
Abstract
Exophiala jeanselmei is an opportunistic pathogenic black yeast growing in humid environments such as water reservoirs of air-conditioning systems. Because this fungal contaminant could be vaporized into the air and subsequently cause health problems, its monitoring is recommended. Currently, this monitoring is based on culture and microscopic identification which are complex, sometimes ambiguous and time-demanding, i.e., up to 21 days. Therefore, molecular, culture-independent methods could be more advantageous for the monitoring of E. jeanselmei. In this study, we developed a SYBR®green real-time PCR assay based on the internal transcribed spacer 2 from the 18S ribosomal DNA complex for the specific detection of E. jeanselmei. The selectivity (100 %), PCR efficiency (95.5 %), dynamic range and repeatability of this qPCR assay were subsequently evaluated. The limit of detection for this qPCR assay was determined to be 1 copy of genomic DNA of E. jeanselmei. Finally, water samples collected from cooling reservoirs were analyzed using this qPCR assay to deliver a proof of concept for the molecular detection of E. jeanselmei in environmental samples. The results obtained by molecular analysis were compared with those of classical methods (i.e., culture and microscopic identification) used in routine analysis and were 100 % matching. This comparison demonstrated that this SYBR®green qPCR assay can be used as a molecular alternative for monitoring and routine investigation of samples contaminated by E. jeanselmei, while eliminating the need for culturing and thereby considerably decreasing the required analysis time to 2 days.
Similar content being viewed by others


Introduction
Exophiala is a fungal genus containing numerous species recognized to be pathogenic (Nucci et al. 2001, 2002; Packeu et al. 2012; Woo et al. 2013a), and it is a member of the black yeasts. This genus is described as ubiquitous and has been isolated from diverse substrates (e.g., wood, soil, sludge, water, feed) (Dixon et al. 1980; Nishimura et al. 1987; Nucci et al. 2002). Within this genus, Exophiala jeanselmei is usually causing cutaneous or subcutaneous infections. However, E. jeanselmei infections are also frequently observed as a systemic infection or as a causal agent of cystic fibrosis in immunosuppressed patients (Nucci et al. 2001, 2002). Nevertheless, although limited data on the number of cases occurring are available and the epidemiology of airways infections caused by E. jeanselmei is poorly documented. Inside buildings, the way of contamination by this species is presumably linked to water reservoirs of air-conditioning units or pipings (Badali et al. 2012; Wang et al. 2001). As this species can cause health problems, its monitoring is needed.
Currently, monitoring of E. jeanselmei contamination of buildings is complicated and requires specific expertise. Indeed, in routine analysis, this monitoring is often based on culture, microscopic visualization, and visual counts (Anaissie et al. 2003). This approach depends on the growth of the culture, which for E. jeanselmei can take up to 21 days (Anaissie et al. 2003). In addition, this culturing is also influenced by the growth media chosen, the culture conditions such as temperature and humidity, or by competition between species (Pitkaranta et al. 2011; Vesper 2011). Moreover, E. jeanselmei is a complex species belonging to a group of morphologically difficult (or impossible) to differentiate species (Kawasaki et al. 2005; Zeng and De Hoog 2008; Zeng et al. 2007), i.e., the Exophiala spinifera clade (De Hoog et al. 2003), which groups among others E. jeanselmei, Exophiala exophialae, Exophiala lecanii-corni, and Exophiala xenobiotica (Kawasaki et al. 1999; Wang et al. 2001; Woo et al. 2013a; Zeng and De Hoog 2008). Also, Exophiala dermatitidis, considered as an important and much studied pathogen inside the Exophiala genus, is morphologically similar to E. jeanselmei (Kawasaki et al. 1990, 2005; Masuda et al. 1989). A molecular approach could bring a solution to these bottlenecks in classification and identification of E. jeanselmei (Haase et al. 1995; Hee and Yoon 2002; Kawasaki et al. 1990).
Actually, the use of molecular tools (such as polymerase chain reaction (PCR), real-time polymerase chain reaction (qPCR) and sequencing) is being increasingly used for the detection, monitoring and the identification of fungi in general (Chemidlin Prevost-Boure et al. 2011; Hospodsky et al. 2010; Melkin et al. 2004; United States Environmental Protection Agency 2015). For example for the genus Exophiala, some PCR (Nagano et al. 2008; Najafzadeh et al. 2013; Sudhadham et al. 2010), qPCR and other molecular tools have been developed for the detection and identification of E. dermatitidis (Wang et al. 2013). Most often, these molecular tools target the 18S rDNA regions and especially the internal transcribed spacer 1 and 2 (ITS1 and ITS2). This ITS region is fully documented and a vast amount of data is available for in silico analysis. In 2012, Schoch and co-workers proposed to define these internal regions as a part of the barcode marker for fungi (Schoch et al. 2012). For E. jeanselmei, these ITS regions offer the possibility to develop molecular tools to discriminate the species, even inside the E. spinifera clade (Wang et al. 2001; Woo et al. 2013a; Zeng and De Hoog 2008). Despite these previous studies investigating the E. jeanselmei genotype (Badali et al. 2012; Kawasaki et al. 2005, 1999; Woo et al. 2013a), molecular tools for the specific detection of this fungus are however still poorly documented.
Based on ITS1 and ITS2, diagnostic molecular tools for E. jeanselmei infections were developed using PCR and classical Sanger sequencing (Nagano et al. 2008; Najafzadeh et al. 2013; Packeu et al. 2012; Wang et al. 2001; Woo et al. 2013b). But although these approaches have proven their effectiveness, they are not really adapted to routine analysis of many environmental samples due to the low specificity of the universal primers used in the PCR assay the time needed for the analysis and the cost. Indeed, environmental samples could contain more than one species. Then, in these samples, the use of universal primers would produce a mix of amplicons which would be poorly discriminated with Sanger sequencing. This problem could be solved by next-generation sequencing (NGS) tools but the associated costs and expertise needed (especially in data analysis) might be too high for routine analysis.
qPCR is an alternative approach offering specificity and a reduction in analysis time and cost, especially when many samples need to be analyzed. Also, a qPCR instrument might be more in the reach of a routine laboratory, as compared to a DNA sequencing instrument. Today, despite the development of qPCR tools for the detection of several fungal contaminants such as the monitoring of airborne molds from outdoor and indoor environment (Hospodsky et al. 2010; Libert et al. 2015; Nagano et al. 2008; Vesper 2011), no qPCR assay specific to E. jeanselmei yet exists.
In this context, this paper describes a SYBR®green qPCR assay for the specific detection of E. jeanselmei. To assess the performance of the developed qPCR assay, the selectivity, sensitivity, and efficiency were evaluated. As previously discussed (Libert et al. 2015), no harmonized guidelines exist for this performance assessment of qPCR assays for fungal detection. Therefore, we followed a similar approach as the one previously reported for Aspergillus versicolor (Libert et al. 2015) which is based on the guidelines developed for the qPCR detection of genetically modified organisms (GMO) (Broeders et al. 2014) and foodborne pathogens (Barbau-Piednoir et al. 2013) and the minimum information for publication of quantitative real-time PCR experiment (MIQE) proposed by Bustin et al. in 2009 (Bustin et al. 2009). Finally, a proof of concept for the detection of E. jeanselmei in environmental samples was provided, comparing the routine protocol based on classical techniques involving culturing steps, and the molecular qPCR method developed and evaluated in this study. Therefore, this study offers a new molecular tool based on SYBR®green chemistry which could be used as a routine protocol for the detection and/or the monitoring of E. jeanselmei in environmental samples, by all actors concerned.
