
Abstract
ABCG5 and ABCG8 are two ATP-binding cassette half-transporters that belong to the G family members. They were identified as proteins that are mutated in a rare human disorder, sitosterolemia, and their identification led to the completion of the physiological pathways by which dietary cholesterol, as well as noncholesterol sterols, traffics in the mammalian body. These proteins are likely to function as heterodimers, and current evidence suggests that these proteins are responsible for the majority of sterol secretions into bile, thus may define the long sought-after biliary sterol transporters. This review will focus on some of the backgrounds of this physiology, the genetics and regulation of these genes, as well as our current understanding of their functions. This review will also highlight the current limitations in our knowledge gap.
Similar content being viewed by others

Historical
The history of how mammals can distinguish between dietary noncholesterol sterols and cholesterol is intertwined with the history of cholesterol itself; whether cholesterol could be synthesized by the body or was wholly absorbed from the diet, whether the body degraded cholesterol, what determines its absorption and biliary secretion, and whether cholesterol was involved in the process of atherosclerosis are all questions that have involved or continue to involve noncholesterol sterols[53, 54]. Cows, for example, eat only foods that contain plant sterols, and yet their bodies contain cholesterol but not plant sterols. Investigations of such observations led to the discovery that plant sterols were excluded by the body, but could compete with bulk cholesterol for entry into the micelles formed during digestion, thus preventing dietary absorption of cholesterol [6, 14, 21, 34, 50, 55, 59]. Into this milieu of understanding, two key landmark observations led to a revolution in our current knowledge of how whole-body sterol balance may be achieved. The context of the first landmark event was a knowledge that plant sterols were poorly absorbed relative to cholesterol [14], and that very high plasma cholesterol levels in humans were caused by a dominant genetic defect in the low-density lipoprotein receptor (and was associated with patients who developed accumulations of sterols in their tendons, called xanthomas). In a classic paper that should epitomize clinical investigation, Bhattacharyya and Connor described a new disease, named β-sitosterolemia, after identification of two sisters who had tendon xanthomas but did not have elevated plasma cholesterol, and who had very high amounts of plasma plant sterols, the major species of which was sitosterol, hence the name [3]. The ∝ conformer of sitosterol is not normally present in nature, and thus it is probably not necessary to use the term ‘β’ preceding sitosterol. In one single publication, these authors showed that a likely single gene defect led to the disruption of the intestinal processes that keeps noncholesterol sterols out, led to tendon xanthomas, an ominous sign of systemic atherosclerosis, and was key to understanding how dietary noncholesterol sterols were (not) absorbed. The second key observation was the identification of Niemann-Pick C1 Like 1 (NPC1L1), the ‘cholesterol receptor’, as the key molecule in determining entry of sterols into enterocytes [1, 31]. The latter will not be discussed in this review, although an overview is provided for better physiological context.
Once the clinical description of sitosterolemia had been reported (clinical and physiological features reviewed in [4]), the next breakthrough was the localization of the sitosterolemia locus, STSL, to chromosome 2p21 to a very narrow region which led to the identification of the genetic defect in sitosterolemia [43]. The surprise was that the STSL locus comprised of not one but two highly homologous genes; complete mutations in either of the two genes are necessary to cause sitosterolemia [2, 37, 39]. These genes encode for the two ABC ‘G’ family half-transporters, ABCG5 and ABCG8, also known as sterolin-1 and sterolin-2, respectively.
