
Abstract.
Apical membrane antigen-1 is a candidate vaccine component for malaria. In the present study, we investigated the polymorphism of the Plasmodium vivax apical membrane antigen-1 gene (PvAMA-1) in 30 Korean isolates. The polymorphic region of the PvAMA-1 gene, corresponding to nucleotides 324–735 (aa 108–245), was amplified using polymerase chain reaction followed by cloning and sequencing. Two genotypes (SKA and SKG) were identified on the basis of amino acid substitution. These were identical to those of the Chinese isolates. The Korean isolates showed sequence variation at six positions on the basis of the sequence of the Sal1 strain. Of these, variation at position 189 (Glu/Lys) was found only in SKA. These two genotypes were related to the genotype of the circumsporozoite and Duffy binding protein of the Korean isolate. These findings suggest that two genotypes of P. vivax coexist in the endemic area and that the re-emerging parasite in Korea may be related to or have originated in East Asia.
Similar content being viewed by others
Introduction
Malaria is one of the most serious diseases affecting people in developing countries with tropical and subtropical climates. Although Plasmodium vivax is not frequently fatal to humans, it is an exhausting, debilitating disease that impairs the quality of life and economic productivity. The enormous toll of mortality caused by Plasmodium falciparum has tended to overshadow the public health importance of P. vivax. For this reason and on account of technical difficulties, relatively little investment has been made in attempting to develop a vaccine against P. vivax. (Snewin et al. 1991). One of the major problems in vaccine development is the antigenic diversity of the vaccine candidate proteins. The critical problem emerges from the fact that the host response to one allele is much less effective against parasites expressing different allelic forms (Crewther et al. 1996; Renia et al. 1997). The polymorphism of an immunodominant antigen, a potential malaria vaccine target, is rather greater for P. vivax than for P. falciparum (Arnot et al. 1990). Therefore, studying the polymorphism of a candidate vaccine protein in P. vivax rather than P. falciparum may be more important in the development of a malaria vaccine.
The study of polymorphism is not only important in establishing the antigenic repertoire of isolates from malaria endemic regions but also in elucidating the mechanisms by which antigenic diversity may be generated.
P. vivax malaria re-emerged in the Republic of Korea after being absent for more than 10 years (Kho et al. 1999a; Chai 1999). In a previous study, P. vivax Duffy binding protein (PvDBP) and circumsporozoite protein (PvCSP) showed genotypes with at least two new phenotypes among the Korean isolates (Kho et al. 1999b, 2001). However, an accurate measure of the extent of genetic diversity in the P. vivax Korean isolates is not available. This is due to the availability of only very few polymorphic markers for studying P. vivax. The P. vivax apical membrane antigen-I (PvAMA-1) is one of the proteins involved in the merozoite invasion of erythrocytes, and is currently considered an important vaccine candidate against the asexual blood-stages of this parasite (Cheng and Saul 1994). Among the different P. vivax isolates, the PvAMA-1 gene exhibits sequence diversity and is a useful marker for typing P. vivax populations. So far, PvAMA-1 is reported to have sequence variations in 22 nucleotide positions from ten different areas. Figtree et al. (2000) classified 69 haplotypes on the basis of amino acid substitution in 22 polymorphic sites.
This study was designed to investigate sequence variation in a polymorphic region of the PvAMA-1 gene of P. vivax isolates from Korea. This will add useful information on the genetic diversity of this species and help in the rational design of a vaccine. Furthermore, understanding the genetic variation of P. vivax Korean isolates will contribute valuable data to elucidating the origin of the current vivax malaria outbreaks in Korea.
Materials and methods
Isolation of parasite genomic DNA
Blood samples were obtained from 30 patients who contracted vivax malaria in Yonchon-gun, Kyonggi-do, in 1998. All patients were diagnosed by microscopic examination in the malaria clinic of Pusan Paik-hospital, Inje University. P. vivax DNA was extracted from 0.1 ml of EDTA-treated blood samples using QIAamp DNA blood kit (Qiagen, Valencia, Calif., USA). The isolated DNA was dissolved in 0.2 ml TE (10 mM Tris-HCl, pH 7.4 and 1 mM EDTA, pH 8.0) buffer and stored at –70°C until used.