Materials and methods
Fungal strains
Table 1 lists all the fungal species (Acrenonium strictum, Alternaria alternata, Aspergillus fumigatus, Cladosporium cladosporioides, Cladosporium herbarum, Cladosporium sphaerospermum, E. dermatitidis, E. exophialae, E. jeanselmei, E. lecanii-corni, E. spinifera, E. xenobiotica, Penicillium chrysogenum, Stachybotrys chartarum, and Ulocladium botrytis) and strains (19 strains in total) used in this study. They were all purchased from the BCCM/IHEM collection (Scientific Institute of Public Health in Brussels, Belgium).
Culture conditions
The fungal strains were grown as described previously (Libert et al. 2015). Briefly, the fungal strains were spiked into a S10 Sabouraud liquid medium (Biorad, Temse, Belgium) and were grown at 25 °C with constant agitation between 3 to 21 days according to the species’ growth conditions.
DNA extraction
DNA of the fungal cultures was prepared as previously reported (Libert et al. 2015). Briefly, after the incubation time, 300 mg of wet sample were transferred to cryotubes containing 0.25 ml of acid-washed glass beads (Sigma Aldrich, Diegem, Belgium) and put at −80 °C during 40 min and freeze-dried overnight with a freeze-dryer Epsilon 1-6D (Martin Christ, Osterode am Harz, Germany). Freeze-dried fungi were then beat-beaten with a Mini bead beater (Biospec Products, OK, USA) during 1 min at maximal speed.
Subsequently, the total DNA was extracted with an adapted phenol chloroform (24:1) protocol (Ashktorab and Cohen, 1992) and purified with the Qiagen CTAB genomic Tip-20 kit (Qiagen Benelux—B.V., KJ Venlo, The Netherlands) according to the manufacturer’s protocol. A 100-μl Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium) was used to elute the DNA. The DNA integrity was verified on a 2 % agarose gel. The DNA concentration and purity were evaluated with a Nanodrop® 2000 (Thermo Scientific, Wilmington, USA).
Primer design
First, a collection of publicly available 18S rDNA sequences from E. jeanslemei strains and other closely related species (namely E. lecanii-corni and E. spinifera) and from the common water contaminant A. fumigatus (Heinemann et al. 1994; Parat et al. 1996), was made (NCBI, GenBank). The following sequences were included: for E. jeanselmei: AB531492.1/AF549447.1/AF050271.1/AJ866273.1/AY857530.1/AY163553.1/AY163549.1/AY163550.1 AY163552.1/AY163556.1/DQ836791.1/DQ836793.1/DQ836795.1/JN625228.1/JX192603.1/JX473278.1/EF025410.1/EF025411.1/EF025412.1/EU910261.1/JX473276.1; for A. fumigatus: KC411924.1/KC237295.1/KC237291.1/KC237292.1/KC142152.1/HE864321.1/KC119199.1/KC119200.1/JX944178.1/JX944118.1; for E. lecanii-corni: GQ426959.1/GQ426975.1/GQ426980.1/JN675374.1/JN675375.1/JX473283.1/JX473285.1/JX681038.1/JX681039.1/JX681040; for E. spinifera: AB025891.1/AB025892.1/AB025857.1/AB025876.1/AY156966.2/AY156970.1/EF551539.1/EF551459.1/KC952672.1/NR_111131.1. These sequences were aligned with the MegAlign software V10.0.1 (Lasergene, Madison, USA) to identify the ITS 1 and ITS 2 sequence regions of interest wherein different primer pairs were designed using the Primer 3V.0.4 software (http://bioinfo.ut.ee/primer3-0.4.0/) (Untergasser et al. 2007). Primer dimers and secondary structure formation was evaluated and predicted during the design with Primer3. The wprimersearch software (https://wemboss.uio.no/wEMBOSS/) (Sarachu and Colet 2005) was used to perform an in silico specificity test, allowing to select the primer pair that only amplifies the target sequences. Also, BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to evaluate the specificity of the primers.
Qualitative SYBR®green qPCR assay
The qPCR assay for the detection of E. jeanselmei (Ejeanselmei_ITS qPCR method) was performed as previously described for A. versicolor (Libert et al. 2015), using the SYBR®green chemistry and a real-time PCR IQ5 ™ system (Biorad, Temse, Belgium). All the primers used in this study were synthetized by Eurogentec (Liège, Belgium).
Briefly, the reaction mix (25 μl final volume) contained 12.5 μl of 2× SYBR®green PCR Mastermix (Diagenode, Liège, Belgium), 0.25 μl of Ej_ITS forward and reverse primers (0.2 μM) (Table 2) and 7 μl of Gibco® DNase, RNase, protease free water (Life Technologies, Gent, Belgium). To this mix, 5 μl of genomic DNA (gDNA) at 200 theoretical genomic copy numbers per microliter was added. The number of genomic DNA copies was calculated according to the formula presented below i.e.,
with C n = genomic copy number; m = the amount of gDNA (grams) and determined by Nanodrop®2000 (Thermo Scientific, Wilmington, USA); Ac the Avogadro’s constant (Mohr et al. 2008). M w = base pair mean molecular weight (649 Da) and G s = Genome size (expressed in bp) of E. jeanselmei = 30,000,000. Because no information on the genome size of E. jeanselmei is currently publicly available, we used an estimation based on the average of the genome size of E. dermatitidis, E. xenobiotica, E. spinifera (Broad Institute 2015). We are also aware that some strain-dependent deviations may exist.
The optimization of the qPCR conditions was performed with the E. jeanselmei strain BCCM/IHEM 4740. Therefore, this species was considered as a reference and used as a positive control added in each run performed in this study.
In each run, a no template control (NTC) was included for the analysis whereby the DNA template was replaced by ultrapure water in the reaction mix. This NTC aimed at verifying that no contamination occurred and that no primer dimers were formed.
The following thermal cycling conditions were used for all runs: 1 cycle of 95 °C for 10 min (Taq activation), followed by 40 amplification cycles of 15 s at 95 °C (denaturing step) and 60 °c for 1 min (annealing and extension step). Subsequently, a melting curve was made with a gradual increase of temperature of 0.5 °C/6 s from 55 to 95 °C during 15 min. The Biorad IQ 5 software V. 2 (Biorad, Temse, Belgium) automatically determined the threshold level for the reaction.
Inhibition test (pure cultures)
Because PCR inhibitors (e.g., co-extracted substances or RNA contaminations) could affect the amplification, the validation results and also the detection of low amount of the targeted DNA, it is important to verify that all of these substances were removed during the DNA extraction step. This was done using an inhibition test. The workflow of this inhibition test was based on that proposed in 2012 by Broeders et al. for the assessment of absence of inhibitors in DNA extracts. Briefly, gDNA was extracted independently from two pure cultures of E. jeanslemei strain BCCM/IHEM 4740. Afterwards, a calibration curve was made based on the analysis of each set of gDNA, diluted 10, 100, 1000, and 5000 fold, with the Ejeanselmei_ITS SYBR®green assay. Two criteria exist to assess the absence of inhibitors in the DNA extracts; i.e., the slope of the calibration curve which should be between −3.6 and −3.1; and the difference (Δ C q) observed between the experimentally obtained C q values and an extrapolated Cq obtained by the regression of the Cq from the undiluted sample (Broeders et al. 2012). In the absence of inhibition, the Δ C q should be ≤0.50 for each dilution (Broeders et al. 2012, European Network of GMO Laboratories 2015).
