
Abstract
Anaerobic ammonium oxidation (anammox) has been recognized as an important process for the global nitrogen cycle. In this study, the occurrence and diversity of anammox bacteria in the deep-sea subsurface sediments of the South China Sea (SCS) were investigated. Results indicated that the anammox bacterial sequences recovered from this habitat by amplifying both 16S rRNA gene and hydrazine oxidoreductase encoding hzo gene were all closely related to the Candidatus Scalindua genus. A total of 96 16S rRNA gene sequences from 346 clones were grouped into five subclusters: two subclusters affiliated with the brodae and arabica species, while three new subclusters named zhenghei-I, -II, and -III showed ≤97.4% nucleic acid sequence identity with other known Candidatus Scalindua species. Meanwhile, 88 hzo gene sequences from the sediments also formed five distant subclusters within hzo cluster 1c. Through fluorescent real-time PCR analysis, the abundance of anammox bacteria in deep-sea subsurface sediment was quantified by hzo genes, which ranged from 1.19 × 104 to 7.17 × 104 copies per gram of dry sediments. Combining all the information from this study, diverse Candidatus Scalindua anammox bacteria were found in the deep-sea subsurface sediments of the SCS, and they could be involved in the nitrogen loss from the fixed inventory in the habitat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anaerobic ammonium oxidation (anammox) has been identified as an important pathway for anaerobic N2 production in marine environment [29, 44]. These initial discoveries have changed the traditional view on the biogeochemical nitrogen cycling and present a new recognition that classical denitrification (heterotrophic process in which nitrogen oxides serve as the terminal electron acceptor for organic carbon metabolism) is not the only pathway for fixed nitrogen loss [12, 21]. The anammox activity in the marine environments was firstly investigated in Danish coastal regions using 15N-labeled NH4 + [50]. The activity and diversity of anammox bacteria have been found in various marine environments, and further study has demonstrated that anammox reaction may be responsible for a large part (>50%) of oceanic N2 production on a global scale [7, 14, 22–24, 34, 50, 52].
Five genera of anammox bacteria have been identified from the surveys of wastewater treatment systems and environmental samples, including Candidatus Brocadia [19, 44], Candidatus Kuenenia [37], Candidatus Anammoxoglobus [18], Candidatus Scalindua, and Candidatus Jettenia [32, 57], which form a monophyletic branch within the phylum Planctomycetes. Although anammox bacteria play an important role in the nitrogen cycle and have a widespread distribution in the marine ecosystem, the phylogenetic diversity of this group is quite low [39]. The anammox bacteria retrieved from the anoxic water columns of Golfo Dulce [7], in Namibian and Peruvian OMZs [13, 14, 24, 51, 58], and a number of temperate estuarine, coastal, and offshore sediments [16, 33, 34, 46, 53] were all affiliated with the Candidatus Scalindua genus, where four subgroups in Candidatus Scalindua sorokinii [22], Candidatus Scalindua brodae, Candidatus Scalindua wagneri [38] and Candidatus Scalindua arabica [56] were recorded and identified from the marine ecosystem.
Marine subsurface sediment is one of the most extensive microbial habitats on the Earth [10, 11, 49]. It had been estimated that the microbial cells in subseafloor sediments constituted 1/2 ~ 5/6 of the earth’s microbial biomass and as much as 1/3 of the earth’s total living biomass [30, 55]. As an essential metabolic process of microorganisms, it can be inferred that the microbial nitrogen transformation should be essential and active in the deep-sea sediment biosphere. Although microorganisms are ubiquitous in the ocean sediments, little is known about nitrogen-utilizing microorganisms in this environment. The diversity of potential nitrogen fixers inhabiting the ocean sediment and hydrothermal vents, based on the unique nitrogenase (nifH) and nifH-like genes, provides a basis for active N transformation in the sediment [8, 28]. However, the removal processes of fixed nitrogen by microorganisms are mainly focused on the denitrification [45], and very little information is available on the anammox bacteria. Understanding the role of fixed nitrogen removal by anammox microbial communities is therefore essential for a more comprehensive understanding of the nitrogen biogeochemical cycling in the deep-sea sediment ecosystem.
