
Abstract
Background
While several lines of evidence prove that elevated concentrations of low-density lipoproteins (LDL) causally contribute to the development of atherosclerosis and its clinical consequences, high-density lipoproteins are still widely believed to exert atheroprotective effects. Hence, HDL cholesterol (HDL-C) is in general still considered as “good cholesterol”. Recent research, however, suggests that this might not always be the case and that a fundamental reassessment of the clinical significance of HDL-C is warranted.
Method
This review article is based on a selective literature review.
Results
In individuals without a history of cardiovascular events, low concentrations of HDL-C are inversely associated with the risk of future cardiovascular events. This relationship may, however, not apply to patients with metabolic disorders or manifest cardiovascular disease. The classical function of HDL is to mobilise cholesterol from extrahepatic tissues for delivery to the liver for excretion. These roles in cholesterol metabolism as well as many other biological functions of HDL particles are dependent on the number as well as protein and lipid composition of HDL particles. They are poorly reflected by the HDL-C concentration. HDL can even exert negative vascular effects, if its composition is pathologically altered. High serum HDL-C is therefore no longer regarded protective. In line with this, recent pharmacological approaches to raise HDL-C concentration have not been able to show reductions of cardiovascular outcomes.
Conclusion
In contrast to LDL cholesterol (LDL-C), HDL-C correlates with cardiovascular risk only in healthy individuals. The calculation of the ratio of LDL-C to HDL-C is not useful for all patients. Low HDL-C should prompt examination of additional metabolic and inflammatory pathologies. An increase in HDL-C through lifestyle change (smoking cessation, physical exercise) has positive effects and is recommended. However, HDL-C is currently not a valid target for drug therapy.
Similar content being viewed by others
Epidemiology
The concept that high-density lipoproteins (HDL) could protect against coronary heart disease (CHD) primarily originated from epidemiological studies of the healthy population, in particular the Framingham study [1]. Patients with manifest CHD frequently exhibit low HDL cholesterol (HDL-C) [2]. The relationship between HDL-C and cardiovascular risk is not linear; for instance, no further improvement in prognosis is seen with HDL-C levels above ~60 mg/dl (1.5 mmol/l) (Fig. 1) [3]. Retrospective investigations of the EPIC Norfolk and of the IDEAL study show that very high concentrations of HDL-C may be associated with increased risk [4]. A recent register study of more than 1 million US veterans found a U-shaped relationship between HDL-C and total mortality with 50 mg/dL (1.25 mmol/L) as the nadir associated with the lowest mortality [5]. A very recent analysis of the Framingham study reports that the predictive value of HDL-C is modified by LDL cholesterol (LDL-C) and triglycerides (TG): Compared to low HDL-C (defined as <50 mg/dl in women and <40 mg/dl in men) in isolation, risk increases when low HDL-C occurs together with high LDL-C and/or TG. Cardiovascular (CV) risk increases by 30% for LDL-C ≥ 100 mg/dl and TG < 100 mg/dl or LDL-C < 100 mg/dl and TG ≥ 100 mg/dl. When both TG and LDL-C are ≥100 mg/dl, CV risk increases by 60% [6].

In summary Low HDL-C is an indicator of increased cardiovascular risk, especially in persons without a history of cardiovascular events; however, the epidemiological relationship between HDL-C and risk is complex. Reduced HDL-C concentrations are frequently confounded with other pro-atherogenic conditions, notably the presence of inflammation and pro-atherogenic triglyceride-rich lipoproteins and their remnants as well as small dense LDL. The continued widespread practice of calculating the ratio of LDL-C to HDL-C is not useful, because high HDL-C is not associated with reduced risk, so that a combination of high LDL-C and HDL-C may lead to the wrong conclusion that risk is not elevated.
Role of HDL in lipoprotein metabolism
HDL are the smallest (5–17 nm) and densest (1.063–1.210 kg/l) lipoproteins in the plasma. Apolipoprotein (Apo) A1, the major protein in HDL, is synthesised in the liver and the small intestine. The liver is the most important organ through which cholesterol is excreted, either directly or after being converted into bile acids. Excess cholesterol is transported from the periphery (e.g. from macrophages in blood vessel walls) to the liver. HDL play a key role in this pathway, known as reverse cholesterol transport (RCT) (Fig. 2) [8–11]. In addition to HDL, LDL also significantly contribute to RCT. The overwhelming majority of HDL-C measured in the blood originates from the liver and the intestine. Therefore, the concentration of HDL-C in the plasma cannot be used as a measure of cholesterol efflux from vessel walls, or of the efficiency of RCT.

Role of HDL in lipoprotein metabolism. Apo A1, the main protein in HDL, is formed in the liver and the small intestine and secreted as lipid-free pre-ß-HDL. Pre-b-HDL can also come from chylomicron metabolism, or from the interconversion of HDL itself. Their interaction with ATP-binding cassette transporter A1 (ABCA1) leads to the efflux of phosphatidylcholine (PC) and free cholesterol (FC), and thus to the formation of disc-shaped HDL. Esterification of the free cholesterol by lecithin–cholesterol acyltransferase (LCAT) leads to the formation of mature spherical HDL. The lipid-rich discoidal and spherical HDL enable passive diffusion out of cells, which is facilitated by ATP-binding cassette transporter G1 (ABCG1), scavenger receptor class B type I (SR-B1) and by cholesterol esterification mediated by LCAT. Cholesteryl ester transfer protein (CETP) transfers cholesteryl esters (CE), triglycerides and phospholipids (PL) between HDL, LDL and VLDL. It thereby contributes to the formation of LDL, which is taken up through LDL receptors (LDLR) in hepatocytes. Phospholipid transfer protein (PLTP) belongs to the same protein family as CETP. Its function is to transport phospholipids between HDL and VLDL, and between various HDL. Mediated by SR-BI, HDL can deliver cholesteryl ester (and free cholesterol) to hepatocytes, steroid-producing cells and adipocytes. With permission of Springer [7]
The RCT begins with the transfer of cholesterol from cell membranes to HDL. To date, four biochemical pathways have been described that are involved in this transfer [8]. As long as the cholesterol content of the cell is normal, more than two-thirds of the (poorly water-soluble, non-esterified) cholesterol leave the cells by passive diffusion along a concentration gradient between the cell membrane and preferably large, globular HDL. This concentration gradient is maintained by extracellular esterification of free cholesterol mediated by the lecithin cholesterol acyltransferase (LCAT).