Sequencing
The identity of the strains of E. jeanselmei IHEM 4740, IHEM 4741, IHEM 22665, and of E. spinifera IHEM 20752 that resulted in a specific amplicon in the qPCR assay was confirmed based on the sequence of their ITS 1 and ITS 2 regions. Hereto, a dideoxy sequence analysis with the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Life Technologies, Gent, Belgium) and an ABI3130xl Genetic Analyzer apparatus (Applied Biosystems, Life Technologies, Gent, Belgium) was used according to the manufacturer’s recommendations. Primers ITS1F (Gardes and Bruns 1993) and ITS4 (White et al. 1990) were first used to amplify the ITS 1 and ITS 2 regions. Primers ITS1 and ITS2 (White et al. 1990) were used for the subsequent sequencing reaction. The consensus sequences of each of the targeted regions (based on the forward and reverse sequence of each target region) were aligned with the MEGA v6.06 (http://www.megasoftware.net/) (Tamura et al. 2013) software and visualized with the CLC sequence viewer v7.0.2 (Qiagen Benelux—B.V., KJ Venlo, The Netherlands). Using BLASTn (http://blast.ncbi.nlm.nih.gov/), the amplification of the targeted DNA regions and their identity was confirmed by comparing them to the sequences available in the NCBI database.
Theoretical T m calculation
The online tool from IDT (http://eu.idtdna.com/calc/analyzer) (IDT, Leuven, Belgium) with the consensus sequences obtained through sequencing as input and under the PCR conditions described above, was used to in silico calculate the theoretical T m of the amplicon obtained in the Ejeanselmei_ITS assay.
Ejeanselmei_ITS assay performance assessment
For the performance assessment of the qPCR assay, the approach as previously outlined by Libert et al. (2015), which on its own is based on the study of Barbau-Piednoir et al. (2013), was followed. Different parameters of the qPCR method were evaluated, i.e., the selectivity (based on inclusivity and exclusivity), the PCR efficiency, the limit of detection (sensitivity test), and the repeatability.
Selectivity test
The selectivity test was composed of a preliminary and a larger selectivity test.
For the preliminary selectivity test, the target species (E. jeanselmei IHEM 4740) and one non-target species (E. dermatitidis IHEM 9780) were used, both at 15,000 theoretical copies of gDNA. The amplicon obtained for the E. jeanselmei strain IHEM 4740, which was taken as a reference in the performance assessment study, was confirmed by sequencing analysis.
The larger selectivity test aimed at evaluating the inclusivity (i.e., the selected primers should amplify the DNA of each tested strain from the target species) and the exclusivity (i.e., DNA of non-target species close to the target or described to frequently occur in the same environment as the target species should not be amplified by the selected primers, with a specific T m) of the Ejeanselmei_ITS qPCR method, using the selected primers and the qPCR conditions as described above. So, a result has been considered as positive when the sample is amplified and the observed T m corresponds to the T m defined in silico for the target. If these two conditions are not observed, the result has been considered as negative.
For the inclusivity, three target strains (i.e., E. jeanselmei) from the BCCM/IHEM collection were selected. The experimental design of the exclusivity test included 14 non-target strains i.e., 5 species closely related to E. jeanselmei (i.e., E. dermatitidis, E. exophialae, E. lecanii-corni, E. spinifera and E. xenobiotica) (Zeng and De Hoog 2008) and 9 other common species in wet environment as described by Heinemann et al. (1994) (i.e., A. strictum, A. alternata, A. fumigatus, C. cladosporioides, C. herbarum, C. sphaerospermum, P. chrysogenum, S. chartarum and U. botrytis).
The conditions described above were used for each qPCR run using a total of 1000 theoretical copies of gDNA per reaction (evaluated for each target with its own corresponding genome size).
Based on the selectivity test, the false positives ratio (FPR) and false negatives ratio (FNR), the sensitivity and the selectivity values (%) were calculated according to the formulas presented by Blakely and Salmond (2002), i.e.,
PCR efficiency estimation
The qPCR analysis of a serial dilution, in duplicate, of gDNA (1000, 500, 100, 50, 10, 5, 2, and 1 theoretical copy number of gDNA obtained by two independent extractions) of E. jeanselmei IHEM 4740 was used to assess the linearity of the SYBR®green qPCR assay. Based on this analysis, two parameters can be evaluated, i.e., the coefficient of determination (R 2) and the PCR efficiency. R 2 is an indicator of the correlation of the data regarding the linear regression curve. The PCR efficiency (E) calculation was previously described by Rutledge and Cote (2003). As previously described for A. versicolor (Libert et al. 2015), according to the most recent guidelines developed for GMO detection with qPCR SYBR®green (European Network of GMO Laboratories 2015), the R 2 and amplification efficiency are not applicable to qualitative methods. However, a R 2 ≥ 0.98 and a PCR efficiency ranging between 80 and 120 % have previously been indicated as performance criteria for the validation of qualitative qPCR methods (Broeders et al. 2014).
Limit of detection (sensitivity test)
The limit of detection (LOD) is defined as the lowest concentration of an analyte which is detected with a probability of 95 % (Barbau-Piednoir et al. 2013; Broeders et al. 2014). Six dilutions (i.e., 100, 50, 10, 5, 2, 1, 0.5, 0.2 and 0.1 theoretical copies of gDNA) of genomic DNA of E. jeanselmei IHEM 4740 were tested in six independent runs (using the qPCR conditions described above), each with six repetitions, to estimate the LOD of the Ejeanselmei_ITS assay. The LOD should be below 25 copies according to definition of minimum performance requirements for analytical methods of GMO testing (European Network of GMO Laboratories 2015; Libert et al. 2015).
PCR repeatability
This repeatability limit (r) is defined as the maximal difference of two results obtained under identical experimental conditions with a probability of 95 % (Barbau-Piednoir et al. 2013). The experimental design used for the LOD evaluation (see above) was also applied for the evaluation of the r value of the Ejeanselmei_ITS qPCR method.
As described previously (Barbau-Piednoir et al. 2013; Libert et al. 2015), the relative standard deviation of the repeatability (RSDr) was calculated as the absolute value of the coefficient variation (%). For these criteria, there is no limit fixed for qualitative qPCR methods (European Network of GMO Laboratories 2015). The RSDr, evaluated for the C q values, should be below 25 % for all dilutions above the LOD for quantitative methods (Barbau-Piednoir et al. 2013; Broeders et al. 2014).
Proof of concept: environmental testing
Sampling
The sampling method for air-conditioning systems was previously described by Nolard et al. (2004). Briefly, 1 l of water was collected in a sterile Duran bottle with a vacuum pump at a distance between 1 and 5 cm of the bottom of the tank of the air-conditioning system. In total, eight tanks from different air-conditioning systems were sampled. The water samples were stored at 4 °C until their analysis.
Classical analysis: culture, microscopic analysis and cell counting
Under laminar flow, a first part of the water sample was diluted 10 and 100 fold and each dilution, as also an undiluted aliquot of 1 ml, was poured into an empty petri dish. Afterwards, malt extract agar (MEA) chloramphenicol liquid medium (Biorad, Temse, Belgium) was put in the petri dish and the plate was kept at room temperature till the medium had solidified. Then, the plate was incubated at 25 °C for 21 days. A first microscopic analysis was made after 5 days of incubation in order to determine the fastest growing species and a second after 21 days of incubation to determine other species such as the Exophiala species.