The South China Sea (SCS) with a maximum depth greater than 5,000 m is the largest marginal sea in Southeast Asia. It covers an area from the equator to 23° N and from 99° E to 121° E, and joins the Pacific Ocean through the Luzon Strait (or named as the Bashi Strait or the Bashi Channel in some literatures) between Taiwan Island and Luzon Island. The SCS is considered an oligotrophic ecosystem; both N and P in its euphotic layer are usually below the detection limits when measured by conventional methods [4]. In the central gyre of the SCS, nitrogen is the limiting factor for phytoplankton growth [58] suggesting that the environment is nitrogen deficient. In the present study, we investigated the diversity and abundance of anammox bacteria in the deep-sea subsurface sediments at different depths in the SCS based on 16S rRNA and hydrazine oxidoreductase encoding gene (hzo) analyses.
Materials and Methods
Sampling and Chemical Analyses
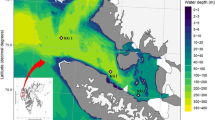
Subsurface sediment samples of the South China Sea were collected in 2008 at four sites: E401 (3.7 mbsf, meter below seafloor), E407 (1.0 mbsf), E525 (4.0 mbsf), and CF5 (7.5 mbsf) during South China Sea Open Cruise by R/V Shiyan 3. The details of the sampling sites are presented in Table 1 and Fig. S1. After surface samples of sediment cores were discarded due to exposure to the air, samples were taken and transferred aseptically into plastic bags and frozen immediately at −20°C. Chemical analysis of ammonium-N, nitrate-N, and nitrite-N in the sediment samples was carried out using 2 M KCl as extractant and measured with an autoanalyzer (QuickChem, Milwaukee, WI) according to standard methods [1].
DNA Extraction and PCR Amplification
Total genomic DNA of the sediment samples were extracted using the SoilMaster DNA Extraction Kit (Epicenter Biotechnologies, Madison, WI) according to the manufacturer’s instructions. PCR amplifications of 16S rRNA genes were based on previous studies with primers Brod541F (5′-GAGCACGTAGGTGGGTTTGT-3′) [31], Amx368F (5′-TTCGCAATGCCCGAAAGG-3′) [38] and Amx820R (5′-AAAACCCCTCTACTTAGTGCCC-3′) [37]. The primers HZOF1 (5′-GTGCATGGTCAATTGAAAG-3′) and HZOR1 (5′-CAACCTCTTCWGCAGGTGCATG-3′) [26] were used for amplification of hzo gene. The optimized PCR reaction mixture in a final volume of 50 μl contained the followings: 2 μl DNA (30 ~ 50 ng μl−1), 0.5 μl bovine serum albumin (100 mg ml−1, Roche), 10 μl 5 × GoTaq Flexi Buffer (Promega) and 4 μl MgCl2 (25 mM, Promega), 1 μl of dNTPs (10 mM, Invitrogen), 1 μl of forward and reverse primers (20 μM), and 0.25 μl of GoTaq Flexi polymerase (5 U μl−1, Promega). PCR programs were as followings: 95°C for 3 min; 30–32 cycles of 95°C for 45 s, 60°C (for Brod541F/Amx820R) or 59°C (for Amx368F/Amx820R) or 53°C (HZOF1/HZOR1) for 1 min, followed by 72°C for 1 min; and finally 72°C for 10 min.
Cloning, Sequencing and Phylogenetic Analysis
The PCR products were purified using the Gel Advance Gel Extraction System (VIOGEME, Taipei) following the manufacturer’s instructions, and cloned into the pMD-18T Vector (Takara, Japan). The insertion of an appropriate-sized DNA fragment was determined by PCR amplification with the primer set M13F and M13R. The clones in each library were screened by restriction fragment length polymorphism (RFLP) (with Sau3AI and NlaIII for 16S rDNA, EcoRI and BamHI for hzo DNA) and selected for sequencing. Sequencing was performed with the BigDye Terminator Kit (Applied Biosystems, Foster City, CA) and an ABI Prism 3730 DNA analyzer.
The DNA sequences were examined and edited by MEGA 4.0 software [47] and then checked for chimera formation using the Check Chimera program of Ribosomal Database Project [5] or manually. For the 16S rRNA gene, DNA sequences were manually compiled and aligned using the ClustalW [48]. For hzo gene, the nucleic acid sequences were firstly translated into amino acids, and the resulting amino acid sequences of the corresponding proteins were aligned using ClustalW. Phylogenetic trees were constructed by MEGA 4.0 with the neighbor-joining and maximum parsimony methods. Bootstrap resampling analysis on 500 replicates was performed to estimate the confidence of the tree nodes.
