
Abstract
Transforming growth factor-β (TGF-β) is a central regulator in chronic liver disease contributing to all stages of disease progression from initial liver injury through inflammation and fibrosis to cirrhosis and hepatocellular carcinoma. Liver-damage-induced levels of active TGF-β enhance hepatocyte destruction and mediate hepatic stellate cell and fibroblast activation resulting in a wound-healing response, including myofibroblast generation and extracellular matrix deposition. Being recognised as a major profibrogenic cytokine, the targeting of the TGF-β signalling pathway has been explored with respect to the inhibition of liver disease progression. Whereas interference with TGF-β signalling in various short-term animal models has provided promising results, liver disease progression in humans is a process of decades with different phases in which TGF-β or its targeting might have both beneficial and adverse outcomes. Based on recent literature, we summarise the cell-type-directed double-edged role of TGF-β in various liver disease stages. We emphasise that, in order to achieve therapeutic effects, we need to target TGF-β signalling in the right cell type at the right time.
Similar content being viewed by others


Introduction
Chronic liver diseases (CLD) and its end-stages, cirrhosis and hepatocellular carcinoma (HCC), are leading causes of morbidity and mortality worldwide with enormous socio-economic costs. Patients with liver cirrhosis are at high risk of deadly hepatic failure and well over 80% of HCC develop on a cirrhotic background. HCC ranks as the fifth most common cancer and, with over 600,000 deaths per annum, it constitutes a major global health problem (Parkin et al. 2005; Venook et al. 2010). The main aetiologies of CLDs are chronic hepatitis C virus (HCV) and hepatitis B virus (HBV) infections, alcohol abuse and, as a result of metabolic syndrome reaching epidemic proportions, an increasing prevalence of non-alcoholic steatohepatitis (NASH).
Liver transplantation is currently the only available therapy for terminal liver failure. With donor organs being limited, preventive measures and the development of new therapies for CLDs are in high demand. Prevention aims at eliminating the source of damage. In alcoholic liver disease, this obviously means avoiding further alcohol consumption. The addictive behaviour of patients, however, puts a serious limitation to the efficiency of this prevention strategy. HBV and HCV infections, representing about one third of CLD aetiologies, are combatted with virostatic treatments, thus improving patient conditions to some extent. The infections can, however, not be fully abrogated, severely reducing the efficiency of such therapy for this subset of CLDs.
Viral activity, chemical toxicity and metabolic overload cause damage and death to hepatocytes. This liver injury triggers a cascade of molecular and cellular reactions geared towards damage limitation, removal or repair of damaged cells, defence against further infection, tissue repair and regeneration. Central to the natural response to injury is inflammation induced by a large battery of signalling molecules and executed by a variety of dedicated cells, repair by activated myofibroblasts, which produce the fibrous tissue and parenchymal cell proliferation, both of which are required for filling the holes caused by damage. Together, these cellular and molecular events release the enormous natural regenerative potential of the liver.
In CLD, hepatocyte damage, wound healing and tissue remodelling go awry, resulting in fibrosis and ultimately cirrhosis, the platform on which HCC and deadly hepatic failure develop. At the cellular and molecular level, the multistep process of progressive CLD is reflected in the complex modulation of intracellular signal transduction circuits, altered cell-cell communications and, more drastically, an altered differentiation state of most liver resident cell types. Evidently, the dissection of these pathways is critical for the development of drugs and therapies. As is well recognised, the multifunctional cytokine transforming growth factor (TGF)-β plays a pivotal role in the sequence of events leading to end-stage CLD, although the complexity of the underlying aberrant responses that, in the various liver cell types and at the organ level, lead to the drastic changes seen in CLD and HCC is not understood in detail (Dooley et al. 2009; Gianelli et al. 2011; Matsuzaki 2009). To set the stage, we will first discuss the current knowledge of cellular communication and molecular mechanisms associated with CLD initiation and progression and thereafter address the role of TGF-β therein.
Cellular mechanisms mediating CLD initiation and progression
The hepatic response to injury undergoes various phases and involves various cell types. The initial event is liver epithelial cell stress, resulting in necrotic and/or apoptotic death. Death-mediated signals induce the activation of Kupffer and stellate cells, which orchestrate an inflammatory and wound-healing response that might lead to tissue regeneration and repair in an acute setting or to fibrogenesis, cirrhosis and cancer when injury occurs chronically. Below, we discuss the diverse phases that trigger disease progression and the process of liver regeneration.
Cell damage and death
CLDs are characterised by persistent hepatocyte damage and death, induced by either chemical toxicity, metabolic overload resulting in high levels of reactive oxygen species (ROS) or viral/microbial activity causing metabolic deregulation (Rombouts and Marra 2010). In non-alcoholic and alcoholic fatty liver disease, the first signs of cell stress are hepatocyte ballooning and lipid droplet storage. This process is known as steatosis, which is (highly) reversible and does not necessarily lead to cell damage. In the case of cholestasis-mediated injury, bile duct epithelial cells (BDECs) rather than hepatocytes are the primary target of damage, bile reflux being the major inducer of this type of injury. Several modes of cell death have been classified in the damaged liver, including apoptosis and necrosis. Hepatocyte death triggers a cascade of reactions initiated actively by specific messengers and signalling molecules or simply by molecules released because of cell damage. The response is aimed towards damage limitation, removal or repair of damaged cells, wound closure, defence against further infection and tissue repair and regeneration. A highly interesting recent observation suggests that, upon liver damage, hedgehog ligand production is upregulated in dying liver epithelial cells to re-initiate a signalling pathway that controls progenitor cell fate and tissue construction during development, thus inducing progenitor cell expansion and liver regeneration (Omenetti et al. 2011).
Liver regeneration
The liver possesses an enormous regenerative capacity. In experimental partial hepatectomy (PHx) models, full liver mass is restored within 5–6 days after two-thirds PHx in rats or mice. This is achieved by one to two rounds of replication of any remaining hepatocytes. Key events and signalling pathways that control hepatocyte proliferation have been extensively investigated and have been summarised (Michalopoulos 2007; Bohm et al. 2010). However, in some settings, the potency of the toxic xenobiotic or the amount of damaged cells might cause the death of a large number of hepatocytes and cholangiocytes. Then, hepatic progenitor cells (HPCs) with bi-potential capacity, residing in the ductules and canals of Hering, differentiate into either hepatocytes or cholangiocytes, depending on the cell compartment that is damaged the most (Michalopoulos 2011). Rapid hepatic failure occurs if hepatic regeneration based on mature hepatocytes/cholangiocytes and HPC proliferation cannot replenish dead liver cells and liver architecture (Duncan et al. 2009).