Molecular analysis: DNA extraction and qPCR analysis
Additionally, aliquots of 15 ml were taken of mixed water samples used for the classical analysis described above. After 15 min of centrifugation at 5000×g, DNA from aliquots was extracted with the DNA extraction protocol used for pure cultures (see above). The DNA concentration and purity was evaluated with a Nanodrop® 2000 (Thermo Scientific, Wilmington, USA). The qPCR reactions were performed on 5 μl of eluted DNA i.e., 10 % of the total DNA eluted from water samples corresponding to the amount of DNA mentioned in the Table 5.
PCR inhibition test (spike test)
To verify that no inhibition from the anti-fungal chemistry used to clean the air-conditioning occurred on the PCR amplification, as part of these chemicals might have been retained in the DNA extract, 1000 theoretical copy numbers of E. jeanselmei gDNA (IHEM 4740) were spiked into 5 μl of DNA extract from sample 3 (considered as an environmental no template control, as by classical analysis, no E. jeanselmei was detected) and analyzed with the Ejeanselmei_ITS qPCR assay. To determine whether inhibition occurred during the qPCR reaction, the obtained C q value with the spiked sample was compared to the C q obtained with 1000 copy numbers of gDNA of E. jeanselmei IHEM 4740 in pure water (Table 1).
Results
Design and selection of qPCR primer pair
For the design of the E. jeanselmei-specific qPCR primers, all the E. jeanselmei 18S rDNA sequences that were publicly available at that time were used. This selection of sequences was extended with those available for the closely related species E. lecanii-corni and E. spinifera, which are belonging together with E. jeanselmei to the E. spinifera clade (Wang et al. 2001; Zeng and De Hoog 2008), and those available for A. fumigatus, a common species observed in water (Heinemann et al. 1994; Parat et al. 1996). Based on an alignment of these sequences, two couples of E. jeanselmei primers targeting a conserved region within the ITS 2 region of the 18S rDNA of E. jeanselmei were designed (Table 2, Fig. 1). It was however not possible to design primers in this region that were exclusively specific to E. jeanselmei, as part of the primers were also conserved in E. spinifera, with some nucleotide variations especially in the forward primer annealing site. The impact of this sequence conservation was addressed during the specificity test as part of the performance assessment (see below).

Alignment of region of ITS sequences of E. jeanselmei, E. lecanii-corni, E. spinifera and A. fumigatus strains, and of selected primers This alignment was made using publicly available ITS sequences of E. jeanselmei, E. lecanii-corni, E. spinifera and A. fumigatus extended with ITS sequences from the strains from the BCCM/IHEM collection used during the performance assessment of the qPCR assay (indicated with IHEM prefix) and with the primers designed in this study (Ej_ITS_f and Ej_ITS_r). Because no nucleotide variation was detected for each of the public sequences used, only one sequence (*) was introduced for this alignment, as a representative for that species. Consensus (last line of the alignment) corresponds to a consensus sequence defined by the software. The conservation level among each sequence (0 to 100 % of conservation) is represented by the pink rectangles at the bottom of the figure. The alignment was made with MEGA 6.06 software (http://www.megasoftware.net/) and visualized with CLC sequence viewer 7 (Qiagen Benelux—B.V., KJ Venlo, The Netherlands)
Preliminary specificity tests with these two primer couples were performed on the ITS 2 regions of E. jeanselmei (IHEM 4740) and E. dermatitidis (IHEM 9780) as a negative control, showing that only the ITS 2 region of E. jeanselmei was amplified (data not shown). Finally, based on the best combination of primers in terms of amplification efficiency (data not shown), the Ej_ITS_f and Ej_ITS r primers (Table 2) were selected for the detection of E. jeanselmei in this qPCR assay (Ejeanselmei_ITS assay).
In order to verify that no inhibition occurred during the PCR amplification, an inhibition test was performed with the selected primers. The slope of the calibration curve obtained with the two sets of gDNA dilutions was determined to be −3.35 (Fig. 2). This slope is in the range of −3.1 to −3.6 as recommended by the ENGL (2011). Δ C q values were calculated for each dilution, i.e., 0.27 for the 5000-fold dilution, 0.43 for the 1000-fold dilution, 0.02 for the 100-fold dilution and 0.33 for the 10-fold dilution. According to the acceptance criteria from the ENGL for GMO for the assessment of the absence of inhibitors in the DNA extracts (ENGL 2011), no inhibition occurred.

Calibration curve (inhibition test). Data were obtained with four replicates of four dilutions (5000, 1000, 100, 10 fold) of gDNA of E. jeanselmei (IHEM 4740). The dotted line corresponds to the trend line
Ejeanselmei_ITS assay performance assessment
To evaluate the quality of this Ejeanselmei_ITS assay, a performance assessment was done as previously described for A. versicolor (Libert et al. 2015), according to the guidelines defined for the validation of qPCR detection and identification methods in other fields (Barbau-Piednoir et al. 2013; Broeders et al. 2014). The following criteria were evaluated: the selectivity of the primers, the LOD, the PCR efficiency, the dynamic range, and the Ejeanselmei_ITS assay repeatability.
Selectivity of the Ejeanselmei_ITS qPCR assay
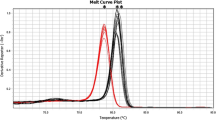
First, an inclusivity test was performed with DNA of 3 E. jeanselmei strains from the BCCM/IHEM collection. We have included all in this collection available E. jeanselmei strains originating from water. DNA of each of the selected strains (E. jeanselmei BCCM/IHEM 4740, BCCM/IHEM 4741, BCCM/IHEM 22665) was amplified (3/3) with a C q of, respectively, 20.86 ± 0.22, 21.81 ± 0.43 and 23.57 ± 0.38 for 1000 copies of gDNA (Table 1). The melting curve analyses showed T m values between 79.25 ± 0.50 and 79.75 ± 0.25 (Table 1, Fig. 3).

Melting curves obtained with the Ejeanselmei_ITS qPCR assay for the E. jeanselmei pure strains listed in Table 1. The melt curves were obtained with the Biorad IQ 5 software V. 2 (Biorad, Temse, Belgium). The X-axis shows the temperature (°C). The Y-axis presents the inverse of the first derivate of the best-fitted curve of the measured fluorescence decrease. The gray curves correspond to the E. jeanselmei listed in Table 1. The blue flat curves represent the NTC
The sequence of these BCCM/IHEM strains matches perfectly with the ones publicly available for the corresponding region and all amplicons showed 100 % identity (Fig. 1). The obtained T m for each amplicon corresponds to the theoretical T m, which was calculated to be 79.50 °C. No false negatives were obtained.