Genetics of ABCG5 and ABCG8
ABCG5 and ABCG8 contain 13 exons and are organized in a head-to-head organization spanning ∼80 kb of the locus [39]. These genes are likely to have evolved from a common ancestral gene, with a tandem duplication and inversion event. The intergenic region between ABCG5 and ABCG8 is very small (<160 bases) and does not contain a conserved TATA motif or motifs that would indicate which transcriptional factors may be involved in regulating this locus (see below). The strongest evidence that ABCG5 and ABCG8 may act as obligate heterodimers comes from the genetic evidence that patients with sitosterolemia are completely mutated for ABCG5 or ABCG8, and mutations in either of these seem to result in an identical phenotype [2, 37, 39]. In addition, in vitro evidence supports their heterodimerization with each other but not other ABC G family members [15, 16]. A compilation of mutations and identified polymorphisms is shown in Fig. 1. There are some notable features. Firstly, founder effects for mutations underlie many of the cases, suggesting that this disease has been present for many generations, perhaps more than 4,000 years [36, 39]. Secondly, mutations in ABCG5 seem to be the cause of sitosterolemia in peoples who have oriental lineage (Chinese, Japanese and Indian) [39]. Currently, this group also represents ∼20% of the cases, though this may be due to under-diagnoses. Thirdly, despite the close proximity and homology between ABCG5 and ABCG8, considerably more polymorphisms are present in ABCG8 compared to ABCG5. This seems to be a phenomenon confined to man, as analyses of the rodent STSL loci indicate an almost equal level of variability [40, 63]. It is not clear if this is an indication that ABCG5 and ABCG8 may also have a function, independent of their role as heterodimers. Other members of the ABCG family are known to function as homodimers (see this issue). Alternatively, it may be that there is a selection for variations at the STSL locus. A number of studies have implicated this locus in disease (or physiological) processes ranging from lipoprotein kinetics, cholesterol absorption, obesity to response to drug therapy [8, 20, 23–25, 42, 44]. At present, it is difficult to envisage a selective advantage for any of these traits. Indeed, the remarkable conservation of the STSL locus between species as diverse as fish, amphibians, rodents, and humans seems to indicate that the polymorphic changes may have a more dramatic effect on function (Fig. 2). Note that almost all of the cSNPs that are nonsynonymous seem to affect amino acid residues that are highly conserved (Fig. 2a,b). One of these is part of the consensus Walker B motif (E238L in ABCG8, Fig. 2b) and thus would be expected to have a dramatic effect on function, though none has been reported thus far. This raises the possibility that each of these cSNPs should alter function in some manner. The key to what these subtle alterations may be may come from the study of individuals who are homozygous for these cSNPs. At present, we do not understand how ABCG5/ABCG8 alter function other than play a role in keeping noncholesterol sterols out of our bodies. These polymorphisms may remain mysterious in the manner they affect physiology for the foreseeable future.

A compilation of reported mutations and polymorphisms affecting the STSL locus. A cartoon representation of ABCG5 (top A) and ABCG8 (bottom B) is shown, each comprising of 13 exons. The sizes of exons and introns are not to scale. Note that the STSL locus comprises of both genes arranged in a head-to-head orientation, with less than 160 bases separating their start transcription sites. Above each gene are shown in filled blue circles all known mutations that cause sitosterolemia. Below each gene are shown polymorphisms. The filled yellow circles depict nonsynonymous changes, whereas the open circles depict synonymous changes. Note that many more polymorphisms have been reported for ABCG8 than for ABCG5, despite extensive resequencing of this locus but a number of different groups

Conservation of ABCG5 and ABCG8 polypeptide sequences in the animal kingdom. Polypeptide sequences for man, chimpanzee, dog, cow, rat, mouse, chicken, toad, and two fishes, Zebra fish and Fugu, were compiled from the publicly available databases. Panel a shows the homology for ABCG5 and panel b that for ABCG8. Homologies around the nonsynonymous changes in humans are shown. It should be noted that the sequences for all but man, mouse, and rat are considered preliminary and need to be parsed for accuracy. For example, the exact start translation sites for some of these have not been established and in some cases, the protein translation is based upon electronically predicted exons, which seem to be incorrect. Despite these reservations, mapping the nonsynonymous changes seen at the STSL locus in humans onto these homology comparisons shows that many of these polymorphisms affect highly conserved residues. It is therefore remarkable that these ‘polymorphisms’ are not ‘disease’-causing (see text for discussion). Note that the variable amino acids not conserved in fish and chicken may turn out to be conserved once the genome sequences for these organisms have been ‘cleaned up’. A complete homology comparison for both genes is available on request from the authors
