–63.
Mohamadnejad M, Alimoghaddam K, Mohyeddin-Bonab M, et al. Phase 1 trial of autologous bone marrow mesenchymal stem cell transplantation in patients with decompensated liver cirrhosis. Arch Iran Med. 2007;10:459–66.
Kharaziha P, Hellstrom PM, Noorinayer B, et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: a phase I–II clinical trial. Eur J Gastroenterol Hepatol. 2009;21:1199–205.
El-Ansary MMS, Abdel-Aziz I, Abdel-Hamid S. Phase I trial: mesenchymal stem cells transplantation in end stage liver disease. J Am Sci. 2010;6:135–44.
Amer ME, El-Sayed SZ, El-Kheir WA, et al. Clinical and laboratory evaluation of patients with end-stage liver cell failure injected with bone marrow-derived hepatocyte-like cells. Eur J Gastroenterol Hepatol. 2011;23:936–41.
Peng L, Xie DY, Lin BL, et al. Autologous bone marrow mesenchymal stem cell transplantation in liver failure patients caused by hepatitis B: short-term and long-term outcomes. Hepatology. 2011;54:820–8.
El-Ansary M, Abdel-Aziz I, Mogawer S, et al. Phase II trial: undifferentiated versus differentiated autologous mesenchymal stem cells transplantation in Egyptian patients with HCV induced liver cirrhosis. Stem Cell Rev. 2012;8:972–81.
Mohamadnejad M, Alimoghaddam K, Bagheri M, et al. Randomized placebo-controlled trial of mesenchymal stem cell transplantation in decompensated cirrhosis. Liver Int. 2013;33:1490–6.
Amin MA, Sabry D, Rashed LA, et al. Short-term evaluation of autologous transplantation of bone marrow-derived mesenchymal stem cells in patients with cirrhosis: Egyptian study. Clin Transplant. 2013;27:607–12.
Jang YO, Kim YJ, Baik SK, et al. Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: a pilot study. Liver Int. 2014;34:33–41.
Salama H, Zekri AR, Medhat E, et al. Peripheral vein infusion of autologous mesenchymal stem cells in Egyptian HCV-positive patients with end-stage liver disease. Stem Cell Res Ther. 2014;5:70.
Xu L, Gong Y, Wang B, et al. Randomized trial of autologous bone marrow mesenchymal stem cells transplantation for hepatitis B virus cirrhosis: regulation of Treg/Th17 cells. J Gastroenterol Hepatol. 2014;29:1620–8.
Lukashyk SP, Tsyrkunov VM, Isaykina YI, et al. Mesenchymal bone marrow-derived stem cells transplantation in patients with HCV related liver cirrhosis. J Clin Transl Hepatol. 2014;2:217–21.
Suk KT, Yoon JH, Kim MY, et al. Transplantation with autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: Phase 2 trial. Hepatology. 2016. doi:10.1002/hep.28693.
Jung KH, Shin HP, Lee S, et al. Effect of human umbilical cord blood-derived mesenchymal stem cells in a cirrhotic rat model. Liver Int. 2009;29:898–909.
Chang YJ, Liu JW, Lin PC, et al. Mesenchymal stem cells facilitate recovery from chemically induced liver damage and decrease liver fibrosis. Life Sci. 2009;85:517–25.
Kim SJ, Park KC, Lee JU, et al. Therapeutic potential of adipose tissue-derived stem cells for liver failure according to the transplantation routes. J Korean Surg Soc. 2011;81:176–86.
Tanimoto H, Terai S, Taro T, et al. Improvement of liver fibrosis by infusion of cultured cells derived from human bone marrow. Cell Tissue Res. 2013;354:717–28.
Jang YO, Kim MY, Cho MY, et al. Effect of bone marrow-derived mesenchymal stem cells on hepatic fibrosis in a thioacetamide-induced cirrhotic rat model. BMC Gastroenterol. 2014;14:198.
Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. 2015;63:1038–9.
Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65.
Ramachandran P, Pellicoro A, Vernon MA, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:E3186–95.
Bird TG, Lu WY, Boulter L, et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. Proc Natl Acad Sci U S A. 2013;110:6542–7.
Boulter L, Govaere O, Bird TG, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–9.
Boulter L, Lu WY, Forbes SJ. Differentiation of progenitors in the liver: a matter of local choice. J Clin Invest. 2013;123:1867–73.
Moore JK, Mackinnon AC, Wojtacha D, et al. Phenotypic and functional characterization of macrophages with therapeutic potential generated from human cirrhotic monocytes in a cohort study. Cytotherapy. 2015;17:1604–16.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (B) (26293175) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the Highway Program for Realization of Regenerative Medicine from the Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Shuji Terai received a research grant from Rohto Pharmaceutical, Interstem, and Nippon Shinyaku Co.
Rights and permissions
About this article

Cite this article
Terai, S., Tsuchiya, A. Status of and candidates for cell therapy in liver cirrhosis: overcoming the “point of no return” in advanced liver cirrhosis. J Gastroenterol 52, 129–140 (2017). https://doi.org/10.1007/s00535-016-1258-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-016-1258-1