Inflammation
Hepatitis is a central mechanism of disease progression and a marker of serious liver disease (Weber et al. 2011). It encompasses a complex inflammatory response induced by a battery of signalling molecules and is executed by a variety of cells. The process is initiated by mutual activation of stellate and Kupffer cells that together provide a cytokine milieu triggering massive infiltration of mononuclear cells, which include macrophages, lymphocytes, eosinophils and plasma cells. Lymphocytes are mobilised and stimulated by contact with antigen to produce lymphokines that further activate macrophages. Cytokines and chemokines from activated macrophages, in turn, stimulate lymphocytes, thus setting the stage for a persistent inflammatory response (Wynn 2004; Heymann et al. 2009).
Evidence is accumulating for the plasticity of liver macrophages, including Kupffer cells, depending on the surrounding cytokine milieu: NF-κB- and AP1-driven pro-inflammatory (M1) or Stat6 and peroxisome proliferation-activated receptor-directed anti-inflammatory (M2) phenotypes are generated and correspondingly impact on subsequent cellular processes (Vats et al. 2006).
Fibrosis
The activated (myo)fibroblast is the cell type responsible for wound closure and fibrosis (reflecting persistent wound-healing activity attributable to chronic damage) in any CLD. Several potential sources for this critical mediator exist, including bone-marrow-derived fibrocytes or circulating mesenchymal cells, which can migrate through the injured liver and become myofibroblasts (Friedman 2008; Bataller and Brenner 2005). Resident cells, e.g. tissue fibroblasts located in the portal tract of the liver or quiescent hepatic stellate cells (HSC) located in the Space of Disse, might also be activated to become myofibroblasts (Friedman 2000). Whether hepatocytes, cholangiocytes or even endothelial cells undergo a transition into activated fibroblasts under certain circumstances remains controversial (Popov and Schuppan 2010). The predominance of evidence still supports a central role for the quiescent HSC becoming activated by cytokine signalling, turning into myofibroblasts and then producing the fibrous scar that can be found in CLD.
Cirrhosis is not simply extensive fibrosis but is characterised by architectural disruption, aberrant hepatocyte regeneration, nodule formation and vascular changes. The chance of a reversal from a cirrhotic to normal liver architecture remains controversial and corresponding data are discussed, for example by Iredale (2007). On the other hand, no doubt exists that the resolution of liver fibrosis can occur, e.g. it is initiated by apoptosis and senescence as orchestrated biological processes to eliminate fibrogenic cells (Friedman 2010; 2008).
Based on a multitude of data from rat or mouse HSCs and animal models of liver damage, several conclusive statements about liver fibrosis can be drawn: (1) oxidative stress induces hepatocyte damage and HSC and Kupffer cell activation, resulting in liver fibrosis, (2) TGF-β is required for liver fibrosis and (3) the blunting of TGF-β signalling reduces fibrogenesis.
Carcinogenesis
In cirrhotic livers, macroregenerative nodules that display foci of hepatocyte dysplasia are considered to be pre-neoplastic lesions of HCC. Histologically, these dysplastic lesions are classified as small cell or large cell lesions or as foci of adenomatous hyperplasia, whereby small cell dysplasia and adenomatous hyperplasia are the predominant preneoplastic lesions (Roskams and Kojiro 2010). Our present lack of a unified comprehensive understanding of liver carcinogenesis is partially attributable to HCC being initiated in multiple genetic and environmental contexts and almost certainly emerging as a consequence of multiple pathways (Whittaker et al. 2010). This limitation regarding the pathogenesis of HCC has also prevented the development of effective, targeted, preventive or therapeutic interventions.
Recent analyses by using genome-wide approaches and improved animal models have initiated new and promising attempts at subclassifying the apparently highly heterogeneous HCC into distinct molecular and prognostic subtypes (Lee and Thorgeirsson 2006). The generally accepted paradigm of hepatocarcinogenesis is that malignant transformation occurs through the stepwise accumulation of genetic and epigenetic alterations that lead to the progressive acquisition of cancer phenotypes. On the other hand, more recent studies have led to the concept that a minimum number of molecular alterations leads to the acquisition of the key cancer phenotype, namely unconstrained cell proliferation (Hoshida et al. 2010; Dooley et al. 2009). This “cancer platform” concept proposes that, in a physiological context, growth-promoting pathways are coupled with the activation of control mechanisms such as cellular senescence or apoptosis that limit their growth effects, producing a natural homeostasis of tissue mass. Inactivation of the latter during oncogenic events then results in unconstrained proliferation. Additionally, the existence of cancer stem cells (CSC) is now being intensely investigated in liver and HCC, since proliferation of stem cells is a frequent and permanent process in this organ and since the accumulation of adverse molecular events, e.g. mutations, bears a high risk of CSC generation (Mishra et al. 2009). Such cancer stem cells, depending on their fate at the time point of carcinogenic conversion, could account for the development of HCC or cholangiocarcinoma (CCC).
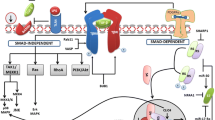
Thus, in CLD upon initial chronic liver damage, various progression phases can be distinguished in which a complexity of cellular crosstalk occurs. TGF-β has a pivotal role in orchestrating and regulating the corresponding phenotypes of CLD (Fig. 1). Upon liver damage, TGF-β production is initiated in non-parenchymal liver cells, e.g. granulocytes, macrophages (especially Kupffer cells) and HSC (Nakatsukasa et al. 1990), whereas fully differentiated epithelial cells do not express TGF-β. Interestingly, hepatocytes appear to absorb and store significant amounts of latent TGF-β in the cytoplasm and this might be activated and available upon damage, thus contributing to the initiation of HSC activation and the wound-healing process (Roth et al. 1998). Of note, hepatocytes under culture conditions lose polarity and activate survival signalling pathways, thus gaining a fibroblastoid phenotype and the facility of autonomous TGF-β production (Dooley et al. 2008).