Regulation of ABCG5 and ABCG8
The major regulation of expression of sterolins appears to be at the transcriptional level, although there is a paucity of data on whether regulation at the post-transcriptional levels occurs. There are robust data to support the role of Lxr-mediated increased mRNA expression of Abcg5 and Abcg8, both in vivo (Table 1) as well as in vitro [2, 11, 47, 48, 65, 68]. LXR ligands have been shown to increase expression of the STSL locus in mouse liver and intestine and in rat hepatoma cells; cholesterol feeding upregulates this locus in part by increased activation of the Lxr pathway [48]. Unfortunately, it is still not clear if this is a direct effect or an indirect effect. To date, scanning for classical cis-acting DNA motifs for LXR recognition in the STSL locus have been negative. The strongest evidence that this is a direct effect is that the changes in RNA expression can be seen with as little as 12 h exposure to the ligands, both in vivo and in vitro [48]. The only direct evidence for both binding sites and an action on transcription has come from the investigation of the role of liver receptor homolog-1 (LRH-1), an orphan receptor, in the pathway that classically involves the FXR-SHP-LRH-1 [12]. The human intergenic sequence contains a LRH-1 binding site, this sequence has been shown to bind directly LRH-1 and activate expression of ABCG5 and ABCG8 in vitro [12]. It is interesting to note that this motif is not conserved in the rodent intergenic region. This is one of the differences between murine and human ‘promoter’ sequences in that the mouse not only has an identifiable TATA box, it also contains sequences that may bind HNF4 (both of which are absent from the human sequence) [12, 40]. This may suggest that the human STSL locus may be fundamentally differently regulated relative to the murine locus, although most of the evidences are based upon the experimental data derived from murine models. Table 1 summarizes data gleaned from the literature, both directly and indirectly summarizing the involvement (or not) of some of the transcriptional factors. Of note is that not only are there species differences that are evident but also the differential effects between liver and intestine for the same transcriptional pathway which deserve some attention. Furthermore, there are some evidences for a post-translational level of regulation. In canine gall bladder epithelial cells, Lxr activation, or exposure to model bile, led to a translocation of Abcg5 and Abcg8 from an intracellular compartment to the apical surface, suggesting a post-translational level of regulation [58]. No changes in RNA expression levels were seen.
Function of ABCG5 and ABCG8
Sterolins are critically involved in regulating the whole-body retention of noncholesterol plant sterols, and that these proteins are also key to secreting cholesterol into the biliary lumen (and likely the intestinal lumen) [30, 32, 35, 46, 64–66]. The genetic disease of sitosterolemia attests to their role in keeping noncholesterol sterols (plant sterols as well as shellfish sterols, etc.) out of the human body [17].
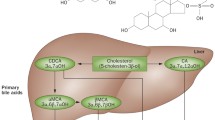
In the intestine, it is clear that the entry of all sterols, but particularly cholesterol, is primarily regulated by a step involving NPC1L1 (see Fig. 3). Based upon the response of patients with sitosterolemia to the drug ezetimibe, blocking NPC1L1 seems to lower plasma plant sterol levels and seems to improve symptoms and signs [52]. From studies involving mice as well as humans, the intestine can secrete cholesterol back into the lumen of the intestine [33, 55]. It is possible that it may also be able to secrete noncholesterol sterols. At present, this has not been tested in animal models, though evidence for this has recently been presented in a mouse model. In humans, it would appear that the liver may be a more important organ for keeping the levels of plant sterols low; a patient with sitosterolemia underwent a liver transplantation, and following this, his plant sterol levels almost normalized despite his genetic defect in the intestine [41]. Thus, ABCG5 and ABCG8 may selectively ‘pump’ sterols out of the intestinal cells as a first-line defense to dietary input, but in the fasting state, the liver can continually pump sterols (cholesterol and noncholesterol sterols) into bile and thus maintain a low noncholesterol sterol level in the body. In the fasting state, the intestine may also be able to reduce whole body sterol pools by continuing to pump sterols (presumably delivered to the intestinal enterocytes via the high-density lipoprotein pathway) [55]. It should be noted that a formal demonstration that these proteins carry out this function actively and utilize adenosine triphosphate (ATP) has not been reported. This is assumed as the ATP-binding domain sequences and both the highly conserved and invariant from the consensus ABC motifs.