Secondly, the passive, aqueous efflux of non-esterified cholesterol can be further enhanced by up-regulation of ATP-binding cassette transporter G1 (ABCG1). ABCG1 mobilises cholesterol from subcellular compartments.
Thirdly, passive, but non-aqueous transfer of free cholesterol from cells to larger HDL can be mediated by the scavenger receptor class B type I (SR-B1). SR-B1 promotes not only the cellular cholesterol efflux, but also the “selective” (i.e. non-endocytotic) delivery of cholesterol from HDL to liver cells [12].
Fourthly, cholesterol and phospholipids from macrophages and foam cells are actively transferred to ATP-binding cassette transporter A1 (ABCA1) to lipid-free apo A1 (pre-ß-HDL). This process is important for the formation of “nascent” HDL particles, (i.e. HDL discs), which contain Apo A1, phosphatidylcholine and non-esterified cholesterol. The importance of ABCA1 for the metabolic maturation of HDL was identified by the elucidation of the genetic cause of Tangier’s disease [13, 14]. The expression of ABCA1 is regulated by the intracellular cholesterol content, and efflux mediated by ABCA1 is therefore important in situations of cellular cholesterol excess.
The directed flux of cholesterol between cells and HDL is dependent on a concentration gradient between the cell membrane and acceptor lipoproteins. This gradient is maintained at the cellular level by the hydrolysis of cholesteryl esters and translocation of cholesterol, extracellularly by lecithin cholesterol acyltransferase (LCAT) and the cholesterol ester transfer protein (CETP). The LCAT esterifies free cholesterol (for example, cholesterol associated with pre-ß-HDL) with a fatty acid from the lecithin. Cholesteryl esters are more hydrophobic than free cholesterol and are located inside the HDL. This transforms HDL from discoidal into spherical, pseudomicellular particles (α-HDL). Maturation of HDL is incomplete in genetic or secondary deficiency of LCAT. In patients with familial LCAT deficiency, spherical HDL are absent.
CETP is a hydrophobic glycoprotein which is mainly secreted by the liver and is mostly bound to HDL in the blood [15]. It facilitates the transfer of cholesteryl esters, triglycerides and, to a smaller extent, phospholipids between HDL, LDL and VLDL. Thus, cholesteryl esters are channelled into the metabolism of LDL and delivered to the liver.
In summary HDL-particles play a crucial role in the reverse transport and excretion of cholesterol. However, HDL-cholesterol measurements do not reflect HDL function in RCT.
Determination of HDL cholesterol in the clinical laboratory
The standard method for the determination of cholesterol in LDL and HDL is the combined method of precipitation and ultracentrifugation (beta quantification) [16]. In the first step, VLDL is recovered by means of ultracentrifugation, then LDL is precipitated and finally HDL-C is determined enzymatically in the supernatant. This method is not suitable for use in the clinical laboratory because of high costs and complexity. In the late 1990s, “homogeneous” methods were introduced which allow the measurement of HDL-C in a single reaction vessel with no prior separation steps [17]. In these methods, the cholesterol in non-HDL particles is “masked” with antibodies, polymers or detergents, and in a second reaction step HDL-C is determined enzymatically. The commercially available homogeneous assays show a good agreement with the reference method in normolipidemic samples and in many cases of hypercholesterolemia. The determination of HDL-C is hardly affected by prior consumption of food and the results can usually well be interpreted in post-prandial samples [18]. Differences to the reference method and between the assays are, however, observed in hypertriglyceridemia (elevated concentrations of chylomicrons and/or VLDL), at low HDL-C (<20 mg/dl), and if atypical lipoproteins are present (type III hyperlipoproteinemia, liver disease, chronic kidney disease) [19–21]. In these cases, HDL-C can be accurately measured by lipoprotein electrophoresis, however by staining lipoproteins using enzymatic cholesterol oxidation rather than unspecific lipophilic chemicals.
Due to the limitations of conventional lipoprotein analysis, researchers are considering the replacement of HDL-C and LDL-C determinations with the determination of Apo A1 and ApoB, respectively. Measurement of apolipoproteins can be readily standardised [22] and is not significantly distorted by high triglycerides. Apo B not only reflects the concentration of LDL, but all atherogenic lipoproteins taken together [VLDL, remnants, LDL, Lp(a)].
In recent years, assays have been introduced which reflect the functionality of HDL better than HDL-C. Of importance are the determination of HDL particle concentrations (and sizes) with nuclear magnetic resonance (NMR) spectroscopy [23], the measurement of HDL proteins (Apo A1, serum amyloid A) and the in vitro measurement of the cholesterol uptake capacity of HDL in cell culture models (cholesterol efflux) [24–27]. While the latter will most likely remain a research method, NMR and apolipoproteins are in principle suitable for routine use, once their standardisation has been accomplished.
In summary Current clinically available methods are able to determine the cholesterol content of HDL particles, but not their biological function. Standardised determinations of HDL-C, and even less so of assays for cholesterol efflux capacity, HDL particle number determination by NMR spectroscopy and HDL proteins, are still not satisfactory.