Pros and cons of transforming growth factor-β (TGF-β) signalling during the progression of chronic liver diseases. Upon liver damage caused by many different aetiologies, active TGF-β ligands show up in the liver and induce downstream signalling in all cell types investigated. TGF-β is recognised as a major profibrogenic cytokine and, thus, TGF-β-directed therapies are being investigated for their capacity to interfere with fibrogenesis and combat disease progression. Although many of these approaches have shown promising results in animal disease models for more than a decade, there is currently still no effective treatment for human disease on the market. This scheme attempts to explain the difficulties one faces when dealing with chronic liver diseases in human patients. In animal models, severe damage from fibrosis and inflammation can be achieved within weeks, whereas the establishment of end-stage liver disease in humans usually takes several decades to establish. During that life span, the liver undergoes many different phases, as shown along the central time line. Strongly depending on the disease stage, TGF-β and thus its targeting might have a good (+) or bad (−) outcome in the organ. TGF-β enhances damage to epithelial cells by inducing apoptosis and oxidative stress, triggers myofibroblast (MFB) activation and a wound-healing response, controls or inhibits liver regeneration by hepatocyte apoptosis and inhibition of proliferation, activates regulatory T cells (T Reg) and Th17 differentiation to calm down inflammatory responses, causes fibrogenesis and liver scarring in chronic disease, inhibits the proliferation of premalignant cells, activates stroma fibroblasts in the neighbourhood of tumour cells, inhibits tumour-directed inflammatory responses, facilitates tumour angiogenesis and induces epithelial-mesenchymal transition (EMT) of tumour cells. This multiplicity of outcomes in one organ during the different stages of one disease clearly reveals the difficulties that we have to face while directing therapeutic approaches towards TGF-β. One must select the accurate therapeutic window, target the right cell type and interfere with the adverse downstream branches of the signalling pathway. To achieve this, a great deal of basic research is still required
TGF-β signal transduction in liver cells
Hepatic stellate cells
HSCs are a primary target for active TGF-β in CLD. Results of experiments in which TGF-β is misexpressed in liver by using adenovirus or transgenic TGF-β mice have revealed an important contribution of TGF-β to HSC activation and fibrogenesis (Kanzler et al. 1999; Hellerbrand et al. 1999; Ueberham et al. 2003). Upon its activation and phenotypic transdifferentiation, the TGF-β response in this cell type changes. Whereas platelet-derived growth-factor-induced proliferation of quiescent HSC is antagonised by TGF-β, transdifferentiated myofibroblasts demonstrate a growth stimulatory effect in response to TGF-β (Dooley et al. 2000). α1 (Nieto et al. 2001) and α2 (Inagaki et al. 1995) type (I) pro-collagen, tissue inhibitor of metalloproteinase-1 and −2 (Herbst et al. 1997) and plasminogen activator inhibitor (PAI)-1 (Knittel et al. 1996) have been identified as direct TGF-β target genes in this cell type, whereas the transcriptional activation of myofibroblast markers α-smooth muscle actin (SMA) and connective tissue factor (CTGF) are induced in a TGF-β-independent manner. Instead, TGF-β signalling is required for α-SMA organisation and stress-fibre formation (Dooley et al. 2003; Uemura et al. 2005).
Quiescent HSC respond to TGF-β treatment by Smad activation and display a functional negative feedback regulation via the induction of Smad7. In contrast, myofibroblasts are fully stimulated via autocrine TGF-β signalling and display a strong intrinsic R-Smad activation and, importantly, lack Smad7 upregulation (Stopa et al. 2000b; 2000a; Dooley et al. 2000; 2001a, 2001b; Liu et al. 2003). Tahashi and co-workers (2002) have confirmed this finding with myofibroblasts isolated from chronically injured rat livers; they conclude that the lack of Smad7 induction as observed in myofibroblasts in CLD could be one reason for excessive TGF-β effects during the progression of liver fibrosis.
Several studies have identified Smad3 as being the main mediator of the fibrogenic response of HSC, especially with respect to the induction of collagen expression (Schnabl et al. 2001; Furukawa et al. 2003; Inagaki et al. 2001a; 2001b; Seyhan et al. 2006). p38/Jun amino-terminal kinase (JNK)/MAP kinase (MAPK)-mediated Smad3 linker phosphorylation has been reported to be associated with HSC migration and disease progression (Yoshida et al. 2005; Furukawa et al. 2003; Matsuzaki et al. 2007). In addition to affecting the activin receptor-like kinase-5 (ALK5)-Smad3 pathway, TGF-β has been found to mediate its profibrogenic action via the activation of an alternative type I receptor pathway in HSCs, i.e. the ALK-1-induced Smad1 pathway mediating Id1 expression. Ectopic overexpression of Id1 enhances HSC activation, whereas its depletion blunts this TGF-β-induced response (Wiercinska et al. 2006). Furthermore, non-Smad TGF-β signalling via Ras, Raf-1, mitogen-activated protein kinase kinase and MAPK p42 and p44 signalling has been reported, although the outcome of these pathways on HSC activation and fibrogenesis has not been extensively studied (Reimann et al. 1997).
Hepatocytes
The first investigations of TGF-β effects on hepatocytes were performed in cultured cells and indicated that TGF-β counteracted the stimulatory action on DNA synthesis by various growth factors such as hepatocyte growth factor (HGF), epidermal growth factor (EGF) or insulin (Hayashi and Carr 1985; Nakamura et al. 1985). This TGF-β-induced growth arrest in hepatocytes was at least in part mediated via the interaction of Smad proteins with Sp1 transcription factors, which induce the expression of the cyclin-dependent kinase (CDK) inhibitor p21 (Moustakas and Kardassis 1998). Consistent with a cytostatic role for TGF-β in the liver, Fausto et al. (1986) showed that TGF-β mRNA increases in a late stage of the regenerating liver after PHx. They further reported that, in the regenerating as in the normal liver, TGF-β mRNA is present in non-parenchymal cells but not in hepatocytes and the authors therefore suggested that TGF-β was a component of a paracrine regulatory loop controlling hepatocyte replication (Fausto et al. 1986). Subsequently, this cytostatic effect could also be shown in cultured hepatocytes from hepatectomised rat livers (Strain et al. 1987).
In addition to being anti-mitogenic, TGF-β (and activin) has been found to induce hepatocyte apoptosis (Schwall et al. 1993; Yasuda et al. 1993; Oberhammer et al. 1991). In one study, the adaptor protein Daxx, which is associated with the Fas receptor that mediates the activation of JNK and programmed cell death induced by Fas, was suggested to provide the connection between TGF-β receptors and the apoptotic machinery (Perlman et al. 2001). In another study, TGF-β-induced apoptosis was shown to be mediated via Smad-mediated induction of death-associated protein (DAP) kinase (Jang et al. 2002). Furthermore, TGF-β has been shown to induce hepatocyte apoptosis via enhanced expression of the pro-apoptotic protein BIM (Ramesh et al. 2008).