Microscopic examination
Thin films were prepared for each patient and stained using the Diff-Quick stain kit (International Reagents, Japan). An expert microscopist examined the slides using an oil immersion lens at 1,000× magnification. Parasites in the smear were counted against 200 leukocytes (WBCs) (Craig and Sharp 1997). For the parasite density estimation, it was assumed that there were 8,000 WBCs in 1 µl of blood (Baker et al. 1992).
PCR conditions and cloning into T-vectors
To investigate sequence variation in a region from 324 bp to 735 bp of the PvAMA-1 gene, two oligonucleotide primers were synthesized (GenoTech, Taejeon, Korea). The primer sets were as follows: AMAF1, 5-AAAGGGGCCTACCGTTGAGA-3 (nucleotides 120–139, positioned after GenBank accession no. AF063138 of the Sal1 strain sequence for P. vivax); AMAR1, 5-CCGAACTTGGCGTTTCCTAA-3 (positions 773–754 of GenBank no. AF063138). The PCR was carried out on a Perkin-Elmer Gene Amp 9700 (Perkin-Elmer, Norwalk, Conn. USA) in a total volume of 20 µl consisting of a mixture of 2 µl of extracted DNA solution, 20 mM of each dNTP, 1 pmol of each primer, 10 mM Tris-HCl (pH 8.0), 50 mM KCl, 1.0 mM MgCl2, and 0.25 units of Taq polymerase (Takara Ex-Taq, Takara, Kyoto, Japan). The reaction involved 35 cycles of 45 s of denaturation at 94°C, 45 s of annealing at 57°C, 45 s of extension at 72°C, and 5 min final extension at 72°C . The amplification products were analyzed by electrophoresis on 1% agarose gel (Seakem LE agarose, FMC, Rockland, Me., USA) in 0.5× TAE running buffer (20 mM Tris-acetate, 0.5 mM EDTA) containing 0.05% ethidium bromide. A total of 4 µl of PCR products were loaded in the lanes of the gels which were then were run at 100 V for 30 min in a Mupid-21 electrophoresis apparatus (Mupid, Cosmobio, Tokyo, Japan).
Each amplified DNA fragment was purified using the QIAEX II gel extraction kit (Qiagen) from agarose gel, ligated into pGEM-T Easy Vector (Promega, Madison, Wis., USA) and transformed into Escherichia coli DH5α. Positive clones were selected using an ampicillin positive selection system and α-complementation of lacZ gene. The plasmid DNA was extracted by QIAprep Spin Miniprep kit (Qiagen), and used in DNA sequencing.
DNA sequencing and data analysis
The nucleotide sequence was determined by a dideoxynucleotide chain termination method using a sequenase kit (ABI PRISM Dye Terminator Cycle Sequencing Core Kit, Perkin Elmer) and an automated DNA sequencer (Applied Biosystems model 377, Perkin Elmer). T7 and SP6 primers were used in the pGEM-T-easy vector for DNA sequencing. For the verification of nucleotide substitutions, a pool of three independent clones was used in the sequence analysis. The DNA sequencing data were analyzed using DNASIS (Hitachi, version 2.5, Japan) and the BLAST program of the NCBI databases (Bethesda, Md., USA).
Results
PCR amplification of the PvAMA-1 gene from Korean isolates
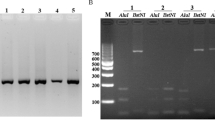
Microscopically, the parasitemias ranged between 0.004% and 0.315% with a mean value of 0.131%. Between 220 and 15,748 parasites were used in each PCR reaction (Table 1 ). Although varying in concentration, the PvAMA-1 gene was successfully amplified in all cases. A single band of about 654 bp was seen after amplification, corresponding to positions 120–773 in the Sal1 strain (GenBank no. AF063138). Neither size polymorphism nor multiple bands were detected in uninfected donor blood used as a negative control (Fig. 1).