Then, an exclusivity test was performed on DNA of non-target species (i.e., A. strictum, A. alternata, A. fumigatus, C. cladosporioides, C. herbarum, C. sphaerospermum, E. dermatitidis, E. exophialae, E. lecanii-corni, E. spinifera, E. xenobiotica, P. chrysogenum, S. chartarum and U. botrytis). These species are closely related to E. jeanselmei and/or are occurring in the same environment (i.e., water reservoir) and/or in indoor environment (Al-gabr et al. 2014; Anaissie et al. 2003; De Hoog et al. 2003; Heinemann et al. 1994; Kawasaki et al. 1999). No false positives were observed (Table 1). Indeed, with this Ejeanselmei_ITS qPCR assay, except for DNA extracted from the E. spinifera IHEM 20752 strain, no DNA from non-targeted species was amplified. The amplification of E. spinifera was not unexpected based on the in silico analysis including the alignment (Fig. 1), which included all the publicly available ITS sequences of E. jeanselmei and E. spinifera. Because the sequence between the two primers was found to be identical for all the sequences available for one species, only one sequence for each species has been represented in the figure.
However, the E. spinifera amplification should be considered as a true negative results based on the SYBR®green characteristic. Indeed, for a same copy number estimation (i.e., 1000 theoretical genomic copy number), the obtained Cq value for E. spinifera was 26.04 ± 0.05 (Table 1) and the T m was 78.50 °C (Table 1, Fig. 4), which are different from the ones obtained for E. jeanselmei. Based on these amplification results and T m values, no false negative values were observed, i.e., FPR and FNR values of 0 % were obtained.

Melting curves obtained with the Ejeanselmei_ITS qPCR assay for the E. jeanselmei and E. spinifera pure strains listed in Table 1. The melt curves were obtained with the Biorad IQ 5 software V. 2 (Biorad, Temse, Belgium). The X-axis shows the temperature (°C). The Y-axis presents the inverse of the first derivative of the best-fitted curve of the measured fluorescence decrease. The blue flat curves represent the NTC
To explain these differences, amplicons of E. jeanselmei and E. spinifera were sequenced and aligned (Fig. 1). The amplicon of E. spinifera differs from that obtained for E. jeanselmei with 11 nucleotides, of which 2 in the annealing site of the forward primer (Fig. 1). These results were expected and met those obtained during the primer design (Fig. 1). These nucleotide differences explained the difference in the observed T m for the two species (Table 1). The BLAST analysis of the ITS 1 and ITS 2 regions confirmed the IHEM 20752 as E. spinifera with 97 % of identity.
Based on these results, a sensitivity of 100 % and a specificity of 100 % were observed. An NTC was included in each assay to verify that no contamination occurred during the qPCR assay preparation (preparation of the mixes, filling of the qPCR plates). In none of the assays, the NTC resulted in a signal. This also showed that no dimerization of primers occurred during the analysis, as predicted during the in silico test (Fig. 3).
Limit of detection (sensitivity test) and PCR repeatability
The limit of detection (LOD) of the Ejeanselmei_ITS assay, based on 6 independent runs with a total of 18 repetitions, was determined to be one theoretical copy number (Tables 3 and 4) (C q = 34.86 ± 0.90). The r and RSDr were 3.45 and 9.73 % respectively for this assay.
Dynamic range and PCR efficiency
A serial dilution of 1000 to 0.1 theoretical genomic copy numbers of E. jeanselmei permitted to define the dynamic range and PCR efficiency of the Ejeanselmei_ITS assay. A linear model with a R 2 of 0.9977 and an efficiency of 95.5 % were obtained with this SYBR®green assay (Fig. 5).

Coefficient of determination and PCR efficiency of Ejeanselmei_ITS qPCR assay Data are obtained with four replicates for each concentration i.e., 1000 to 0.1 theoretical copy number of genome of E. jeanselmei (IHEM 4740)
Environmental testing
To evaluate the performance of this E. jeanselmei_ITS method on real-life samples, a test was performed on different water samples collected from air-cooled systems in office buildings. This proof of concept allows to test the Ejeanselmei_ITS assay on environmental samples and to compare this method of detection with a classical routine analysis method (Table 5).
The detection method used in routine (culture, counting, and microscopic identification) allowed to detect E. jeanselmei in three water samples with an amount ranging between 2 CFU/ml and 4 CFU/ml (Table 5). All of these samples contained one or two other contaminants frequently recovered from water reservoirs (Table 5) i.e., Acremonium sp., A. fumigatus and a sterile mycelium (undetermined).
The results from the Ejeanselmei_ITS assay were in accordance with those of the classical method for the detection of E. jeanselmei. Indeed, this qPCR assay gave positive signals for the same water samples where E. jeanselmei was detected using the classical methods, i.e., sample nos. 1, 2, and 3 (Table 5). The obtained C q values were all ranging around 35 and the T m values around 79.5 °C, as expected. No signal was observed for the samples where no E. jeanselmei was detected on plate. The spike test confirmed that no inhibition from potentially remaining anti-fungal chemistry products occurred during the qPCR analysis as a C q of 22.50 ± 0.68 was obtained which corresponds to the value shown in Table 1 for E. jeanselmei IHEM 4740.
Discussion
E. jeanselmei is frequently found inside buildings in canalizations, reservoirs of drinking water, non-drinking water reservoirs for air-conditioning. As this species is an opportunistic pathogen causing health problems (Al-gabr et al. 2014; Nucci et al. 2001; Nucci et al. 2002; Zeng et al. 2007), its monitoring is important. Classical approaches used for the detection of this pathogenic fungus are difficult because of morphologically closely related species and are moreover time consuming. Indeed E. jeanselmei requires up to 21 days of incubation at 35 °C before microscopic identification (Najafzadeh et al. 2013; Nishimura et al. 1987; Nolard et al. 2004). This is why molecular analysis, such as the in this study developed Ejeanselmei_ITS assay, could be attractive for a more efficient monitoring and diagnosis of a contamination by E. jeanselmei. The real-time PCR analysis, and especially the SYBR®green qPCR chemistry, offers the advantage to be fast, sensitive, instrumental-wise more accessible and more cost-effective than other molecular techniques like direct sequencing, especially when many samples need to be analyzed. Indeed, the SYBR®green chemistry is cheaper than the chemistry used for sequencing, with a cost of less than 1 euro per reaction. In comparison with the classical tools, this technique is more expensive. However, the time saving (2 days against 21 for the classical approach) is a key advantage in terms of monitoring and diagnosis.
Another advantage is that the specificity is based on a primer couple and on the associated T m value of the generated amplicon, thereby avoiding a sequencing step which is needed when using classical PCR approaches (Klein, 2002). In this assay, the primers were designed in the ITS 2 region from the 18S rDNA complex.
Because no guidelines nor norms exist for the development of qPCR methods for the detection of fungi, the performance assessment flow was based on guidelines and recommendations given for GMO detection and foodborne pathogen (Barbau-Piednoir et al. 2013; Broeders et al. 2014), as was previously done for a qPCR assay for the specific detection of A. versicolor (Libert et al. 2015). To evaluate the performance of this Ejeanselmei_ITS SYBR®green assay, the selectivity, PCR efficiency, dynamic range, sensitivity and repeatability parameters were investigated.
First, the inclusivity tests revealed that the Ejeanselmei_ITS assay detects all the tested E. jeanselmei strains. Nevertheless, between these two amplified species, a variation of approximately 3 C q was observed. As shown by sequencing analysis (Fig. 1), no variation occurred between the amplicon obtained for each of these strains, and the sequence between the primers matched 100 % with the corresponding one retrieved for all publicly available E. jeanselemi ITS 2 sequences. This variation in the C q could however be explained by a dissimilarity of the 18S rDNA copy number which is known to have an interspecies and an intraspecies variability (Black et al. 2013; Corradi et al. 2007; Iwen et al. 2002; Schoch et al. 2012), and which was also suggested for A. versicolor (Libert et al. 2015). Because no variation rates for the 18S rDNA copy number are known for E. jeanselmei or closely related species, this Ejeanselmei_ITS assay is a qualitative qPCR. To develop a quantitative tool based on the ITS sequence, the range of variation of the 18S rDNA copy number should be determined for each targeted strain in order to extrapolate the quantity expressed in genome copy numbers.