Lesions from genetic abnormality of HDL metabolism
Table 1 provides an overview of rare monogenic disorders in the HDL metabolism. According to the results from Danish and American population studies, approximately 10% of individuals with HDL-C levels below the 5th percentile are heterozygous for mutations in the genes of APOA1, ABCA1 or LCAT [28, 29]. Data on the risk of atherosclerosis in these individuals are contradictory [30]. Some mutations of APOA1 have been associated with increased risk of myocardial infarction [31], and another one, Apo A1-Milano, may reduce the risk. Some mutations of the APOA1 gene also cause familial amyloidosis. In large Danish population studies, heterozygosity for HDL-C-lowering mutations of the ABCA1 or APOA1 gene was not associated with an increased risk of myocardial infarction. Interestingly, ABCA1 mutations increase the risk for Alzheimer’s disease. Data from large Dutch family studies suggest an increased cardiovascular risk of HDL-C-lowering mutations in the genes of APOA1, ABCA1 or LCAT. Italian studies found no evidence for an increased risk of atherosclerosis in heterozygous carriers of LCAT mutations [32–34].
Homozygous and heterozygous carriers of mutations in these three genes are very rare and present themselves with pronounced or even complete HDL deficiency and characteristic clinical syndromes: patients with complete LCAT deficiency develop corneal opacities, anaemia and progressive renal failure, which ultimately requires renal replacement therapy. Patients with partial LCAT deficiency (“fish eye disease”) develop corneal opacities, but neither renal disease nor anaemia. In both cases, the remaining HDL are discoidal in shape. In classical LCAT deficiency, LDL include particles which are similar to Lipoprotein X and may be the cause of the characteristic nephropathy. The risk of atherosclerosis of mutation carriers is not increased or at most slightly increased [35], possibly because HDL can also transport free cholesterol directly to the liver [36].
Homozygosity for mutations in the ABCA1 gene leads to Tangier disease [13, 14]. As a result of the strongly reduced cellular cholesterol efflux, cholesterol accumulates in macrophages in various organs (enlarged tonsils, hepatosplenomegaly and peripheral neuropathy). Patients with Tangier disease often also have low LDL-C, which could attenuate the atherogenic effect of the low HDL concentration [Schaefer, 2010 # 21]. Homozygous nonsense mutations in the APOA1 gene were found to [28] be the cause of HDL deficiency in patients with severe xanthomatosis and early atherosclerosis.
Most patients with a deficiency of cholesteryl ester transfer protein (CETP) have been observed in Japan. Affected persons have very high concentrations of HDL-C. Pharmacological inhibition of CETP has therefore been tested as an approach to increase HDL. It is unclear whether the HDL produced through CETP inhibition have a normal function. We observed a slightly increased cardiovascular mortality at low concentrations of CETP in the blood despite high HDL-C [37].
Only recently, mutations of scavenger receptor (SR-BI) (P279S, P376L) have been described which are associated with both significant increases in HDL-C and increased cardiovascular risk [38–40]. These mutations impair the selective uptake of cholesteryl esters by hepatocytes. In mice, the overexpression of SR-B1 leads to an increase in RCT, despite a drop in HDL-C [41]. On the other hand, lack of SR-BI increases HDL-C, but is atherogenic [42]. High HDL-C therefore does not provide protection from atherosclerosis in all cases.
This stands in contrast to genetic factors which increase LDL cholesterol, because monogenetic (familial hypercholesterolemia) and polygenetic factors increasing LDL cholesterol consistently lead to concordant changes in the CV risk (positive Mendelian randomisation) [43, 44].
In summary Current genetic data do not support the concept of a general protective role for HDL-C with regard to coronary heart disease.
Disorders with reduced HDL concentration
Compared to genetically determined abnormalities in HDL metabolism, low HDL-C much more frequently occur in patients with metabolic syndrome or diabetes mellitus. Low HDL-C levels are also associated with systemic inflammation, e.g. with cigarette smoking, chronic inflammatory diseases or chronic kidney disease (Fig. 3) [45, 46]. In cases of extremely low HDL-C, rare diagnosis may be considered, e.g. neoplasia or an increased risk for sepsis [47, 48].

Aetiologies of low HDL cholesterol. With permission of Springer [7]
In summary Low HDL-C is an indicator that the affected individual should be examined for metabolic and inflammatory pathology.
Vascular protective function of HDL
Evidence has long been available from animal experiments for anti-atherosclerotic effects of HDL. Administration of homologous HDL in addition to an atherogenic diet inhibits the formation of fatty streaks in rabbits [49]. Infusion of Apo A1 Milano (R173C, complexed with phospholipids) inhibits atherogenesis in cholesterol-fed rabbits [50] and apo E-deficient mice [51].
Alongside its prominent role in RCT, anti-oxidative and anti-inflammatory effects are also attributed to HDL, as well as improvement of endothelial function [9, 52, 53]. They inhibit the pathological attachment of monocytes to the endothelium and support endothelial repair mechanisms. Many of the endothelial effects of HDL are mediated by the endothelial SR-B1 receptor and by the bioactive lipid sphingosine-1-phosphate that can act via the S1P3 receptor on endothelial cells [54]. Investigations of the composition of HDL [55, 56] have found enzymes, acute-phase proteins, components of the complement system and protease inhibitors in addition to the characteristic apolipoproteins associated with HDL. The lipidome of HDL is even more heterogeneous, and HDL can also function as the vehicle for microRNA in the blood [57]. Paraoxonase 1 (PON1) is an esterase associated with HDL, which inhibits the formation of lipid peroxides in LDL [58, 59] and HDL. Compared to healthy people, the activity of HDL-associated PON1 is reduced in patients with CHD [60]. An anti-oxidative effect is also attributed to Apo A1 itself [61]. HDL are anti-inflammatory in several ways: they inhibit the expression of adhesion molecules (VCAM-I and ICAM-I) in the endothelium, inhibit the terminal complement complex, inhibit the synthesis of chemokines (MCP-1) and induce the expression of the anti-inflammatory cytokine transforming growth factor beta 2.
In addition, HDL can have antithrombotic and profibrinolytic effects [62]. HDL protect from cell damage, necrosis and apoptosis. These protective effects have also been demonstrated in pancreatic beta cells, implying that HDL might be able to improve insulin secretion and protect from the development of diabetes mellitus [63–68].
In summary The anti-inflammatory, cytoprotective and wound-healing effects of HDL make them a part of the innate host defense system [69]. From an evolutionary point of view, the potential anti-atherogenic effects, if any of them exist, would likely be secondary.