TGF-β has been found, as for many other epithelial cell types, to have a biphasic role in HCC. Initially, in the primary tumour, TGF-β has tumour-suppressive effects. In agreement with this notion, the reduced availability of TGF-β type II receptor (TβRII) by the ectopic expression of soluble TβRII in hepatocytes (Kanzler et al. 2001) or by using heterozygous mice with reduced TβRII expression (Im et al. 2001) results in enhanced susceptibility to HCC, confirming the tumour-suppressor function of the TGF-β signalling pathway. Ectopic expression of mutated Ha-Ras but not c-myc, has been found to abrogate the cytostatic effect of TGF-β on hepatocytes (Houck et al. 1989). HCC cells might become selectively resistant to the cytostatic effects of TGF-β (Braun et al. 1990) and not lose all responses to TGF-β, such as epithelial-to-mesenchymal transition (EMT). The latter process in hepatocarcinogenesis was first described by Grotzmann et al. (2002) in a study of immortalised murine hepatocytes. These hepatocytes display a high degree of differentiation and undergo cell cycle arrest in the G1 phase following exposure to TGF-β. They maintain epithelial polarisation, despite the expression of oncogenic Ha-Ras; however, upon TGF-β stimulation, they convert into a migratory cell type with fibroblastoid morphology and proliferation is no longer inhibited by TGF-β. Without oncogenic Ras, in a preneoplastic setting, TGF-β might activate the epidermal growth factor receptor (EGFR) and induce c-Src phosphorylation leading to Akt activation and cell survival. The blocking of EGFR signalling then amplifies the apoptotic response to TGF-β, whereas TGF-β-mediated EMT in hepatocytes is unaffected (Murillo et al. 2005). This indicates that the activation of EGFR is required for impairing cytostatic TGF-β activity but is dispensable for the EMT process.
Using monolayer and sandwich culture systems, we have shown that hepatocytes can exist in differentiated and dedifferentiated states that are reversible and can be switched by manipulating the responsible key factors of the signalling network. For example, focal adhesion kinase-mediated Akt and extracellular signal-regulated kinase 1/2 signalling interferes with the cytostatic effects of TGF-β, thus facilitating fibroblastoid transdifferentiation (EMT). Abrogating survival signalling resensitises hepatocytes to TGF-β-induced apoptosis (Godoy et al. 2009; Dooley et al. 2008; Weng et al. 2007). In addition, immortalised, highly differentiated hepatocytes, when treated with TGF-β, maintain their epithelial morphology and undergo dramatic alterations in adhesion, leading to detachment, re-adhesion and spreading. These alterations in adhesive behaviour are caused by sequential changes in the expression of α5β1 integrin and its ligand fibronectin (Biname et al. 2008). CTGF is potently induced by TGF-β and both factors are thought to play an important role and to cooperate in fibrogenesis and EMT (Wang et al. 2011; Gressner and Gressner 2008).
The results described in the preceding paragraph and from others (Kaimori et al. 2007; Zeisberg et al. 2007) show that TGF-β, e.g. via the induction of the transcriptional repressor Snail, induces adult mouse hepatocytes to undergo phenotypic and functional changes typical of EMT. Whether, however, hepatocyte EMT occurs in vivo during fibrogenesis is currently under discussion. Zeisberg et al. (2007) have performed lineage-tracing experiments with AlbCre-R26RstoplacZ double-transgenic mice and demonstrated that hepatocytes undergoing EMT substantially contribute to the population of fibroblast-specific protein-1 (FSP1)-positive fibroblasts in carbon tetrachloride (CCl4)-induced liver fibrosis. Moreover, Rowe et al. (2011) have demonstrated that hepatocytes up-regulate the expression of Snail in vivo during tissue remodelling. Hepatocyte-specific ablation of Snail demonstrates that this transcription factor plays a key role in liver fibrosis progression in vivo by triggering multiple aspects of fibrogenesis, including growth factor expression, extracellular matrix (ECM) synthesis and chronic inflammatory responses (Rowe et al. 2011). In contrast, however, elegant studies from Taura et al. (2010) have strongly challenged the concept that hepatocytes in vivo acquire a mesenchymal phenotype through EMT to produce ECM in liver fibrosis. When triple-transgenic mice expressing ROSA26 stop β-galactosidase, albumin Cre and collagen α1(I) green fluorescent protein (GFP) and in which hepatocyte-derived cells are permanently labelled by β-galactosidase and type I collagen-expressing cells are labelled by GFP are subjected to the induction of liver fibrosis by repeated CCl4 injections, no cells with double-positivity for GFP and β-galactosidase can be found, although all β-galactosidase-positive cells exhibit abundant cytoplasm and the typical morphology of hepatocytes and express none of the mesenchymal markers including α-SMA, FSP1, desmin and vimentin. The authors conclude that type I collagen-producing cells do not originate from hepatocytes and that hepatocytes in vivo neither acquire mesenchymal marker expression nor exhibit a morphological change that is distinguishable from normal hepatocytes (Taura et al. 2010). In another similar lineage tracing approach, Wells and coworkers present even more convincing results that, in Alfp-Cre 3 Rosa26-yellow fluorescent protein (YFP) mice, neither hepatocytes, nor cholangiocytes (or their bipotential progenitors) display evidence for in vivo colocalisation of YFP with mesenchymal markers S100A4, vimentin, α-SMA or procollagen 1 α2 in three animal models of chronic liver disease (bile duct ligation, CCl4 and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (Chu et al. 2011).
Reconciling these apparently contradictory findings will require further investigations. Nevertheless, if transgenic mice with upregulated Smad7 expression only in their hepatocytes are challenged with chronic exposure to CCl4, they demonstrate significantly diminished liver damage and fibrosis when compared with controls (Dooley et al. 2008), indicating that TGF-β signalling in hepatocytes in vivo is required for fibrogenesis progression, as it is in HSC.
Other cell types of the liver
Much less intensely investigated are the effects of TGF-β on other cell types of the liver, e.g. BDEC, immune cells and liver sinusoidal endothelial cells, although a major influence can be expected in liver physiology and during CLD. Similar to hepatocytes, TGF-β might provide cytostatic and tumorigenic effects towards BDEC, in particular during carcinogenic progression in cholangiocarcinoma. From many studies in other tissues, an inhibitory role of inflammation is expected, especially in its role as a differentiation factor for regulatory T cells (TRegs; Becker et al. 2006; Korn et al. 2009) and Th17 cells (Tallima et al. 2009). Finally, the impact of TGF-β as a pro-angiogenic factor has been well-described (ten Dijke and Arthur 2007) and will be relevant in the branched sinusoid network of the liver in the settings of regeneration, cirrhosis and carcinogenesis.