Physiological pathways involved in dietary sterol trafficking. Upon ingestion, dietary sterols are unesterified, mixed with biliary sterols, phospholipids, and bile salts to form mixed micelles, a necessary step for absorption. These interact with the apical surface of the proximal small intestine enterocyte (process as yet undefined), whereby the sterols are allowed to enter the enterocyte. Although the exact molecular processes remain to be defined, it is clear that this process involves the multipass transmembrane protein NPC1L1; blocking this protein with the drug ezetimibe blocks entry of all sterols. In a process not understood, noncholesterol sterols seem to be excluded from progressing further and may be pumped back into the lumen by sterolins, ABCG5, and ABCG8. Cholesterol is allowed to progress to the ER where it is esterified, packaged with triglyceride and apolipoprotein B48 into chylomicrons to be secreted at the basolateral surface into the lymphatic channels for transport to the venous circulation. The enterocyte also synthesizes ApoA1 and may play a role in the maturation of HDL via ABCA1. HDL is a particle involved in the removal of cholesterol (and noncholesterol sterols) from the body. Thus, intestinal uptake of HDL may result in the net secretion of sterols. Although other ABC transporters are also expressed at the apical membrane, their role in cholesterol trafficking is not defined (indicated by the question mark). In addition, although bile acids are a necessary constituent of the micelles, these are not absorbed in the proximal jejunum but are transported to the terminal ileum where they are specifically taken up via interscapular brown adipose tissue. It is not clear if bile acids enter the jejunal enterocyte at all, or entered and are pumped back out by a specific transporter. At the hepatocyte level, entry of sterols adds to the pool of sterols that are rapidly channeled for secretion via ABC transporters ABCG5/ABCG8, with bile salt and phospholipid secretion mediated by bile salt export pump (BSEP) (ABCB11) and MDR2/3 (ABCB4). It has been demonstrated in humans that the liver preferentially excretes noncholesterol sterols and that HDL, via SR-B1-mediated sterol entry, is important in this channeling process. Note that cholesterol can be secreted directly into bile (via sterolins), as well as broken down to a molecule of bile acid and secreted into bile (via BSEP). Furthermore, although sterol secretion is greatly attenuated in sitosterolemia, as well as in knockout mice, some sterols are still secreted, suggesting that there are other pathways for sterol secretion. ACAT-2 acyl-CoA: cholesterol acyltransferase-2, Chol cholesterol, MTP microsomal triglyceride transfer protein, MRP2 multidrug resistance-associated protein 2 (ABCC2), NTCP sodium taurocholate cotransporting polypeptide, PL phospholipids, SR-B1 scavenger receptor B1. Adapted from Lu et al. [38], copyright 2001 with permission from Elsevier
From genetic manipulations in mice, it is clear that these proteins are key to secreting cholesterol into bile. The liver is the dominant organ for whole-body sterol balance and uses the biliary system for sterol loss, whether as direct sterol secretion or by breakdown of cholesterol into bile acids and secretion into bile. Over-expression of human ABCG5 and ABCG8 in mice led to a supersaturation of cholesterol in bile [66], whereas knockout of either Abcg5/Abcg8 or Abcg8 genes led to a failure to secrete sterols [30, 65]. However, knockout of Abcg5 led to a sterol-poor bile, but sterol secretion seemed to be restored upon Lxr activation [46]. Lxr activation in Abcg5/Abcg8 double knockout did not lead to a stimulation of biliary sterol secretion [65, 68]. There are no data reported for Abcg8 knockout in response to Lxr agonists. From both genetic data, as well as in vitro experimental data, ABCG5 and ABCG8 are likely to function as obligate heterodimers. It is also unlikely that they can heterodimerize with other ABCG family members. Thus, this observation, as well as the difference in the accumulations of polymorphisms in ABCG5 gene relative to ABCG8, is perplexing. Studies in humans have shown that there is preferential excretion of sitosterol compared to cholesterol when these are directly infused into their veins [51]. Studies in knockout mice confirm that this response, at least for biliary sterol secretion, is mediated by Abcg5/Abcg8 and is increased by Lxr activation [67]. While it seems that sterols in general seem to be substrates for ABCG5/ABCG8, limited studies indicate that oxysterols are not substrates [67]. The full spectrum of substrates for ABCG5/ABCG8 is not known. Finally, despite their role in secreting sterols into the lumen, the exact mechanism of action remains a matter of controversy. One hypothesis argues that ABCG5/ABCG8 may act as ‘extruders’, exposing sterols in the outer leaflet of the membrane for facilitated extraction into the lumen by sterol acceptors [56], such as bile acid:phospholipid complexes, whereas others have proposed that sterolins may act as ‘flippases’, akin to the flipping of phospholipids from the inner to the outer leaflet of the apical membranes [18]. In the absence of robust in vitro assays, it is not possible to discern if either of these models is valid. Finally, it should be noted that some sterol secretions continue, both in man, as well as mice deficient in sterolins, suggesting that there are other pathways whereby sterols can gain entry into the biliary tract, albeit at very low levels. Moreover, the ‘rate-limiting’ step for biliary sterol secretion has not been identified. ABCG5/ABCG8 may not be rate-limiting, because parents of sitosterolemic patients have no biochemical phenotype under physiological conditions, although they must have half-normal functioning sterolins. In this context, a recent study of ABCG5/ABCG8 expression in liver transplant patients found no correlation between mRNA expression of these transporters and biliary cholesterol secretion (sitosterol secretion was not reported) [13].