HDL dysfunction
Interestingly, several studies have provided evidence that the vascular effects of HDL are variable and hardly correlate with HDL-C concentrations in the plasma. In patients with diabetes mellitus, coronary disease, chronic renal insufficiency, cardiovascular risk factors and disorders, the function of HDL is impaired [9, 61, 70–74]. In patients who underwent coronary angiography, we have observed that HDL-C correlates inversely with cardiovascular mortality in the absence of CHD, that the relationship of HDL-C to long-term prognosis was weakened in stable coronary heart disease, whereas in clinically unstable patients the relationship was completely abolished [74].
The protein composition of HDL changes significantly in the course of an acute-phase reaction. The “acute-phase HDL” are characterised, for example, by increased concentrations of serum amyloid A (SAA), secretory phospholipase A2 (sPLA2-IIa) and ceruloplasmin, and a lower proportion of Apo A1 [73]. Unlike HDL from healthy subjects, HDL from patients with CHD, kidney disease or diabetes mellitus have no protective vascular effects or may even cause paradoxical harmful effects [55, 56, 61, 70, 71, 75, 76]. The characterisation of the HDL’s “milieu” may offer an opportunity for determining the function of HDL. For example, HDL-C corrected by the SAA concentration correlates with risk better than HDL-C alone in several independent cohorts [77].
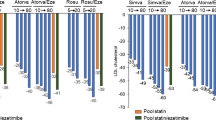
Another way to determine the functionality of HDL in the laboratory, although one that requires more efforts, is measuring cellular cholesterol efflux. In this method, cultivated cells are enriched with labelled cholesterol and incubated with Apo B-free serum from the patient, and the rate of transfer of labelled cholesterol into the patient serum is determined. The rate of cholesterol efflux has been associated with the prevalence of CHD, independent of the HDL-C concentration [24], with cardiovascular events and deaths (Fig. 4) [25, 26]. In another study, cellular efflux was inversely correlated with prevalent atherosclerosis, but not with the long-term risk of cardiovascular events [78].

Cellular cholesterol efflux and cardiovascular mortality. Kaplan–Meier curves and hazard ratios by quartiles of cholesterol efflux capacity. Model 1: adjusted for age and gender; Model 2: also adjusted for use of lipid-lowering drugs, CHD (none, stable, unstable CHD, NSTEMI, STEMI), body mass index, hypertension, smoking, LDL-C/HDL-C ratio, triglycerides, metabolic syndrome/type 2 diabetes mellitus [26]. With permission of Springer [7]
In summary The biological function of HDL is in part dependent on their protein composition. The properties of HDL are poorly reflected by their cholesterol content. In disease states, HDL may turn dysfunctional and can even have negative vascular effects.
Should HDL-C be increased by therapeutic measures?
The positive vascular effects of HDL have been an attractive target for the treatment of chronic or acute vascular disease. Approaches to increase HDL include lifestyle modifications, conventional pharmacological therapy and novel therapeutic modalities which are still under scrutiny.
Lifestyle
HDL-C can be increased through a number of lifestyle changes [79] (Table 2). The most important among these are increased physical activity and dietary changes. Lifestyle changes that increase HDL-C are also associated with reduced risk of cardiovascular disease [80]. The actual contributions made by increases in HDL-C are however not clear, because these interventions do not increase HDL-C in isolation, but influence metabolism in a variety of ways. Physical activity not only leads to an increase in HDL-C, but also to changes in HDL functionality [69]. At the same time, the metabolism of triglyceride-rich lipoproteins (fasting and postprandial) is improved [81, 82]. Yet, a number of other pathologies are also subject to positive changes (e.g. insulin sensitivity). We therefore conclude that the increase in HDL-C observed with lifestyle changes represents a marker of reduced cardiovascular risk.
Statins
Statins competitively inhibit HMG-CoA reductase, the rate-limiting enzyme for cholesterol biosynthesis, and they reduce LDL-C. Statins also lower triglycerides and slightly raise HDL-C. It is unlikely that this increase in HDL-C makes a substantial contribution to the reduction of events mediated by statins [83–85].
Fibrates
Fibrates are agonists of the nuclear receptor PPAR-α (peroxisome proliferator-activated receptor alpha). Fibrates stimulated the expression of Apo A1 and via LXR the expression of ABCA1. Fibrates also up-regulate SR B1, which mitigates their HDL-raising effect, but is synergistic in activating RCT. Fibrates also activate lipolytic enzymes, lipoprotein lipase (LPL) in particular, which leads to increased catabolism of triglyceride-rich lipoproteins and an indirect increase in HDL-C.
A meta-analysis of 18 prospective, randomised outcome studies concluded that fibrates reduce the rate of severe cardiovascular events by 10% and the progression of albuminuria by 14% [86]. It is unclear whether the clinical effects of fibrates are due primarily to the increase in HDL-C, the decrease in triglycerides or the slight reduction in LDL. For patients with diabetes mellitus and statin therapy, fibrates have not been shown to provide any further reduction of risk [87–89]. Retrospective analyses of fibrate trials raise the possibility that reductions in risk can be achieved in subgroups with atherogenic dyslipidemia (high triglycerides and low HDL-C) [88, 90, 91]. In Phase 3 studies with new fibrates, this hypothesis is being tested prospectively. If these studies were successful, the use of fibrates in patients with high triglycerides and/or low HDL-C would be warranted.
Niacin
Niacin and derivatives reduce triglycerides, VLDL cholesterol, LDL-C and lipoprotein (a), and they increase HDL-C. Among other factors, the effects are based on the inhibition of hepatic diacylglycerol acyltransferase 2, a key enzyme in the synthesis of triglycerides. Meta-analyses of studies with niacin (mostly without simultaneous administration of statins) indicate a decrease in cardiovascular events [92–94]. A subsequent meta-analysis of 11 outcome studies, which were mainly conducted in secondary prevention and included the AIM-HIGH-study [92], indicated that niacin reduced cardiovascular events and adverse coronary endpoints by 34 and 25%, respectively [94].