Targeting TGF-β signalling in animal models for CLD
Various strategies have been pursued to accomplish the inhibition of TGF-β signalling in fibrosis (Fig. 2), first in animal studies and then with the objective of being translated into humans (see next paragraph). These include (1) sequence-specific anti-sense oligonucleotides that inhibit TGF-β mRNA expression, (2) isoform-selective neutralising antibodies, soluble TβRII fragments or synthetic peptides that interfere with ligand binding to the endogenous receptor complex, (3) overexpression of the natural TGF-β signalling inhibitor Smad7 or cytokines such as interferon-γ (IFN-γ) that induce Smad7 expression, (4) neutralising antibodies to integrins that interfere with the activation of latent TGF-β and (5) low-molecular-weight inhibitors antagonising the intracellular kinase activity of TGF-β receptors (Iyer et al. 2005; Pennison and Pasche 2007; Yingling et al. 2004; Dooley et al. 2003; Petersen et al. 2008; Weng et al. 2007; Hawinkels and ten Dijke 2011). Such anti-TGF-β approaches have been established and successfully used for the treatment of experimental fibrogenesis. For example, a synthetic peptide that blocks the interaction of TGF-β with its receptor has been established to be effective in protection against CCl4-induced liver fibrosis (Ezquerro et al. 2003). Further, dominant-negative or soluble TβRIIs have been applied to suppress fibrosis in mice and rats upon dimethylnitrosamine-, CCl4- or bile duct ligation-mediated liver damage (Qi et al. 1999; Ueno et al. 2000; Nakamura et al. 2000; George et al. 1999; Yata et al. 2002). In part, the inhibitory effect of soluble TβRII is achieved by interference with oxidative stress, including the generation of ROS (Cui et al. 2003). Similarly, TGF-β-binding proteins, such as decorin and antagonistic cytokines, such as bone morphogenetic protein-7, hepatocyte growth factor, interleukin-10 or IFN-γ were as efficient as camostat mesilate, a protease inhibitor that possibly abrogates the proteolytic activation of TGF-β (Breitkopf et al. 2005). Moreover, adenovirus-mediated overexpression of Smad7 in the liver potently blunted bile duct ligation-induced liver fibrosis and achieved efficient inhibition of intracellular TGF-β signalling. Bile duct ligation induced profibrogenic effects in cultured HSCs and in vivo were inhibited (Dooley et al. 2003). A similar approach with adenovirus-mediated overexpression of Smad7 successfully interfered with bleomycin-induced lung fibrosis in mice (Nakao et al. 1999).

TGF-β signal transduction pathway and targets for therapeutic intervention. TGF-β signals via heteromeric transmembrane complexes of type I and type II receptors (TβR) that are endowed with intrinsic serine/threonine kinase activity (ALK activin receptor-like kinase). Upon type-II-mediated phosphorylation of the type I receptor, the activated type I receptor initiates intracellular signalling by phosphorylating receptor regulated (R)-Smad2 and Smad3. Activated R-Smads form heteromeric complexes with Smad4 and these complexes accumulate in the nucleus where they mediate transcriptional responses. Inhibitory Smad7 antagonises TGF-β/Smad signalling by competing with R-Smads for receptor interaction and by recruiting E3 ubiquitin ligases to the activated receptor complex and mediating its degradation. This pathway has been targeted by anti-sense molecules that inhibit TGF-β mRNA expression, by neutralising antibodies against TGF-β or TGF-β receptors that interfere with ligand-receptor interactions, by antibodies that interfere with the activation of latent TGF-β and by soluble extracellular domains of the type II receptor that sequester ligand binding to endogenous receptors and small ATP mimetics of TGF-β receptor kinases. Antagonising pathways, such as interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α) and epidermal growth factor (EGF), can inhibit TGF-β/Smad-induced responses by stimulating Smad7 expression
As mentioned above, experimental evidence exists showing that Smad3 is the predominant mediator of fibrogenic TGF-β downstream signalling. Therefore, strategies to interfere specifically with Smad3 are promising. In line with this assumption, the targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice (Latella et al. 2009) and against cutaneous injury induced by ionising radiation (Flanders et al. 2002). However, small molecule inhibitors announced as specific for Smad3 (Jinnin et al. 2006; Liu et al. 2006) have not yet yielded breakthrough results and seem to need further improvement.
With regard to interference with cancer, recent studies in G. Giannelli’s lab (Giannelli et al. 2011) have shown that the inhibition of TGF-β signalling results in multiple synergistic downstream effects that probably improve the clinical outcome in HCC. Further, the small molecule inhibitor LY2109761, which targets TβRI/ALK5 and TβRII induces a complete abrogation of Smad-dependent and -independent signalling in human colon carcinoma cells harbouring activated K-RAS, resulting in reduced tumour cell invasion and liver metastasis (Zhang et al. 2009).
Therapeutic targeting of TGF-β signalling in CLD patients
Contrary to the stimulating beneficial outcome of anti-TGF-β treatment in animal disease models (Breitkopf et al. 2006), only limited positive or even adverse results exist for human disease. For example, metelimumab, a monoclonal antibody against TGF-β1, has been used to treat systemic sclerosis. The study was stopped as four patients died (Denton and Abraham 2004). Other studies involving the use of lerdelimumab, a monoclonal antibody against TGF-β2, to treat eye scarring (Mead et al. 2003), or GC1008, a pan-antibody against TGF-β1-3 for lung fibrosis are still ongoing. Intriguingly, an antisense strategy against TGF-β2 has been successful in the treatment of glioma and is currently being tested in other malignant tumours such as pancreatic carcinoma, colon carcinoma and melanoma (Schlingensiepen et al. 2008).
A major reason that most of these promising results from animal disease models have not yet robustly been translated into clinical use is probably the multiplicity of the possible biological-context-dependent functions of TGF-β. In the time course of CLD progression, various phases occur, such as initiation, regeneration, perpetuation, fibrogenesis, tumorigenesis and metastasis. Depending on the specific disease stage, TGF-β may have adverse or beneficial outcomes as outlined briefly below. Initially, TGF-β enhances hepatocyte damage. On the other hand, it triggers the transdifferentiation of HSC to myofibroblasts and thus mediates a wound-healing response. During regeneration and hepatocyte proliferation, TGF-β has an important tissue-mass-limiting cytostatic effect and controls inflammation by generating TRegs. During perpetuation and fibrogenesis in chronic disease stages, the overwhelming scar-forming wound-healing reaction is adverse for the liver. In the pre-malignant stage, the cytostatic effect that controls epithelial cell proliferation may prevent carcinogenesis. In this context, the negative outcome of TGF-β on inflammation might inhibit the immune response against arising tumour cells. In carcinogenesis, when TGF-β-cytostatic effects are lost and the signalling branch is redirected to EMT, TGF-β may favour cancer progression and metastasis. Furthermore, the pro-angiogenic action of TGF-β towards endothelial cells may also be important for tumour progression (Meyer et al. 2010). Thus, an important consideration for TGF-β-directed treatment of fibroproliferative diseases, such as CLD, is to select the right time point and cell type for targeted intervention (Fig. 1). Moreover, an additional challenge is that the selective inhibition of some but not all TGF-β-induced cellular responses might be beneficial.