Conclusions
There are now robust data to support the contention that sterolins (ABCG5 and ABCG8) are the major sterol transporters active at the apical surface of hepatocytes and enterocytes. Evidence in support of this comes from the genetic basis of sitosterolemia, from the development of animal models that specifically manipulate their function, and from experiments that show to alter biliary sterol secretion. The STSL genetic locus is highly conserved from fish, toad, chicken, rodent, cow, dog, ape, and man, preserving not only the exon–intron organization but also shows a remarkably high degree of polypeptide conservation, raising the possibility that the true function of these proteins may still remain to be defined. A number of questions remain to be fully determined. One of these is defining fully the mode of transcriptional control; another is how much post-transcriptional regulation may occur as well as how these putative transporters ‘pump’ sterols out of the cell. Finally, the conservation of the polypeptides suggests a conservation in function. Thus, it is not clear what the selective advantage would be in this remarkable evolution, unless these proteins evolved to limit the entry of as yet unknown dietary sterol toxins.
References
Altmann SW, Davis HR Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N, Graziano MP (2004) Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 303:1201–1204
Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH (2000) Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290:1771–1775
Bhattacharyya AK, Connor WE (1974) Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest 53:1033–1043
Bjorkhem I, Boberg KM, Leitersdorf E (2005) Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease http://www.ommbid.com/ McGraw-Hill, New York, Ch 123, Sept 23
Bloks VW, Bakker-Van Waarde WM, Verkade HJ, Kema IP, Wolters H, Vink E, Groen AK, Kuipers F (2004) Down-regulation of hepatic and intestinal Abcg5 and Abcg8 expression associated with altered sterol fluxes in rats with streptozotocin-induced diabetes. Diabetologia 47:104–112
Borgstrom B (1968) Quantitative aspects of the intestinal absorption and metabolism of cholesterol and beta-sitosterol in the rat. J Lipid Res 9:473–481
Calpe-Berdiel L, Escola-Gil JC, Ribas V, Navarro-Sastre A, Garces-Garces J, Blanco-Vaca F (2005) Changes in intestinal and liver global gene expression in response to a phytosterol-enriched diet. Atherosclerosis 181:75–85
Chan DC, Watts GF, Barrett PH, Whitfield AJ, van Bockxmeer FM (2004) ATP-binding cassette transporter G8 gene as a determinant of apolipoprotein B-100 kinetics in overweight men. Arterioscler Thromb Vasc Biol 24:2188–2191
Dieter MZ, Maher JM, Cheng X, Klaassen CD (2004) Expression and regulation of the sterol half-transporter genes ABCG5 and ABCG8 in rats. Comp Biochem Physiol C Toxicol Pharmacol 139:209–218
Duan LP, Wang HH, Ohashi A, Wang DQ (2005) Role of intestinal sterol transporters Abcg5, Abcg8, and Npc1l1 in cholesterol absorption in mice: gender and age effects. Am J Physiol Gastrointest Liver Physiol
Duan LP, Wang HH, Wang DQ (2004) Cholesterol absorption is mainly regulated by the jejunal and ileal ATP-binding cassette sterol efflux transporters Abcg5 and Abcg8 in mice. J Lipid Res 45:1312–1323
Freeman LA, Kennedy A, Wu J, Bark S, Remaley AT, Santamarina-Fojo S, Brewer HB Jr (2004) The orphan nuclear receptor LRH-1 activates the ABCG5/ABCG8 intergenic promoter. J Lipid Res 45:1197–1206