A combination of niacin with laropiprant was tested on 25,673 patients in the HPS2-THRIVE study [95]. No reduction in vascular events was observed, but the rates of severe side effects such as bleeding, myopathy, infections and diabetes mellitus increased. Since then, niacin preparations are no longer available in Europe. Based on the AIM-HIGH, ACCORD and HPS2-THRIVE studies, the Federal Drug Administration has come to the conclusion that there is no sufficient evidence for the reduction of cardiovascular events through the use of fibrates and niacin [96, 97], especially in conjunction with statin therapy.
CETP inhibition
Cholesteryl ester transfer protein (CETP) mediates the exchange of cholesterol and triglycerides between various lipoproteins and thus plays an important role in RCT (Fig. 2) [98, 99]. The concept of CETP inhibition is based on the following observations: rodents are naturally CETP deficient, display high HDL-C and are resistant to diet-induced atherosclerosis. Patients with genetically reduced or absent CETP activity have high HDL-C. Inhibition of CETP has a positive influence on the lipoprotein profile in humans and could therefore show atheroprotective effects.
However, animal experiments have yielded contradictory results. In rabbits, CETP inhibition led to a reduction in atherosclerosis. In mice, both atheroprotective and atherogenic effects have been reported. Studies in humans have also been contradictory. On the one hand, patients with genetically reduced CETP serum concentrations appear to have a slightly reduced risk of coronary heart disease [99–102]. On the other hand, many studies in which CETP was tested in the plasma showed an inverse correlation between CETP and cardiovascular risk [37, 103]. Drug-based inhibition of CETP in humans leads to a significant increase in HDL-C and a drop in LDL-C [104, 105]. However, three out of four prospective studies with CETP inhibitors had to be terminated due to increased mortality in the treatment group or due to futility (Table 3). (https://investor.lilly.com/releasedetail.cfm?ReleaseID=936130) [106, 107]. The results of the large HPS-3/TIMI55 study, including over 30,000 patients, are pending.
One reason for the disappointing performance of CETP inhibitors thus far could be the fact that the transfer of cholesteryl esters from HDL to LDL, and thus a pivotal step in RCT, is being inhibited. CETP inhibition probably also extends the lifespan of dysfunctional HDL. This may not only eliminate the postulated atheroprotective effect but even be harmful if HDL gained adverse properties [9, 74, 108]. Future strategies may include the identification of patients who benefit from CETP inhibition. Another alternative approach would be the development of new inhibitors that reduce CETP production or modification of CETP function [109].
Other HDL-boosting drugs in development
Recent approaches primarily aim to optimise the function of HDL in RCT [110, 111]. Although HDL likely influences atherogenesis in a variety of ways, the focus is on the HDL-dependent aspects of RCT (Table 4). For instance, some such approaches use artificial discoidal HDL which, in addition to phospholipids, contain Apo A1 (CSL112) isolated from plasma, recombinant Apo A1 (CER001) or variant Apo A1 (Apo A1 Milano). Others generate pre-beta HDL via delipidation of the patient’s own plasma [111]. Both in vitro and in vivo, these approaches can achieve a transient acceleration of RCT, sometimes without causing any significant change in HDL-C. One problem is at present, however, that there is still no simple biomarker available for HDL functionality. The further development of Apo A1 Milano has been stopped in 2016 due to lack of efficacy [112]. However, the search for other possible ways to intensify RCT remains the subject of ongoing research.
In summary An increase in HDL-C through lifestyle changes has positive effects and is recommended. This is especially true for smoking cessation and physical activity. Outcome studies with fibrates and with niacin have been conducted to evaluate the potential of HDL-C as a therapeutic target. However, since these drugs also cause drops in triglycerides and (to a moderate extent) LDL-C, neither fibrates nor niacin can conclusively value HDL-C as a drug target. There is no evidence for a reduction in clinical events through niacin or fibrates in combination with statins. In the studies available to date, increasing HDL-C with CETP inhibitors has not led to a reduction in clinical events. We conclude that HDL-C is not a target for drug-based therapy at the present time.
Practical consequences
HDL play a central role in RCT. Large epidemiological studies have found an inverse relationship between the concentration of HDL-C in plasma and CHD. Mendelian randomisation studies show no protective effects of a genetically high HDL-C concentration. Cholesterol transported with HDL neither has any protective function nor reflects the functionality of HDL. HDL particles as such, as well as specific proteins and lipids in HDL, contribute to the classical functions of HDL in RCT, or mediate their anti-oxidative and anti-inflammatory properties. HDL composition is modified in complex ways in acute and chronic diseases. In this sense, it is misleading to interpret HDL-C as being equivalent to HDL or “good cholesterol”. The determination of pathophysiologically relevant lipids or proteins associated with HDL may come to replace HDL-C in the future, but at present such assays are not yet available for routine clinical use [9, 115]. The following clinical consequences result from these new findings:
-
Low HDL-C is often a clinical indicator of disturbed metabolism of triglyceride-rich lipoproteins (e.g. in diabetes mellitus) or a chronic inflammation.
-
The prognosis with regard to cardiovascular disease cannot be determined via HDL-C; the ratio of HDL-C to LDL-C may be misleading if HDL-C is high and if comorbidities like CHD, diabetes mellitus or chronic kidney disease exist.
-
Increasing HDL-C through lifestyle factors such as physical activity and smoking cessation is associated with vascular protective effects.
-
At present, HDL-C is not a target for drug-based treatment. The primary goal of lipid therapy is currently the reduction of LDL cholesterol.
Clearly, the “HDL-Cholesterol story” is not over but the recent findings set the stage for future research on this complex lipopoprotein.