Abbreviations
- ALK:
-
Activin receptor-like kinase
- BDEC:
-
Bile duct epithelial cells
- CCl4:
-
Carbon tetrachloride
- CDK:
-
Cyclin-dependent kinase
- CLD:
-
Chronic liver disease
- CSC:
-
Cancer stem cells
- CTGF:
-
Connective tissue growth factor
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial to mesenchymal transition
- ECM:
-
Extracellular matrix
- FSP1:
-
Fibroblast-specific protein-1
- GFP:
-
Green fluorescent protein
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular carcinoma
- HCV:
-
Hepatitis C virus
- HPC:
-
Hepatic progenitor cells
- HSC:
-
Hepatic stellate cells
- IFN:
-
Interferon
- JNK:
-
c-Jun amino terminal kinase
- NASH:
-
Non-alcoholic steatohepatitis
- PHx:
-
Partial hepatectomy
- ROS:
-
Reactive oxygen species
- SMA:
-
Smooth muscle actin
- TGF-β:
-
Transforming growth factor-β
- TβR:
-
TGF-β receptor
- YFP:
-
Yellow fluorescent protein
References
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218
Becker C, Fantini MC, Neurath MF (2006) TGF-β as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev 17:97–106
Biname F, Lassus P, Hibner U (2008) Transforming growth factor β controls the directional migration of hepatocyte cohorts by modulating their adhesion to fibronectin. Mol Biol Cell 19:945–956
Bohm F, Kohler UA, Speicher T, Werner S (2010) Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med 2:294–305
Braun L, Gruppuso P, Mikumo R, Fausto N (1990) Transforming growth factor β1 in liver carcinogenesis: messenger RNA expression and growth effects. Cell Growth Differ 1:103–111
Breitkopf K, Haas S, Wiercinska E, Singer MV, Dooley S (2005) Anti-TGF-β strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res 29:121S–131S
Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S (2006) TGF-β/Smad signaling in the injured liver. Z Gastroenterol 44:57–66
Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, Alpini G, Stanger BZ, Wells RG (2011) Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 53:1685–1695
Cui X, Shimizu I, Lu G, Itonaga M, Inoue H, Shono M, Tamaki K, Fukuno H, Ueno H, Ito S (2003) Inhibitory effect of a soluble transforming growth factor β type II receptor on the activation of rat hepatic stellate cells in primary culture. J Hepatol 39:731–737
Denton CP, Abraham DJ (2004) Transgenic analysis of scleroderma: understanding key pathogenic events in vivo. Autoimmun Rev 3:285–293
Dijke P ten, Arthur HM (2007) Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol 8:857–869
Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM (2000) Modulation of transforming growth factor β response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 31:1094–1106
Dooley S, Delvoux B, Streckert M, Bonzel L, Stopa M, Ten DP, Gressner AM (2001a) Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFβ signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett 502:4–10
Dooley S, Streckert M, Delvoux B, Gressner AM (2001b) Expression of Smads during in vitro transdifferentiation of hepatic stellate cells to myofibroblasts. Biochem Biophys Res Commun 283:554–562
Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, Dijke ten P, Gressner AM (2003) Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 125:178–191
Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, Gebhardt R, Kanzler S, Geier A, Breitkopf K, Weng H, Mertens PR (2008) Hepatocyte-specific Smad7 expression attenuates TGF-β-mediated fibrogenesis and protects against liver damage. Gastroenterology 135:642–659
Dooley S, Weng H, Mertens PR (2009) Hypotheses on the role of transforming growth factor-β in the onset and progression of hepatocellular carcinoma. Dig Dis 27:93–101
Duncan AW, Dorrell C, Grompe M (2009) Stem cells and liver regeneration. Gastroenterology 137:466–481
Ezquerro IJ, Lasarte JJ, Dotor J, Castilla-Cortazar I, Bustos M, Penuelas I, Blanco G, Rodriguez C, Lechuga MC, Greenwel P, Rojkind M, Prieto J, Borras-Cuesta F (2003) A synthetic peptide from transforming growth factor β type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury. Cytokine 22:12–20
Fausto N, Mead JE, Braun L, Thompson NL, Panzica M, Goyette M, Bell GI, Shank PR (1986) Proto-oncogene expression and growth factors during liver regeneration. Symp Fundam Cancer Res 39:69–86
Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB, Roberts AB (2002) Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol 160:1057–1068
Friedman SL (2000) Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275:2247–2250
Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–1669
Friedman SL (2010) Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol 7:425–436
Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y, Nishizawa M, Fujisawa J, Inoue K (2003) p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 38:879–889
George J, Roulot D, Koteliansky VE, Bissell DM (1999) In vivo inhibition of rat stellate cell activation by soluble transforming growth factor β type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA 96:12719–12724
Giannelli G, Mazzocca A, Fransvea E, Lahn M, Antonaci S (2011) Inhibiting TGF-β signaling in hepatocellular carcinoma. Biochim Biophys Acta 1815:214–223
Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Muller A, Tuschl G, Mueller SO, Dooley S (2009) Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor β-induced apoptosis. Hepatology 49:2031–2043
Gotzmann J, Huber H, Thallinger C, Wolschek M, Jansen B, Schulte-Hermann R, Beug H, Mikulits W (2002) Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-β1 and Ha-Ras: steps towards invasiveness. J Cell Sci 115:1189–1202
Gressner OA, Gressner AM (2008) Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int 28:1065–1079
Hawinkels LJ, Dijke P ten (2011) Exploring anti TGF-β therapies in cancer and fibrosis. Growth Factors 29:140–152
Hayashi I, Carr BI (1985) DNA synthesis in rat hepatocytes: inhibition by a platelet factor and stimulation by an endogenous factor. J Cell Physiol 125:82–90
Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA (1999) The role of TGFβ1 in initiating hepatic stellate cell activation in vivo. J Hepatol 30:77–87
Herbst H, Wege T, Milani S, Pellegrini G, Orzechowski HD, Bechstein WO, Neuhaus P, Gressner AM, Schuppan D (1997) Tissue inhibitor of metalloproteinase-1 and −2 RNA expression in rat and human liver fibrosis. Am J Pathol 150:1647–1659
Heymann F, Trautwein C, Tacke F (2009) Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm Allergy Drug Targets 8:307–318
Hoshida Y, Toffanin S, Lachenmayer A, Villanueva A, Minguez B, Llovet JM (2010) Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin Liver Dis 30:35–51