Geuken E, Visser DS, Leuvenink HG, de Jong KP, Peeters PM, Slooff MJ, Kuipers F, Porte RJ (2005) Hepatic expression of ABC transporters G5 and G8 does not correlate with biliary cholesterol secretion in liver transplant patients. Hepatology 42:1166–1174
Gould RG, Jones RJ, LeRoy GV, Wissler RW, Taylor CB (1969) Absorbability of beta-sitosterol in humans. Metabolism 18:652–662
Graf GA, Li WP, Gerard RD, Gelissen I, White A, Cohen JC, Hobbs HH (2002) Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J Clin Invest 110:659–669
Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC, Hobbs HH (2003) ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem 2003
Gregg RE, Connor WE, Lin DS, Brewer H Jr (1986) Abnormal metabolism of shellfish sterols in a patient with sitosterolemia and xanthomatosis. J Clin Invest 77:1864–1872
Groen AK, Oude Elferink RP (2005) Lipid transport into bile and role in bile formation. Curr Drug Targets Immune Endocr Metabol Disord 5:131–135
Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB Jr, Kliewer SA, Gonzalez FJ, Sinal CJ (2003) Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem 278:45062–45071
Gylling H, Hallikainen M, Pihlajamaki J, Agren J, Laakso M, Rajaratnam RA, Rauramaa R, Miettinen TA (2004) Polymorphisms in the ABCG5 and ABCG8 genes associate with cholesterol absorption and insulin sensitivity. J Lipid Res
Hernandez HH, Chaikoff IL, Dauben WG, Abraham S (1954) The absorption of C14-labeled epicholesterol in the rat. J Biol Chem 206:757–765
Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 100:12027–12032
Hubacek JA, Berge KE, Stefkova J, Pitha J, Skodova Z, Lanska V, Poledne R (2004) Polymorphisms in ABCG5 and ABCG8 transporters and plasma cholesterol levels. Physiol Res 53:395–401
Kajinami K, Brousseau ME, Nartsupha C, Ordovas JM, Schaefer EJ (2004) ATP binding cassette transporter G5 and G8 genotypes and plasma lipoprotein levels before and after treatment with atorvastatin. J Lipid Res 45:653–656
Kajinami K, Brousseau ME, Ordovas JM, Schaefer EJ (2004) Interactions between common genetic polymorphisms in ABCG5/G8 and CYP7A1 on LDL cholesterol-lowering response to atorvastatin. Atherosclerosis 175:287–293
Kamisako T, Ogawa H (2003) Effect of obstructive jaundice on the regulation of hepatic cholesterol metabolism in the rat. Disappearance of abcg5 and abcg8 mRNA after bile duct ligation. Hepatol Res 25:99–104
Kamisako T, Ogawa H (2003) Regulation of biliary cholesterol secretion is associated with abcg5 and abcg8 expressions in the rats: effects of diosgenin and ethinyl estradiol. Hepatol Res 26:348–352
Kamisako T, Ogawa H (2005) Alteration of the expression of adenosine triphosphate-binding cassette transporters associated with bile acid and cholesterol transport in the rat liver and intestine during cholestasis. J Gastroenterol Hepatol 20:1429–1434
Kaneko E, Matsuda M, Yamada Y, Tachibana Y, Shimomura I, Makishima M (2003) Induction of intestinal ATP-binding cassette transporters by a phytosterol-derived liver X receptor agonist. J Biol Chem 278:36091–36098
Klett EL, Lu K, Kosters A, Vink E, Lee MH, Altenburg M, Shefer S, Batta AK, Yu H, Chen J, Klein R, Looije N, Oude-Elferink R, Groen AK, Maeda N, Salen G, Patel SB (2004) A mouse model of sitosterolemia: absence of Abcg8/sterolin-2 results in failure to secrete biliary cholesterol. BMC Med 2:5
Klett EL, Patel SB (2004) Biomedicine. Will the real cholesterol transporter please stand up. Science 303:1149–1150
Kosters A, Frijters RJ, Kunne C, Vink E, Schneiders MS, Schaap FG, Nibbering CP, Patel SB, Groen AK (2005) Diosgenin-induced biliary cholesterol secretion in mice requires Abcg8. Hepatology 41:141–150