References
Gordon DJ, Probstfield JL, Garrison RJ et al (1989) High density lipoprotein cholesterol and cardiovascular disease. Four Prospective American Studies. Circulation 79:8–15
Genest JJ, McNamara JR, Salem DN, Schaefer EJ (1991) Prevalence of risk factors in men with premature coronary artery disease. Am J Cardiol 67(15):1185–1189
Di Angelantonio E, Sarwar N, Perry P et al (2009) Major lipids, apolipoproteins, and risk of vascular disease. JAMA 302(18):1993–2000
van der Steeg WA, Holme I, Boekholdt SM et al (2008) High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: significance for cardiovascular risk: the IDEAL and EPIC-Norfolk studies. J Am Coll Cardiol 51(6):634–642
Bowe B, Xie Y, Xian H, Balasubramanian S, Zayed MA, Al-Aly Z (2016) High density lipoprotein cholesterol and the risk of all-cause mortality among US Veterans. Clin J Am Soc Nephrol. doi:10.2215/CJN.00730116
Bartlett J, Predazzi IM, Williams SM et al (2016) Is isolated low high-density lipoprotein cholesterol a cardiovascular disease risk factor? New insights from the Framingham offspring study. Circ Cardiovasc Qual Outcomes 9(3):206–212
März W, Kleber ME, Scharnagl H et al (2017) Clinical importance of HDL cholesterol. Herz 42(1):58–66
Phillips MC (2014) Molecular mechanisms of cellular cholesterol efflux. J Biol Chem 289(35):24020–24029
Annema W, von Eckardstein A (2016) Dysfunctional high-density lipoproteins in coronary heart disease: implications for diagnostics and therapy. Transl Res 173:30–57
Lee-Rueckert M, Escola-Gil JC, Kovanen PT (2016) HDL functionality in reverse cholesterol transport–Challenges in translating data emerging from mouse models to human disease. Biochim Biophys Acta 1861(7):566–583
Rosenson RS, Brewer HB Jr, Davidson WS et al (2012) Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125(15):1905–1919
Rohrl C, Stangl H (2013) HDL endocytosis and resecretion. Biochim Biophys Acta 1831(11):1626–1633
Bodzioch M, Orso E, Klucken J et al (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet 22(4):347–351
Rust S, Rosier M, Funke H et al (1999) Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet 22(4):352–355
Quintao EC, Cazita PM (2010) Lipid transfer proteins: past, present and perspectives. Atherosclerosis 209(1):1–9
Bachorik PS, Ross JW (1995) National Cholesterol Education Program recommendations for measurement of low-density lipoprotein cholesterol: executive summary. The National Cholesterol Education Program Working Group on Lipoprotein Measurement. Clin Chem 41(10):1414–1420
Warnick GR, Nauck M, Rifai N (2001) Evolution of methods for measurement of HDL-cholesterol: from ultracentrifugation to homogeneous assays. Clin Chem 47(9):1579–1596
Nordestgaard BG, Langsted A, Mora S et al (2016) Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J 37(25):1944–1958
Miller WG, Myers GL, Sakurabayashi I et al (2010) Seven direct methods for measuring HDL and LDL cholesterol compared with ultracentrifugation reference measurement procedures. Clin Chem 56(6):977–986
Miida T, Nishimura K, Okamura T et al (2014) Validation of homogeneous assays for HDL-cholesterol using fresh samples from healthy and diseased subjects. Atherosclerosis 233(1):253–259
Langlois MR, Descamps OS, van der Laarse A et al (2014) Clinical impact of direct HDLc and LDLc method bias in hypertriglyceridemia. A simulation study of the EAS-EFLM Collaborative Project Group. Atherosclerosis 233(1):83–90
van den Broek I, Romijn FP, Nouta J et al (2016) Automated multiplex LC-MS/MS assay for quantifying serum apolipoproteins A-I, B, C-I, C-II, C-III, and E with qualitative apolipoprotein E phenotyping. Clin Chem 62(1):188–197
Kaess B, Fischer M, Baessler A et al (2008) The lipoprotein subfraction profile: heritability and identification of quantitative trait loci. J Lipid Res 49(4):715–723
Khera AV, Cuchel M, de la Llera-Moya M et al (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364(2):127–135
Rohatgi A, Khera A, Berry JD et al (2014) HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med 371(25):2383–2393
Ritsch A, Scharnagl H, Marz W (2015) HDL cholesterol efflux capacity and cardiovascular events. N Engl J Med 372(19):1870–1871
März W, Ritsch A (2016) Cholesterol efflux capacity: choke point of reverse cholesterol traffic? J Am Coll Cardiol 67(21):2488–2491
Schaefer EJ, Santos RD, Asztalos BF (2010) Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol 21(4):289–297
von Eckardstein A (2006) Differential diagnosis of familial high density lipoprotein deficiency syndromes. Atherosclerosis 186(2):231–239
Modesto KM, Dispenzieri A, Gertz M et al (2007) Vascular abnormalities in primary amyloidosis. Eur Heart J 28(8):1019–1024
Hovingh GK, Brownlie A, Bisoendial RJ et al (2004) A novel apoA-I mutation (L178P) leads to endothelial dysfunction, increased arterial wall thickness, and premature coronary artery disease. J Am Coll Cardiol 44(7):1429–1435
Tietjen I, Hovingh GK, Singaraja R et al (2012) Increased risk of coronary artery disease in Caucasians with extremely low HDL cholesterol due to mutations in ABCA1, APOA1, and LCAT. Biochim Biophys Acta 1821(3):416–424
Frikke-Schmidt R (2010) Genetic variation in the ABCA1 gene, HDL cholesterol, and risk of ischemic heart disease in the general population. Atherosclerosis 208(2):305–316
Haase CL, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A (2012) Population-based resequencing of APOA1 in 10,330 individuals: spectrum of genetic variation, phenotype, and comparison with extreme phenotype approach. PLoS Genet 8(11):e1003063
Calabresi L, Baldassarre D, Castelnuovo S et al (2009) Functional lecithin: cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation 120(7):628–635
Tanigawa H, Billheimer JT, Tohyama J et al (2009) Lecithin: cholesterol acyltransferase expression has minimal effects on macrophage reverse cholesterol transport in vivo. Circulation 120(2):160–169