Houck KA, Michalopoulos GK, Strom SC (1989) Introduction of a Ha-ras oncogene into rat liver epithelial cells and parenchymal hepatocytes confers resistance to the growth inhibitory effects of TGF-β. Oncogene 4:19–25
Im YH, Kim HT, Kim IY, Factor VM, Hahm KB, Anzano M, Jang JJ, Flanders K, Haines DC, Thorgeirsson SS, Sizeland A, Kim SJ (2001) Heterozygous mice for the transforming growth factor-β type II receptor gene have increased susceptibility to hepatocellular carcinogenesis. Cancer Res 61:6665–6668
Inagaki Y, Truter S, Greenwel P, Rojkind M, Unoura M, Kobayashi K, Ramirez F (1995) Regulation of the α2(I) collagen gene transcription in fat-storing cells derived from a cirrhotic liver. Hepatology 22:573–579
Inagaki Y, Mamura M, Kanamaru Y, Greenwel P, Nemoto T, Takehara K, ten Dijke P, Nakao A (2001a) Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol 187:117–123
Inagaki Y, Nemoto T, Nakao A, Dijke P ten, Kobayashi K, Takehara K, Greenwel P (2001b) Interaction between GC box binding factors and Smad proteins modulates cell lineage-specific α2(I) collagen gene transcription. J Biol Chem 276:16573–16579
Iredale JP (2007) Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Investig 117:539–548
Iyer S, Wang ZG, Akhtari M, Zhao W, Seth P (2005) Targeting TGFβ signaling for cancer therapy. Cancer Biol Ther 4:261–266
Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH (2002) TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol 4:51–58
Jinnin M, Ihn H, Tamaki K (2006) Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-β1-induced extracellular matrix expression. Mol Pharmacol 69:597–607
Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A (2007) Transforming growth factor-β1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem 282:22089–22101
Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, Rose-John S, Buschenfelde KH zum, Blessing M (1999) TGF-β1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol 276:G1059–G1068
Kanzler S, Meyer E, Lohse AW, Schirmacher P, Henninger J, Galle PR, Blessing M (2001) Hepatocellular expression of a dominant-negative mutant TGF-β type II receptor accelerates chemically induced hepatocarcinogenesis. Oncogene 20:5015–5024
Knittel T, Fellmer P, Ramadori G (1996) Gene expression and regulation of plasminogen activator inhibitor type I in hepatic stellate cells of rat liver. Gastroenterology 111:745–754
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27:485–517
Latella G, Vetuschi A, Sferra R, Catitti V, D'Angelo A, Zanninelli G, Flanders KC, Gaudio E (2009) Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int 29:997–1009
Lee JS, Thorgeirsson SS (2006) Comparative and integrative functional genomics of HCC. Oncogene 25:3801–3809
Liu C, Gaca MD, Swenson ES, Vellucci VF, Reiss M, Wells RG (2003) Smads 2 and 3 are differentially activated by transforming growth factor-β (TGF-β) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-beta-independent. J Biol Chem 278:11721–11728
Liu X, Wang W, Hu H, Tang N, Zhang C, Liang W, Wang M (2006) Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-β1 in cultured rat hepatic stellate cells. Pharm Res 23:82–89
Matsuzaki K (2009) Modulation of TGF-β signaling during progression of chronic liver diseases. Front Biosci 14:2923–2934
Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M, Fujisawa J, Okazaki K, Seki T (2007) Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 46:48–57
Mead AL, Wong TT, Cordeiro MF, Anderson IK, Khaw PT (2003) Evaluation of anti-TGF-β2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest Ophthalmol Vis Sci 44:3394–3401
Meyer C, Meindl-Beinker NM, Dooley S (2010) TGF-β signaling in alcohol induced hepatic injury. Front Biosci 15:740–749
Michalopoulos GK (2007) Liver regeneration. J Cell Physiol 213:286–300
Michalopoulos GK (2011) Liver regeneration: alternative epithelial pathways. Int J Biochem Cell Biol 43:173–179
Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR, Shetty K, Johnson L, Reddy EP (2009) Liver stem cells and hepatocellular carcinoma. Hepatology 49:318–329
Moustakas A, Kardassis D (1998) Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc Natl Acad Sci USA 95:6733–6738
Murillo MM, del Castillo G, Sanchez A, Fernandez M, Fabregat I (2005) Involvement of EGF receptor and c-Src in the survival signals induced by TGF-β1 in hepatocytes. Oncogene 24:4580–4587
Nakamura T, Tomita Y, Hirai R, Yamaoka K, Kaji K, Ichihara A (1985) Inhibitory effect of transforming growth factor-β on DNA synthesis of adult rat hepatocytes in primary culture. Biochem Biophys Res Commun 133:1042–1050
Nakamura T, Sakata R, Ueno T, Sata M, Ueno H (2000) Inhibition of transforming growth factor β prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 32:247–255
Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I (1999) Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest 104:5–11
Nakatsukasa H, Nagy P, Evarts RP, Hsia CC, Marsden E, Thorgeirsson SS (1990) Cellular distribution of transforming growth factor-β1 and procollagen types I, III, and IV transcripts in carbon tetrachloride-induced rat liver fibrosis. J Clin Invest 85:1833–1843
Nieto N, Dominguez-Rosales JA, Fontana L, Salazar A, Armendariz-Borunda J, Greenwel P, Rojkind M (2001) Rat hepatic stellate cells contribute to the acute-phase response with increased expression of α1(I) and α1(IV) collagens, tissue inhibitor of metalloproteinase-1, and matrix-metalloproteinase-2 messenger RNAs. Hepatology 33:597–607
Oberhammer F, Bursch W, Parzefall W, Breit P, Erber E, Stadler M, Schulte-Hermann R (1991) Effect of transforming growth factor β on cell death of cultured rat hepatocytes. Cancer Res 51:2478–2485
Omenetti A, Choi S, Michelotti G, Diehl AM (2011) Hedgehog signaling in the liver. J Hepatol 54:366–373
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108
Pennison M, Pasche B (2007) Targeting transforming growth factor-β signaling. Curr Opin Oncol 19:579–585
Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA (2001) TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol 3:708–714
Petersen M, Thorikay M, Deckers M, Dinther M van, Grygielko ET, Gellibert F, Gouville AC de, Huet S, Dijke P ten, Laping NJ (2008) Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int 73:705–715
Popov Y, Schuppan D (2010) Epithelial-to-mesenchymal transition in liver fibrosis: dead or alive? Gastroenterology 139:722–725
Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H (1999) Blockade of type β transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci USA 96:2345–2349