Kruit JK, Plosch T, Havinga R, Boverhof R, Groot PH, Groen AK, Kuipers F (2005) Increased fecal neutral sterol loss upon liver X receptor activation is independent of biliary sterol secretion in mice. Gastroenterology 128:147–156
Kuksis A, Huang TC (1962) Differential absorption of plant sterols in the dog. Can J of Biochem Physiol 40:1493–1504
Langheim S, Yu L, von Bergmann K, Lutjohann D, Xu F, Hobbs HH, Cohen JC (2005) ABCG5 and ABCG8 require MDR2 for secretion of cholesterol into bile. J Lipid Res 46:1732–1738
Lee M-H, Gordon D, Ott J, Lu K, Ose L, Miettinen T, Gylling H, Stalenhoef AF, Pandya A, Hidaka H, Brewer JB, Kojima H, Sakuma N, Pegoraro R, Salen G, Patel SB (2001) Fine mapping of a gene responsible for regulating dietary cholesterol absorption; founder effects underlie cases of phytosterolemia in multiple communities. Eur J Hum Gen 9:375–384
Lee M-H, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Kojima H, Allikmets R, Sakuma N, Pegoraro R, Srivastava AK, Salen G, Dean M, Patel SB (2001) Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet 27:79–83
Lu K, Lee M, Patel SB (2001) Dietary cholesterol absorption; more than just bile. Trends Endocrinol Metab 12:314–320
Lu K, Lee M-H, Hazard S, Brooks-Wilson A, Hidaka H, Kojima H, Ose L, Stanlenhoef AFH, Mietinnen T, Bjorkhem I, Brukert EPA, Brewer HB, Salen G, Dean M, Srivastava A, Patel SB (2001) Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am J Hum Gen 69:278–290
Lu K, Lee MH, Yu H, Zhou Y, Sandell SA, Salen G, Patel SB (2002) Molecular cloning, genomic organization, genetic variations, and characterization of murine sterolin genes Abcg5 and Abcg8. J Lipid Res 43:565–578
Miettinen TA, Klett EL, Gylling H, Isoniemi H, Patel SB (2005) Liver transplantation in a patient with sitosterolemia and liver cirrhosis. Gastroenterology
Miwa K, Inazu A, Kobayashi J, Higashikata T, Nohara A, Kawashiri M, Katsuda S, Takata M, Koizumi J, Mabuchi H (2005) ATP-binding cassette transporter G8 M429V polymorphism as a novel genetic marker of higher cholesterol absorption in hypercholesterolaemic Japanese subjects. Clin Sci (Lond) 109:183–188
Patel SB, Salen G, Hidaka H, Kwiterovich PO, Stalenhoef AF, Miettinen TA, Grundy SM, Lee MH, Rubenstein JS, Polymeropoulos MH, Brownstein MJ (1998) Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J Clin Invest 102:1041–1044
Plat J, Bragt MC, Mensink RP (2005) Common sequence variations in ABCG8 are related to plant sterol metabolism in healthy volunteers. J Lipid Res 46:68–75
Plat J, Nichols JA, Mensink RP (2005) Plant sterols and stanols: effects on mixed micellar composition and LXR (target gene) activation. J Lipid Res 46:2468–2476
Plosch T, Bloks VW, Terasawa Y, Berdy S, Siegler K, Van Der Sluijs F, Kema IP, Groen AK, Shan B, Kuipers F, Schwartz M (2004) Sitosterolemia in ABC-transporter G5-deficient mice is aggravated on activation of the liver-X receptor. Gastroenterology 126:290–300
Plosch T, Kok T, Bloks VW, Smit MJ, Havinga R, Chimini G, Groen AK, Kuipers F (2002) Increased hepatobiliary and fecal cholesterol excretion upon activation of the liver X receptor is independent of ABCA1. J Biol Chem 277:33870–33877
Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ (2002) Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J Biol Chem 277:18793–18800
Roglans N, Vazquez-Carrera M, Alegret M, Novell F, Zambon D, Ros E, Laguna JC, Sanchez RM (2004) Fibrates modify the expression of key factors involved in bile-acid synthesis and biliary-lipid secretion in gallstone patients. Eur J Clin Pharmacol 59:855–861