PCR generated fragments from the PvAMA-1 gene migrated on 1% agarose gel and stained with ethidium bromide. Lane M lDNA/EcoRI+HindIII (MBI Fermentas, Amherst, N.Y., USA) was used as a DNA size marker. Lanes 1–4 show PCR products of PvAMA-1 for the Korean isolates and lane 5 is the negative control
Analysis of the PvAMA-1 polymorphic region in Korean isolates
Of each PCR product, a 412 bp region (excluding primer regions and adjacent bases which did not provide a uniformly reliable sequence) was analyzed for polymorphism. The DNA sequences of the 30 isolates were determined and deduced from the amino acid sequences (Fig. 2). Two genotypes, SKA (GenBank no. AF357212) and SKG (GenBank no. AF357213), were identified on the basis of grouping mutations in the nucleotides and their corresponding amino acids. Previously, unreported polymorphic nucleotides were not found in this study. Of the 30 isolates, 15 showed SKA and 15 SKG genotypes. These two genotypes were identical to those of Chinese isolates; namely, SKA and SKG were identical to AAP and AAN haplotypes of the Chinese isolates, respectively (Fig. 2) (Figtree et al. 2000). A difference between the two genotypes was detected in only one nucleotide sequence variation at 567 (guanine/adenine), resulting in either an E (Glu) or a K (Lys). We previously studied the PvCSP and PvDBP genotypes using isolated DNA. The two genotypes of PvAMA-1 were related to the genotypes of PvCSP and PvDBP of Korean isolate (Table 1 ).

Alignment of deduced amino acid sequences of the Korean isolates (Ko) (SKA and SKG) comparing the Sal1 strain with isolates from polymorphic region from Af (Africa), Ch (China), In (India), Ind (Indonesia), Ph (Philippines), Pn (Papua New Guinea), So (Solomon Islands) and Th (Thailand). Genotypes are marked according to the Figtree's classification (Figtree et al. 2000). The geographic origin of genotype is shown in the right panel. Variation between SKA and SKG genotypes is shown by an arrowhead
Discussion
The identification, characterization and quantification of Plasmodium spp. genetic polymorphisms is becoming increasingly important in vaccine development. In addition to vaccine development, the information on the genetic variation of an antigen can be utilized in various fields including genetics and molecular epidemiology. The genetic variation of some of the genes in Plasmodium spp. is geographically related. Therefore, the information on genetic variation may be used in tracking the origin of Plasmodium and studying its geophylogenetics. We previously reported on the SKA and SKB genotypes of PvCSP of the Korean isolates (Kho et al. 1999b). It was suggested that the re-emerging P. vivax in Korea may have originated from East Asia. In this study, we found that the genotypes of PvAMA-1 in the Korean isolates were identical to those of Chinese isolates. This finding supports the hypothesis that the present parasite in Korea may have originated from East Asia. However, the characteristic geographical linkage of PvAMA-1 is unknown because it is relatively poorly understood at the molecular level. To understand PvAMA-1 in relation to geographical distribution, further investigations of isolates from Southeast Asia, China, and neighboring countries are indispensable.
Attempts are currently being made to measure transmission intensity using genetic information; genetic variation in hyper-endemic areas has been found to be larger than in hypo-endemic areas (Babiker and Walliker 1997; Paul et al. 1998). Previous studies of the genetic variation within the polymorphic region of PvAMA-1 show several patterns among isolates. A large genetic variation in high transmission intensity areas was also observed (Figtree et al. 2000). The extent of the genetic polymorphism of PvAMA-1 varies according to the geographical region. Two genotypes were found in 30 patients in Korea, while 15 genotypes came from 23 patients in Papua New Guinea (PNG) and six genotypes came from six patients in Thailand (Figtree et al. 2000). The difference, both in the number of genotypes and the changed positions in amino acid sequences, between Korea and the other two countries may be due to the difference in the intensity of malaria transmission. It is believed that the genetic variation in Plasmodium spp. genes is frequent in areas of high transmission because it is thought to occur mainly due to a continuous immune pressure or selection. Both countries, PNG and Thailand, are malaria endemic areas and have shown high transmission intensity for a long time. Although the recent intensity of malaria transmission in Korea has not been scientifically measured, it is believed that the likelihood of malaria being transmitted by mosquitoes might be strictly limited since transmission occurs during the limited season from May to October, and the transmission ability of the vector mosquito may be extremely low (Ree 2000). Taking into account the epidemic characteristics of malaria, genetic changes may occur more slowly in Korea than in highly endemic areas. In the Republic of Korea, one case of indigenous vivax malaria, which had disappeared in the late 1970s, was reported in 1993 (Chai et al. 1994). Since then, the number of cases has increased exponentially year after year. Currently, about 3,000–4,000 cases are reported annually (Chai 1999; Kho et al. 1999a). The result of this study may also be utilized as baseline data for monitoring the genetic changes in the malaria endemic area in Korea.