The exclusivity test, the second step of the specificity test, showed that no non-targeted strains from indoor environment, including the air-conditioning system, were detected with the Ejeanselmei_ITS assay. This demonstrates the good specificity of the Ejeanselmei_ITS assay. For none of the closely related and morphologically difficult-to-discriminate species was the DNA was amplified with the Ejeanselmei_ITS assay, except for E. spinifera. However, despite this amplification, the melting curve analysis allowed the discrimination between E. spinifera and E. jeanselmei, as the T m differs with 1 °C between the two species (T m E. spinifera = 78.50 °C, T m E. jeanselmei = 79.50 °C) (Fig. 4). This difference in T m can be explained by the interspecies diversity observed in the ITS region of species from the E. spinifera clade which includes E. jeanselmei (Wang et al. 2001; Woo et al. 2013a; Zeng and De Hoog 2008) (Fig. 1). The post-analysis based on the melting temperature to discriminate a species is currently used with the SYBR®green chemistry also in other fields to discriminate two different strains amplified by the same couple of primers (Barbau-Piednoir et al. 2013), without the need for sequencing confirmation.
In the future, high-resolution melting (applied to the Ejeanselmei_ITS assay) might be used to obtain an even more pronounced discrimination of these two Exophiala species based on the T m of the amplicon, if needed. Nevertheless, the Ejeanselmei_ITS assay allows to discriminate between species that are frequently confused with E. jeanselmei with culture-dependent analysis and some molecular methods like classical PCR.
Then, efficiency and PCR linearity were evaluated for this SYBR®green assay. To evaluate these parameters, the strain E. jeanselmei IHEM 4740 was selected as the reference strain. Because the Cq values obtained for these strains were the highest among those for the strains tested, the results obtained for these parameters correspond to the worst case scenario for this assay. With an efficiency of 95.5 %, this SYBR®green assay is efficient according to the criteria defined for qualitative analysis of GMO (Broeders et al. 2014). Moreover, the obtained R 2 value (0.9977) shows the linearity and the accuracy of this qPCR SYBR®green assay.
Furthermore, with an LOD defined at one theoretical genomic copies per reaction, this Ejeanselmei_ITS is sensitive and well within the criteria put forward by the GMO community (European Network of GMO Laboratories 2015). This Ejeanselmei_ITS assay is also repeatable with r (3.45) and RSDr (9.73 %) values below the limits defined by the guidelines used in this study (Barbau-Piednoir et al. 2013; Broeders et al. 2014). Because no information was available on the genome size of E. jeanselmei, the copy number estimation was done with an average of the genome size based on the size of three Exophiala species (i.e., E. dermatitidis, E. spinifera, and E. xenobiotica). Therefore, this estimation should be reviewed when the real genome size of E. jeanselmei will be available. Nevertheless, if the genome size of E. dermatitidis (i.e., 26.35 Mb) is used as worst case scenario to define the copy number of gDNA of E. jeanselmei, the underestimation of the copy number at LOD is evaluated to be 1 % only, which should not influence drastically our results.
These performance assessment tests were done using DNA extracted from pure strains from the BCCM/IHEM collection, which are well characterized. In order to test this Ejeanselmei_ITS assay under environmental conditions, water samples from air-conditioning were submitted to our qPCR method. The obtained results were compared with those obtained with the routine protocol based on the classical analysis method (i.e., culture, microscopic visualization, and counting). As similar results were obtained for both classical and qPCR method, this proof of concept demonstrated that the Ejeanselmei_ITS assay could be useful for the monitoring of E. jeanslemei. Concerning the time period to obtain these results, this Ejeanselmei_ITS assay seems to be a better alternative to the classical analysis. Indeed, using the classical methods, the incubation period was 21 days, while our SYBR®green analysis (lyophilization step and DNA extraction included) required 2 days. A low diversity, maximum two species per sample, was observed with the classical method as it was previously observed in other study (Hamada and Fujita 2002; Kelkar et al. 2005; Parat et al. 1996). In total, five species were observed (Acremonium sp., A. fumigatus, E. jeanselmei, P. chrysogenum) (Table 5). This implies that the water reservoirs were regularly cleaned with anti-fungal chemistry to avoid health problems such as allergies, asthma, or sick building syndrome. Therefore, it was verified that this anti-fungal chemicals did not inhibit the qPCR reaction, which was found not to be the case. Based on the C q values obtained for the LOD estimation, an extrapolation of the DNA theoretical copy number was performed in order to compare the results from the classical and the molecular methods. According to this estimation, the theoretical (estimated) copy number of genomic DNA for E. jeanselmei was evaluated to 1 and 2 copy number of DNA per ml of analyzed water. These estimations were in the range of those obtained with classical methods (between 2 and 4 CFU/ml). However, some considerations should be made. Firstly, this extrapolation was done based on the values obtained for the reference strain (with the estimated genome size of 30 Mb), and it is not so unlikely that another strain of E. jeanselmei was present in the real-life samples. Therefore the effect of the variation in 18S rDNA copy number should be taken into account. Secondly, this comparison supposed that one CFU observed on plate corresponds to one copy of gDNA. However, a colony can originate from more than one copy of gDNA if aggregates were present. Therefore, although the range of detected E. jeanselmei contamination was comparable for both methods, and therefore the qPCR assay could be regarded as a semi-quantitative method, the absolute quantities of E. jeanselmei are difficult to be compared between the two analysis methods. However, this was expected, because as elaborated above, the ITS-based qPCR method is a qualitative method. If an absolute quantification is needed, the classical methods should still be used.
The results of this proof of concept demonstrate that molecular methods such as qPCR could also be used for the detection of other contaminants present in the water reservoir of air-conditioners. In order to reduce even more the analysis time, it should also be interesting to develop a multiplex tool for the detection of several contaminants simultaneously. This will allow achieving the same advantage of the classical method, which is not limited to the detection of E. jeanselmei only.
In addition, the use of this SYBR®green qPCR assay should not be reduced to water environments. It would also be useful for the detection of E. jeanselmei in other environmental or in clinical samples. As qPCR methods, being based on DNA amplification, offer the advantage of detecting also non-culturable/dead organisms, it would be interesting to apply this method to the monitoring of E. jeanselmei in environments where its occurrence has not yet been reported. Indeed, because E. jeanselmei requires a humid environment to be kept alive and the currently used monitoring methods for this pathogen are based on culturing (which implies that the to-be-detected organism should be alive and culturable), its presence in for instance indoor air, where the organism could suffer from desiccation impacting its growth, could not yet be demonstrated.
In conclusion, this paper reports on a novel SYBR®green qPCR assay for the specific detection of E. jeanselmei, called Ejeanselmei_ITS. Because classical methods are time consuming, and E. jeanselmei has demanding growth conditions, this qualitative assay, and molecular tools like qPCR in general, offers the possibility to reduce the analysis time period and to extend the monitoring to other environments. This will contribute to an improved response against fungal contamination and a better insight into the causal link between E. jeanselmei and health problems.