Ritsch A, Scharnagl H, Eller P et al (2010) Cholesteryl ester transfer protein and mortality in patients undergoing coronary angiography: the Ludwigshafen Risk and Cardiovascular Health study. Circulation 121(3):366–374
Vergeer M, Korporaal SJ, Franssen R et al (2011) Genetic variant of the scavenger receptor BI in humans. N Engl J Med 364(2):136–145
Ljunggren SA, Levels JH, Hovingh K et al (2015) Lipoprotein profiles in human heterozygote carriers of a functional mutation P297S in scavenger receptor class B1. Biochim Biophys Acta 1851(12):1587–1595
Zanoni P, Khetarpal SA, Larach DB et al (2016) Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351(6278):1166–1171
Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ (2005) Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest 115(10):2870–2874
Huby T, Doucet C, Dachet C et al (2006) Knockdown expression and hepatic deficiency reveal an atheroprotective role for SR-BI in liver and peripheral tissues. J Clin Invest 116(10):2767–2776
Voight BF, Peloso GM, Orho-Melander M et al (2012) Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet 380(9841):572–580
Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD (2015) Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol 65(15):1552–1561
Parhofer KG (2015) Interaction between glucose and lipid metabolism: more than diabetic dyslipidemia. Diabetes Metab J 39(5):353–362
Taskinen MR, Boren J (2015) New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis 239(2):483–495
van Gorselen EO, Diekman T, Hessels J, Verhorst PM, von Birgelen C (2010) Artifactual measurement of low serum HDL-cholesterol due to paraproteinemia. Clin Res Cardiol Off J Ger Card Soc 99(9):599–602
Shor R, Wainstein J, Oz D et al (2008) Low HDL levels and the risk of death, sepsis and malignancy. Clin Res Cardiol Off J Ger Card Soc 97(4):227–233
Badimon JJ, Badimon L, Fuster V (1990) Regression of atherosclerostic lesions by high density lipoprotein lasma fraction in the cholesterol-fed rabbit. J Clin Invest 85:1234–1241
Ameli S, Hultgardh-Nilsson A, Cercek B et al (1994) Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation 90(4):1935–1941
Shah PK, Nilsson J, Kaul S et al (1998) Effects of recombinant apolipoprotein A-I(Milano) on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation 97(8):780–785
Annema W, von Eckardstein A (2013) High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J 77(10):2432–2448
Luscher TF, Landmesser U, von Eckardstein A, Fogelman AM (2014) High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target. Circ Res 114(1):171–182
Nofer JR, van der Giet M, Tolle M et al (2004) HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest 113(4):569–581
Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A (2009) Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol 29(6):870–876
Vaisar T, Pennathur S, Green PS et al (2007) Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest 117(3):746–756
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13(4):423–433
Mackness MI, Durrington PN, Mackness B (2000) How high-density lipoprotein protects against the effects of lipid peroxidation. Curr Opin Lipidol 11(4):383–388
Regieli JJ, Jukema JW, Doevendans PA et al (2009) Paraoxonase variants relate to 10-year risk in coronary artery disease: impact of a high-density lipoprotein-bound antioxidant in secondary prevention. J Am Coll Cardiol 54(14):1238–1245
Besler C, Luscher TF, Landmesser U (2012) Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med 4(4):251–268
Sorrentino SA, Besler C, Rohrer L et al (2010) Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 121(1):110–122
Griffin JH, Kojima K, Banka CL, Curtiss LK, Fernandez JA (1999) High-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein C. J Clin Invest 103(2):219–227
Drew BG, Duffy SJ, Formosa MF et al (2009) High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 119(15):2103–2111
Barter PJ, Rye KA, Tardif JC et al (2011) Effect of torcetrapib on glucose, insulin, and hemoglobin A1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Circulation 124(5):555–562
Siebel AL, Natoli AK, Yap FY et al (2013) Effects of high-density lipoprotein elevation with cholesteryl ester transfer protein inhibition on insulin secretion. Circ Res 113(2):167–175
Mortensen SP, Boushel R (2013) High-density lipoprotein: a new therapeutic target for glucose intolerance? Circulation 128(22):2349–2350
von Eckardstein A, Widmann C (2014) High-density lipoprotein, beta cells, and diabetes. Cardiovasc Res 103(3):384–394
Lehti M, Donelan E, Abplanalp W et al (2013) High-density lipoprotein maintains skeletal muscle function by modulating cellular respiration in mice. Circulation 128(22):2364–2371
Adams V, Besler C, Fischer T et al (2013) Exercise training in patients with chronic heart failure promotes restoration of high-density lipoprotein functional properties. Circ Res 113(12):1345–1355
Besler C, Heinrich K, Rohrer L et al (2011) Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest 121(7):2693–2708
Speer T, Rohrer L, Blyszczuk P et al (2013) Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 38(4):754–768
Zewinger S, Speer T, Kleber ME et al (2014) HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J Am Soc Nephrol JASN 25(5):1073–1082
Weichhart T, Kopecky C, Kubicek M et al (2012) Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol JASN 23(5):934–947
Silbernagel G, Schottker B, Appelbaum S et al (2013) High-density lipoprotein cholesterol, coronary artery disease, and cardiovascular mortality. Eur Heart J 34(46):3563–3571
Riwanto M, Rohrer L, Roschitzki B et al (2013) Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation 127(8):891–904
Huang Y, Wu Z, Riwanto M et al (2013) Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest 123(9):3815–3828
Zewinger S, Drechsler C, Kleber ME et al (2015) Serum amyloid A: high-density lipoproteins interaction and cardiovascular risk. Eur Heart J 36(43):3007–3016
Li XM, Tang WH, Mosior MK et al (2013) Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol 33(7):1696–1705