Interestingly, the two genotypes of PvAMA-1 found in Korea were related to the two genotypes of PvCSP and PvDBP of the Korean isolates (Kho et al. 1999b, 2001). This finding strongly suggests that the two strains of P. vivax coexist in the endemic area of Korea.
These results suggest the Korean PvAMA-1 isolates have little polymorphism and that two genotypes of P. vivax coexist in the endemic areas of Korea. Also, the re-emerged Korean P. vivax isolates were most closely related to isolates from East Asia.
References
Arnot DE, Stewart MJ, Barnwell JW (1990) Antigenic diversity in Thai Plasmodium vivax circumsporozoite proteins. Mol Biochem Parasitol 43:147–149
Babiker HA, Walliker D (1997) Current views on the population structure of Plasmodium falciparum: implications for control. Parasitol Today 13:262–267
Baker BHJr, Branchongaskorn T, Courval JM, Suwonkerd W, Rimwungtragoon K, Wirth DF (1992) A simple method to detect Plasmodium falciparum directly from blood samples using the polymerase chain reaction. Am J Trop Med Hyg 46:416–426
Chai IH, Lim GI, Yoon SN, Oh WI, Kim SJ, Chai JY (1994) Occurrence of tertian malaria in a male patient who has never been abroad. Korean J Parasitol 32:195–200
Chai JY (1999) Re-emerging Plasmodium vivax malaria in the Republic of Korea. Korean J Parasitol 37:129–143
Cheng Q, Saul A (1994) Sequence analysis of the apical membrane antigen I (AMA-1) of Plasmodium vivax. Mol Biochem Parasitol 65:183–187
Craig MH, Sharp BL (1997) Comparative evaluation of four techniques for the diagnosis of Plasmodium falciparum infections. Trans R Soc Trop Med Hyg 91:279–282
Crewther PE, Matthew ML, Flegg RH, Anders RF (1996) Protective immune responses to apical membrane antigen 1 of Plasmodium chabaudi involve recognition of strain-specific epitopes. Infect Immun 64:3310–3307
Figtree M, Pasay CJ, Slade R, Cheng Q, Cloonan N, Walker J, Saul A (2000) Plasmodium vivax synonymous substitution frequencies, evolution and population structure deduced from diversity in AMA 1 and MSP 1 genes. Mol Biochem Parasitol 108:53–66
Kho WG, Jang JY, Hong ST, Lee HW, Lee WJ, Lee JS (1999a) Border malaria characters of reemerging vivax malaria in the Republic of Korea. Korean J Parasitol 37:71–76
Kho WG, Park YH, Chung JY, Kim JP, Hong ST, Lee WJ, Kim TS, Lee JS (1999b) Two new genotypes of Plasmodium vivax circumsporozoite protein found in the Republic of Korea. Korean J Parasitol 37:265–270
Kho WG, Chung JY, Sim EJ, Kim DW, Chung WC (2001) Analysis of polymorphic regions of Plasmodium vivax Duffy binding protein of Korean isolates. Korean J Parasitol 39:143–150
Paul REL, Hackford I, Brockman A, Muller-Graf C, Price R, Luxemburger C, White NJ, Nosten F, Day KP (1998) Transmission intensity and Plasmodium falciparum diversity on the northwestern border of Thailand. Am J Trop Med Hyg 58:195–203
Ree HI (2000) Unstable vivax malaria in Korea. Korean J Parasitol 38:119–138
Renia L, Ling IT, Marussig M, Miltgen F, Holder AA, Mazier D (1997) Immunization with a recombinant C-terminal fragment of Plasmodium yoelii merozoite surface protein 1 protects mice against homologous but not heterologous P. yoelii sporozoite challenge. Infect Immun 65:4419–4423
Snewin VA, Longacre S, David PH (1991) Plasmodium vivax: older and wiser? Res Immunol 142:631–636
Acknowledgements.
This work was supported by the 2000 Inje University research grant. We acknowledge the technical expertise provided by Eun-Jeong Sim and Seung-Young Hwang. The authors would like to thank Hyewon Lee for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chung, JY., Chun, EH., Chun, JH. et al. Analysis of the Plasmodium vivax apical membrane antigen-1 gene from re-emerging Korean isolates. Parasitol Res 90, 325–329 (2003). https://doi.org/10.1007/s00436-002-0777-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-002-0777-2