References
Al-gabr HM, Zheng T, Yu X (2014) Occurence and quantification of fungi and detection of mycotoxigenic fungi in drinking water in Xiamen City, China. Sci Total Environ 466(467):1103–1111. doi:10.1016/j.scitotenv.2012.12.060
Anaissie E, Stratton SL, Dignagi MC, Lee C-K, Summerbell RC, Rex JH, Monson TP, Walsh TJ (2003) Pathogenic molds (including Aspergillus species) in hospital water distribution systems: a 3-year prospective study and clinical implications for patients with hematologic malignancies. Blood 101(7):2542–2546. doi:10.1182/blood-2002-02-0530
Ashktorab H, Cohen RJ (1992) Facile isolation of genomic DNA from filamentous fungi. Biotechniques 13(2):198–200
Badali H, Chander J, Bayat M, Seyedmousavi S, Sidhu S, Rani H, Attri A, Handa U, Meis JF, De Hoog GS (2012) Multiple subcutaneous cysts due to Exophiala spinifera in an immunocompetent patient. Med Mycol 50(2):207–213. doi:10.3109/13693786.2011.603367
Barbau-Piednoir E, Botteldoorn N, Yde M, Mahillon J, Roosens NH (2013) Development and validation of qualitative SYBR(R)Green real-time PCR for detection and discrimination of Listeria spp. and Listeria monocytogenes. Appl Microbiol Biotechnol 97(9):4021–4037. doi:10.1007/s00253-012-4477-2
Black J, Dean T, Byfield G, Foarde K, Menetrez M (2013) Determining Fungi rRNA Copy Number by PCR. JBT 24(1):1–7
Blakely T, Salmond C (2002) Probabilistic record linkage and a method to calculate the positive predictive value. Int J Epidemiol 31:1246–1252. doi:10.1093/ije/31.6.1246
Broad Institute (2015) Genome index. Broad Institute. http://www.broadinstitute.org/annotation/genome/Black_Yeasts/GenomesIndex.html. Accessed 24 July 2015
Broeders S, Huber I, Grohmann L, Berben G, Taverniers I, Mazzara M, Roosens NH, Morisset D (2014) Guidelines for validation of quantitative real-time PCR methods. Trends Food Sci Tech 37115-126. doi:10.1016/j.tifs.2014.03.008
Broeders S, Papazova N, Van Den Bulke M, Roosens NH (2012) Development of a Molecular Platform for GMO Detection in Food and Feed on the Basis of “Combinatory qPCR” Technology. Intech. http://www.intechopen.com/books/polymerase-chain-reaction/development-of-a-molecular-platform-for-gmo-detection-in-food-and-feed-on-the-basis-of-combinatory-q. Accessed 06/11/2015
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley J, Vandesompele J, Wittwer CT (2009) The MIQE guildelines: minimum information for publication of quantitative real-time PCR experiment. Clin Chem 55(4):611–622. doi:10.1373/clinchem.2008.11279
Chemidlin Prevost-Boure N, Christen R, Dequiedt S, Mougel C, Lelievre M, Jolivet C, Shahbazkia HR, Guillou L, Arrouays D, Ranjard L (2011) Validation and application of a PCR primer set to quantify fungal communities in the soil environment by real-time quantitative PCR. PLoS ONE 6(9):e24166. doi:10.1371/journal.pone.0024166
Corradi N, Croll D, Colard A, Kuhn G, Ehinger M, Sanders IR (2007) Gene copy number polymorphisms in an arbuscular mycorrhizal fungal population. Appl Environ Microbiol 73(1):366–369
De Hoog GS, Vicente V, Caligiorne RB, Kantarcioglu S, Tintelnot K, Van Den Ende G, Haase G (2003) Species diversity and polymorphism in the Exophiala spinifera clade containing opportunistic black yeast-like fungi. J Clin Microbiol 41(10):4767–4778. doi:10.1128/JCM.41.10.4767-4778.2003
Dixon DM, Shadomy HJ, Shadomy S (1980) Dematiaceous fungal pathogens isolated from nature. Mycopathologia 70(3):153–161. doi:10.1007/BF00443026
European Network of GMO Laboratories (2011) Verification of analytical methods for GMO testing when implementing interlaboratory validated methods. Joint Research Centre, Luxembourg, Luxembourg
European Network of GMO Laboratories (2015) JCR technical report : Definition of minimum performance requirements for analytical methods of GMO testing. Joint Research Centre, Luxembourg, Luxembourg
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol 2113-118. doi:10.1111/j.1365-294X.1993.tb00005.x/pdf
Haase G, Sonntag L, van de Peer Y, Uijthof JM, Podbielski A, Melzer-Krick B (1995) Phylogenetic analysis of ten black yeast species using nuclear small subunit rRNA gene sequences. A van Leeuw J Microb 68(1):19–33. doi:10.1007/BF00873289
Hamada N, Fujita T (2002) Effect of air-conditionner on fungal contamination. Atmos Environ 365443-5448. doi:10.1016/S1352-2310(02)00661-1
Hee YC, Yoon KK (2002) Molecular analysis of Exophiala species using molecular markers. Mycobiology 30(1):1–4. doi:10.4489/MYCO.2002.30.1.001
Heinemann S, Beguin H, Nolard N (1994) Biocontamination in air-conditioning. In: Samson RA, Flannigan B, Flannigan ME, Verhoeff AP, Adan OCG, Hoekstra ES (eds) Health implications of fungi in indoor environments. Elsevier Science, Amsterdam, The Netherlands,pp
Hospodsky D, Yamamoto N, Peccia J (2010) Accuracy, precision, and method detection limits of quantitative PCR for airborne bacteria and fungi. Appl Environ Microbiol 76(21):7004–7012. doi:10.1128/AEM.01240-10
Iwen PC, Hinrichs SH, Rupp ME (2002) Utilization of the internal transcribed spacer regions as molecular targets to detect and identify human fungal pathogens. Med Mycol 40(1):87–109. doi:10.1080/mmy.40.1.87.109
Kawasaki M, Ishizaki H, Nishimura K, Miyaji M (1990) Mitochondrial DNA analysis of Exophiala jeanselmei and Exophiala dermatitidis. Mycopathologia 110(2):107–112. doi:10.1007/BF00446999
Kawasaki M, Anzawa K, Tanabe, H, Ochizuki T, Ishizaki H, Nishimura K (2005) Intra-species variation of genotypes of Exophiala jeanselmei isolated from patients in Japan. Jpn J Med Mycol 45261–45265. doi:10.3314/jjmm.46.261
Kawasaki M, Ishizaki H, Matsumoto T, Matsuda T, Nishimura K, Miyaji M (1999) Mitochondrial DNA analysis of Exophiala jeanselmei var. lecanii-corni and Exophiala castellanii. Mycopathologia 146(2):75–77. doi:10.1023/A:1007057713782
Kelkar U, Bal AM, Kulkarni S (2005) Fungal contamination of air conditionning units in operating theatres in India. JHI 6081-84. doi:10.1016/j.jhin.2004.10.011
Klein D (2002) Quantification using real-time PCR technology: applications and limitations. Trends Mol Med 8(6):257–260
Libert X, Chasseur C, Bladt S, Packeu A, Bureau F, Roosens NH, De Keersmaecker SJC (2015) Development and validation of a qualitative SYBR®green real-time PCR assay for the detection of Aspergillus versicolor from indoor air. Appl Microbiol Biotechnol 99(7):7267–7282. doi:10.1007/s00253-015-6785-9