Ramesh S, Qi XJ, Wildey GM, Robinson J, Molkentin J, Letterio J, Howe PH (2008) TGFβ-mediated BIM expression and apoptosis are regulated through SMAD3-dependent expression of the MAPK phosphatase MKP2. EMBO Rep 9:990–997
Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW (1997) Transforming growth factor-β1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett 403:57–60
Rombouts K, Marra F (2010) Molecular mechanisms of hepatic fibrosis in non-alcoholic steatohepatitis. Dig Dis 28:229–235
Roskams T, Kojiro M (2010) Pathology of early hepatocellular carcinoma: conventional and molecular diagnosis. Semin Liver Dis 30:17–25
Roth S, Michel K, Gressner AM (1998) (Latent) transforming growth factor β in liver parenchymal cells, its injury-dependent release, and paracrine effects on rat hepatic stellate cells. Hepatology 27:1003–1012
Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, Weiss SJ (2011) Hepatocyte-derived snail1 propagates liver fibrosis progression. Mol Cell Biol 31:2392–2403
Schlingensiepen KH, Fischer-Blass B, Schmaus S, Ludwig S (2008) Antisense therapeutics for tumor treatment: the TGF-β2 inhibitor AP 12009 in clinical development against malignant tumors. Recent Results Cancer Res 177:137–150
Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA (2001) The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 34:89–100
Schwall RH, Robbins K, Jardieu P, Chang L, Lai C, Terrell TG (1993) Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology 18:347–356
Seyhan H, Hamzavi J, Wiercinska E, Gressner AM, Mertens PR, Kopp J, Horch RE, Breitkopf K, Dooley S (2006) Liver fibrogenesis due to cholestasis is associated with increased Smad7 expression and Smad3 signaling. J Cell Mol Med 10:922–932
Stopa M, Anhuf D, Terstegen L, Gatsios P, Gressner AM, Dooley S (2000a) Participation of Smad2, Smad3, and Smad4 in transforming growth factor β (TGF-β)-induced activation of Smad7. The TGF-β response element of the promoter requires functional Smad binding element and E-box sequences for transcriptional regulation. J Biol Chem 275:29308–29317
Stopa M, Benes V, Ansorge W, Gressner AM, Dooley S (2000b) Genomic locus and promoter region of rat Smad7, an important antagonist of TGFβ signaling. Mamm Genome 11:169–176
Strain AJ, Frazer A, Hill DJ, Milner RD (1987) Transforming growth factor β inhibits DNA synthesis in hepatocytes isolated from normal and regenerating rat liver. Biochem Biophys Res Commun 145:436–442
Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K (2002) Differential regulation of TGF-β signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 35:49–61
Tallima H, Salah M, Guirguis FR, El Ridi R (2009) Transforming growth factor-β and Th17 responses in resistance to primary murine schistosomiasis mansoni. Cytokine 48:239–245
Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, Brenner DA (2010) Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 51:1027–1036
Ueberham E, Low R, Ueberham U, Schonig K, Bujard H, Gebhardt R (2003) Conditional tetracycline-regulated expression of TGF-β1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology 37:1067–1078
Uemura M, Swenson ES, Gaca MD, Giordano FJ, Reiss M, Wells RG (2005) Smad2 and Smad3 play different roles in rat hepatic stellate cell function and α-smooth muscle actin organization. Mol Biol Cell 16:4214–4224
Ueno H, Sakamoto T, Nakamura T, Qi Z, Astuchi N, Takeshita A, Shimizu K, Ohashi H (2000) A soluble transforming growth factor β receptor expressed in muscle prevents liver fibrogenesis and dysfunction in rats. Hum Gene Ther 11:33–42
Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A (2006) Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab 4:13–24
Venook AP, Papandreou C, Furuse J, Guevara LL de (2010) The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist 15(Suppl 4):5–13
Wang Q, Usinger W, Nichols B, Gray J, Xu L, Seeley TW, Brenner M, Guo G, Zhang W, Oliver N, Lin A, Yeowell D (2011) Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair 4:4
Weber A, Boege Y, Reisinger F, Heikenwalder M (2011) Chronic liver inflammation and hepatocellular carcinoma: persistence matters. Swiss Med Wkly 141:w13197
Weng HL, Ciuclan L, Liu Y, Hamzavi J, Godoy P, Gaitantzi H, Kanzler S, Heuchel R, Ueberham U, Gebhardt R, Breitkopf K, Dooley S (2007) Profibrogenic transforming growth factor-β/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology 46:1257–1270
Whittaker S, Marais R, Zhu AX (2010) The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 29:4989–5005
Wiercinska E, Wickert L, Denecke B, Said HM, Hamzavi J, Gressner AM, Thorikay M, Dijke P ten, Mertens PR, Breitkopf K, Dooley S (2006) Id1 is a critical mediator in TGF-β-induced transdifferentiation of rat hepatic stellate cells. Hepatology 43:1032–1041
Wynn TA (2004) Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4:583–594
Yasuda H, Mine T, Shibata H, Eto Y, Hasegawa Y, Takeuchi T, Asano S, Kojima I (1993) Activin A: an autocrine inhibitor of initiation of DNA synthesis in rat hepatocytes. J Clin Invest 92:1491–1496
Yata Y, Gotwals P, Koteliansky V, Rockey DC (2002) Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-β soluble receptor: implications for antifibrotic therapy. Hepatology 35:1022–1030
Yingling JM, Blanchard KL, Sawyer JS (2004) Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov 3:1011–1022
Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K (2005) Transforming growth factor-β and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol 166:1029–1039
Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R (2007) Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282:23337–23347
Zhang B, Halder SK, Zhang S, Datta PK (2009) Targeting transforming growth factor-β signaling in liver metastasis of colon cancer. Cancer Lett 277:114–120
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Our studies of the role of TGF-β in fibrosis are supported by the Netherlands Institute for Regenerative Medicine (NIRM), the Netherlands Organisation for Scientific Research (NWO-MW), the Centre for Biomedical Genetics (PtD), the German Research Foundation with the programs SFB TRR77 Liver Cancer TP A6 and Do373/8-1 and BMBF grants (The Virtual Liver and Cell Therapy in Liver Regeneration).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Dooley, S., ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res 347, 245–256 (2012). https://doi.org/10.1007/s00441-011-1246-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1246-y