Salen G, Ahrens E Jr, Grundy SM (1970) Metabolism of beta-sitosterol in man. J Clin Invest 49:952–967
Salen G, Tint GS, Shefer S, Shore V, Nguyen L (1992) Increased sitosterol absorption is offset by rapid elimination to prevent accumulation in heterozygotes with sitosterolemia. Arterioscler Thromb 12:563–568
Salen G, von Bergmann K, Lutjohann D, Kwiterovich P, Kane J, Patel SB, Musliner T, Stein P, Musser B (2004) Ezetimibe effectively reduces plasma plant sterols in patients with sitosterolemia. Circulation 109:966–971
Schoenheimer R (1929) Uber die Bedeutung der Pflanzensterine fur den tierischen Organismus. Hoppe-Seyler’s Z für physiol Chem 180:1–5
Schoenheimer R, Breusch F (1933) Synthesis and destruction of cholesterol in the organism. J Biol Chem 103:439–448
Simmonds WJ, Hofmann AF, Theodor E (1967) Absorption of cholesterol from a micellar solution: intestinal perfusion studies in man. J Clin Invest 46:874–890
Small DM (2003) Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci U S A 100:4–6
Sonoda J, Chong LW, Downes M, Barish GD, Coulter S, Liddle C, Lee CH, Evans RM (2005) Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc Natl Acad Sci U S A 102:2198–2203
Tauscher A, Kuver R (2003) ABCG5 and ABCG8 are expressed in gallbladder epithelial cells. Biochem Biophys Res Commun 307:1021–1028
Treadwell CR, Vahouny GV (1968) Cholesterol absorption. In: Code CF, Heide W (eds) Handbook of physiology, vol III. American Physiological Society, Washington, DC, pp 1407–1438
van der Veen JN, Kruit JK, Havinga R, Baller JF, Chimini G, Lestavel S, Staels B, Groot PH, Groen AK, Kuipers F (2005) Reduced cholesterol absorption upon PPARdelta activation coincides with decreased intestinal expression of NPC1L1. J Lipid Res 46:526–534
Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, Zatloukal K, Denk H, Trauner M (2005) CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 42:420–430
Yang C, Yu L, Li W, Xu F, Cohen JC, Hobbs HH (2004) Disruption of cholesterol homeostasis by plant sterols. J Clin Invest 114:813–822
Yu H, Pandit B, Klett E, Lee MH, Lu K, Helou K, Ikeda I, Egashira N, Sato M, Klein R, Batta A, Salen G, Patel SB (2003) The rat STSL locus: characterization, chromosomal assignment, and genetic variations in sitosterolemic hypertensive rats. BMC Cardiovasc Disord 3:4
Yu L, Gupta S, Xu F, Liverman AD, Moschetta A, Mangelsdorf DJ, Repa JJ, Hobbs HH, Cohen JC (2005) Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J Biol Chem 280:8742–8747
Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2002) Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A 99:16237–16242
Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH (2002) Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest 110:671–680
Yu L, von Bergmann K, Lutjohann D, Hobbs HH, Cohen JC (2004) Selective sterol accumulation in ABCG5/ABCG8-deficient mice. J Lipid Res 45:301–307
Yu L, York J, von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2003) Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J Biol Chem 278:15565–15570
Acknowledgements
We would like to thank all the members of the Patel lab, past and present, for their contributions to this work. We apologize to all of our colleagues whose works may not have referenced by us, as time and space conspired against us.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hazard, S.E., Patel, S.B. Sterolins ABCG5 and ABCG8: regulators of whole body dietary sterols. Pflugers Arch - Eur J Physiol 453, 745–752 (2007). https://doi.org/10.1007/s00424-005-0040-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-005-0040-7