Masuda M, Naka W, Tajima S, Harada T, Nishikawa T, Kaufman L, Standard P (1989) Deoxyribonucleic acid hybridization studies of Exophiala dermatitidis and Exophiala jeanselmei. Microbiol Immunol 33(8):631–639. doi:10.1111/j.1348-0421.1989.tb02013
Melkin T, Haugland RA, Reponen T, Varma M, Lummus Z, Bernstein D, Wymer LJ, Vesper SJ (2004) Quantitative PCR analysis of houses dust can reveal abnormal mold conditions. J Environ Monit 6(7):615–620. doi:10.1039/B400250D
Mohr PJ, Taylor BN, Newell DB (2008) CODATA recommended values of the fundamental physical constants: 2006. Rev Mod Phys 80(2):633–730. doi:10.1103/RevModPhys.80.633
Nagano Y, Elborn JS, Millar BC, Goldsmith CE, Rendall J, Moore JE (2008) Development of a novel PCR assay for the identification of the black yeast, Exophiala (Wangiella) dermatitidis from adult patients with cystic fibrosis (CF). J Cyst Fibros 7(6):576–580. doi:10.1016/j.jcf.2008.05.004
Najafzadeh MJ, Dolatabadi S, Saradeghi Keisari M, Naseri A, Feng P, De Hoog GS (2013) Detection and identification of opportunistic Exophiala species using the rolling circle amplification of ribosomal internal transcribed spacers. J Microbiol Methods 94(3):338–342. doi:10.1016/j.mimet.2013.06.026
Nishimura K, Miyaji M, Taguchi H, Tanaka R (1987) Fungi in bathwater and sludge of bathroom drainpipes. 1. Frequent isolation of Exophiala species. Mycopathologia 97(1):17–23. doi:10.1007/BF00437326
Nolard N, Chasseur C, Marlier M, Lognay G (2004) Validation des méthodes microbiologiques et chimes de contrôle des lieux de travail. Belspo. http://www.belspo.be/belspo/organisation/publ/pub_ostc/PS/rPS19_fr.pdf. Accessed 22 July 2015
Nucci M, Akiti T, Barreiros G, Silveira F, Revankar SG, Sutton DA, Patterson TF (2001) Nosocomial fungemia due to Exophiala jeanselmei var. jeanselmei and a Rhinocladiella species: newly described causes of bloodstream infection. J Clin Microbiol 39(2):514–518. doi:10.1128/JCM.39.2.514-518.2001
Nucci M, Akiti T, Barreiros G, Silveira F, Revankar SG, Wickes BL, Sutton DA, Patterson TF (2002) Nosocomial outbreak of Exophiala jeanselmei fungemia associated with contamination of hospital water. Clin Infect Dis 34(11):1475–1480. doi:10.1086/340344
Packeu A, Lebecque P, Rodriguez-Villalobos H, Boeras A, Hendrickx M, Bouchara JP, Symoens F (2012) Molecular typing and antifungal susceptibility of Exophiala isolates from patients with cystic fibrosis. J Med Microbiol 61(Pt 9):1226–1233. doi:10.1099/jmm.0.042317-0
Parat S, Fricker-Hidalgo H, Perdrix A, Bemer D, Pelissier N, Grillot R (1996) Airborne fungal contamination in air-conditionning systems: Effect of filtering and humidifying devices. AIHA journal 57996-1001. doi:10.1080/15428119691014323
Pitkaranta M, Meklin T, Hyvarinen A, Nevalainen A, Paulin L, Auvinen P, Lignell U, Rintala H (2011) Molecular profiling of fungal communities in moisture damaged buildings before and after remediation--a comparison of culture-dependent and culture-independent methods. BMC Microbiol 11235-doi:10.1186/1471-2180-11-235
Rutledge RG, Cote C (2003) Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res 31(16):e93. doi:10.1093/nar/gng093
Sarachu M, Colet M (2005) wEMBOSS: a web interface for EMBOSS. Bioinformatics. 21:540–541. doi:10.1093/bioinformatics/bti031
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A 109(16):6241–6246. doi:10.1073/pnas.1117018109
Sudhadham M, de Hoog GS, Menken SBJ, van den Ende AH G, Sihanonth P (2010) Rapid screening for genotypes as possible markers of virulence in the neurotropic black yeast Exophiala dermatitidis using PCR-RFLP. J Microbiol Methods 80(138):142. doi:10.1016/j.mimet.2009.11.007
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. MBE doi:10.1093/molbev/mst197
United States Environmental Protection Agency (2015) The Environmental Relative Moldiness Index: A Research Tool. United States Environmental Protection Agency. http://www2.epa.gov/sites/production/files/2014-09/documents/moldiness_index_1.pdf. Accessed October 2015
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35:W71–W74. doi:10.1093/nar/gkm306
Vesper SJ (2011) Traditional mould analysis compared to a DNA-based method of mould analysis. Crit Rev Microbiol 37(1):15–24. doi:10.3109/1040841X.2010.506177
Wang F, Mu C, Zhao J-Y, Gao L, Hu X-H, Li H, Tian S-G (2013) Establishment of a real-time fluorescence quantitative PCR method for the detection of clinical Exophiala dermatitidis. Chin J Mycol 8(6):321–324
Wang L, Yokoyama K, Miyaji M, Nishimura K (2001) Identification, classification, and phylogeny of the pathogenic species Exophiala jeanselmei and related species by mitochondrial cytochrome b gene analysis. J Clin Microbiol 39(12):4462–4467. doi:10.1128/JCM.39.12.4462-4467.2001
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand GH, Sninsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications. Academic Press, New York, USA, pp
Woo PC, Ngan AH, Tsang CC, Ling IW, Chan JF, Leung SY, Yuen KY, Lau SK (2013a) Clinical spectrum of Exophiala infections and a novel Exophiala species, Exophiala hongkongensis. J Clin Microbiol 51(1):260–267. doi:10.1128/JCM.02336-12
Woo PC, Ngan AH, Tsang CC, Ling IW, Chan JF, Leung SY, Yuen KY, Lau SK (2013b) Clinical spectrum of Exophiala infections and a novel Exophiala species, Exophiala hongkongensis. J Clin Microbiol 51(1):260–267. doi:10.1128/JCM.02336-12
Zeng JS, De Hoog GS (2008) Exophiala spinifera and its allies: diagnostics from morphology to DNA barcoding. Med Mycol 46(3):193–208. doi:10.1080/13693780701799217
Zeng JS, Sutton DA, Fothergill AW, Rinaldi MG, Harrak MJ, De Hoog GS (2007) Spectrum of clinically relevant Exophiala species in the United States. J Clin Microbiol 45(11):3713–3720. doi:10.1128/JCM.02012-06
Acknowledgments
We would like to thank Hakima El Khabbabi, Pascal Kanyandekwe and Danny Philips for the classical analysis of water samples. Sequencing was performed at the Platform Biotechnology and Molecular Biology at the Scientific Institute of Public Health (WIV-ISP). This work was supported by RP/PJ WIV-ISP (mycoMOLAIR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Libert, X., Chasseur, C., Packeu, A. et al. A molecular approach for the rapid, selective and sensitive detection of Exophiala jeanselmei in environmental samples: development and performance assessment of a real-time PCR assay. Appl Microbiol Biotechnol 100, 1377–1392 (2016). https://doi.org/10.1007/s00253-015-7175-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7175-z