Singh IM, Shishehbor MH, Ansell BJ (2007) High-density lipoprotein as a therapeutic target: a systematic review. JAMA 298(7):786–798
Eckel RH, Jakicic JM, Ard JD, et al (2013) AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 129(25 Suppl 2):S76–S99
Cai M, Zou Z (2016) Effect of aerobic exercise on blood lipid and glucose in obese or overweight adults: a meta-analysis of randomised controlled trials. Obes Res Clin Pract 10(5):589–602. doi:10.1016/j.orcp.2015.10.010
Ghafouri K, Cooney J, Bedford DK, Wilson J, Caslake MJ, Gill JM (2015) Moderate exercise increases affinity of large very low-density lipoproteins for hydrolysis by lipoprotein lipase. J Clin Endocrinol Metab 100(6):2205–2213
Briel M, Ferreira-Gonzalez I, You JJ et al (2009) Association between change in high density lipoprotein cholesterol and cardiovascular disease morbidity and mortality: systematic review and meta-regression analysis. BMJ 338:b92
Soran H, Dent R, Durrington P (2017) Evidence-based goals in LDL-C reduction. Clin Res Cardiol. doi:10.1007/s00392-016-1069-7
Laufs U, Karmann B, Pittrow D (2016) Atorvastatin treatment and LDL cholesterol target attainment in patients at very high cardiovascular risk. Clin Res Cardiol Off J Ger Card Soc 105(9):783–790
Jun M, Foote C, Lv J et al (2010) Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet 375(9729):1875–1884
Keech A, Simes RJ, Barter P et al (2005) Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366(9500):1849–1861
Ginsberg HN, Elam MB, Lovato LC et al (2010) Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 362(17):1563–1574
Rajamani K, Colman PG, Li LP et al (2009) Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): a prespecified analysis of a randomised controlled trial. Lancet 373(9677):1780–1788
Fruchart JC, Sacks FM, Hermans MP (2010) Implications of the ACCORD lipid study: perspective from the Residual Risk Reduction Initiative (R(3)i). Curr Med Res Opin 26(8):1793–1797
Bruckert E, Labreuche J, Deplanque D, Touboul PJ, Amarenco P (2011) Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: a systematic review and meta-analysis. J Cardiovasc Pharmacol 57(2):267–272
Boden WE, Probstfield JL, Anderson T et al (2011) Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 365(24):2255–2267
Bruckert E, Labreuche J, Amarenco P (2010) Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis 210(2):353–361
Lavigne PM, Karas RH (2013) The current state of niacin in cardiovascular disease prevention: a systematic review and meta-regression. J Am Coll Cardiol 61(4):440–446
Landray MJ, Haynes R, Hopewell JC et al (2014) Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 371(3):203–212
Administration FaD (2016) Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. Fed Reg 81(74)
Administration FaD (2016) Withdrawal of approval of new drug applications for Advicor and Simcor. Fed Reg 81(74)
Fielding CJ, Fielding PE (1995) Molecular physiology of reverse cholesterol transport. J Lipid Res 36(2):211–228
Tall AR (1993) Plasma cholesteryl ester transfer protein. J Lipid Res 34(8):1255–1274
Johannsen TH, Frikke-Schmidt R, Schou J, Nordestgaard BG, Tybjaerg-Hansen A (2012) Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J Am Coll Cardiol 60(20):2041–2048
Li YY, Wu XY, Xu J, Qian Y, Zhou CW, Wang B (2013) Apo A5 -1131 T/C, FgB – 455G/A, -148C/T, and CETP TaqIB gene polymorphisms and coronary artery disease in the Chinese population: a meta-analysis of 15,055 subjects. Mol Biol Rep 40(2):1997–2014
Niu W, Qi Y (2015) Circulating cholesteryl ester transfer protein and coronary heart disease: Mendelian randomization meta-analysis. Circ Cardiovasc Genet 8(1):114–121
Hovingh GK, Ray KK, Boekholdt SM (2015) Is cholesteryl ester transfer protein inhibition an effective strategy to reduce cardiovascular risk? CETP as a target to lower CVD risk: suspension of disbelief? Circulation 132(5):433–440
Brousseau ME, Schaefer EJ, Wolfe ML et al (2004) Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med 350(15):1505–1515
Kastelein JJ (2007) Refocusing on use of cholesteryl ester transfer protein inhibitors. Am J Cardiol 100(11 A):n47–n52
Barter PJ, Caulfield M, Eriksson M et al (2007) Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357(21):2109–2122
Schwartz GG, Olsson AG, Abt M et al (2012) Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 367(22):2089–2099
von Eckardstein A, Rohrer L (2016) HDLs in crises. Curr Opin Lipidol 27(3):264–273
Morton RE, Izem L (2015) Modification of CETP function by changing its substrate preference: a new paradigm for CETP drug design. J Lipid Res 56(3):612–619
Parhofer KG (2015) Increasing HDL-cholesterol and prevention of atherosclerosis: A critical perspective. Atheroscler Suppl 18:109–111
Kingwell BA, Chapman MJ, Kontush A, Miller NE (2014) HDL-targeted therapies: progress, failures and future. Nat Rev Drug Discov 13(6):445–464
Nicholls SJ, Nissen SE, Kallend D, Winjgaard P, Borgman M (2016) Impact of infusion of an ApoA-I-Milano HDL mimetic on regression of coronary atherosclerosis in acute coronary syndrome patients: the MILANO-PILOT Study. American Heart Association Scientific Sessions 2016, New Orleans, Louisiana, USA, Nov 12–16. http://www.abstractsonline.com/pp8/#!/4096/presentation/58822. Accessed 21 March 2017
Nicholls SJ, Puri R, Wolski K et al (2016) Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: results of the Phase 2b, randomized, double-blind, multicenter, ASSURE Trial. Am J Cardiovasc Drugs Drugs Devices Other Interv 16(1):55–65
Tardif JC, Ballantyne CM, Barter P et al (2014) Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J 35(46):3277–3286
Siddiqi HK, Kiss D, Rader D (2015) HDL-cholesterol and cardiovascular disease: rethinking our approach. Curr Opin Cardiol 30(5):536–542
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
März, W., Kleber, M.E., Scharnagl, H. et al. HDL cholesterol: reappraisal of its clinical relevance. Clin Res Cardiol 106, 663–675 (2017). https://doi.org/10.1007/s00392-017-1106-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-017-1106-1