Quantitative PCR Assay
The copy numbers of hzo gene in all samples were determined in triplicate using an ABI 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). The quantification was based on the fluorescent dye SYBR Green I. Each reaction was performed in a 25-μl volume containing 1 μl of DNA template, 0.5 μl BSA (0.1%), 0.5 μl of each primer (20 μM, HZOF1 and HZOR1) and 12.5 μl of Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). The PCR cycle was started with 2 min at 50°C and 10 min at 95°C, followed by a total of 48 cycles of 1 min at 95°C, 1 min at 53°C, and 1.5 min at 72°C. A standard plasmid carrying the hzo gene fragment was generated by amplifying the hzo gene from the extracted DNA from sediments and cloned into pMD18-T Vector (Takara, Japan). The plasmid DNA concentration was determined on a Biophotometer (Eppendorf, Germany), and the copy number of the target gene was calculated directly from the concentration of the extracted plasmid DNA. Tenfold serial dilutions of a known copy number of the plasmid DNA (1.83 × 102 to 1.83 × 1010 copies) were subjected to fluorescent real-time PCR assay in triplicate to generate an external standard curve (r 2 = 0.991).
Statistical Analysis
The Distance-based OTU and Richness (DOTUR) program [36] was employed to compare diversity for anammox 16S rRNA and hzo gene sequences from each sampling site. Operational taxonomic units (OTUs) for community analysis were defined by 3% differences in nucleic acid or protein sequences, as determined by using the furthest neighbor algorithm in DOTUR [36]. DOTUR was also used to generate diversity indices, such as Shannon and Simpson for each clone library. The correlation analysis between diversity and environmental variables was conducted using Microsoft Excel. To examine the geographic distribution of the phylogenetic structure of anammox bacteria in deep-sea subfloor sediments, 16S rRNA gene and Hzo protein sequences were analyzed with the online software UniFrac (http://bmf2.colorado.edu/unifrac/index.psp) by using the principal coordinates analysis (PCoA) as suggested previously [27].
Nucleic Acid Sequence Accession Numbers
The GenBank accession numbers for the 16S rRNA gene sequences reported here are GQ331139 to GQ331201 (with primer pair Brod541F/Amx820R) and GQ1202 to GQ331244 (with primer pair Amx368F/Amx820R), and the GenBank accession numbers for the hzo gene sequences are GQ331245 to GQ331332.
Results
Detection of Anammox Bacteria and Phylogenetic Analysis with 16S rRNA Gene
Anammox bacterial communities in the deep-sea subsurface sediments of the SCS were detected successfully with 16S rRNA gene. From the eight clone libraries (two clone libraries were constructed for each sampling site with anammox-specific primer pairs Brod541F/Amx820R and Amx368F/Amx820R), a total of 346 clones (from more than 1,000 clones with RFLP) were sequenced. The BLAST results revealed that 96 clones (27.8%) were closely affiliated with known anammox bacteria. Frequencies of positive clones for anammox bacterial 16S rRNA gene were highly variable and site dependent (Table 2). The positive clone (affiliated into the anammox cluster in phylogeny) frequency was 59%, 19%, 27%, and 12% for site E401, E407, E525, and CF5 in this study respectively.
The phylogenetic tree was built with the 96 positive anammox clones and other known anammox 16S rRNA gene sequences using neighbor-joining and maximum parsimony algorithms (Fig. 1). The tree revealed that all anammox bacterial-like sequences of this study were affiliated with the genus Candidatus Scalindua. Two phylogenetic trees constructed using different algorithms supported the grouping of anammox bacterial sequences into six coherent subclusters: brodae subcluster, arabica subcluster, wagneri subcluster, zhenghei-I subcluster, zhenghei-II subcluster, and zhenghei-III subcluster. The 96 anammox-like sequences identified from the deep-sea subsurface sediments of SCS were distributed into five of the six subclusters; none of the sequences that were identified were affiliated with the wagneri group. The percentage of anammox-like sequences affiliated with the brodae subcluster was 62.5%. This subcluster includes sequences from all four sampling sites. In addition, this subcluster contains identified anammox bacteria Candidatus Scalindua brodae, Candidatus Scalindua sorokinii as well as some clones from Peruvian and Namibian OMZs. Only one clone from the E525 sampling site was included within the arabica subcluster, which is a newly proposed anammox bacteria phylotype [56].

Phylogenetic tree based on an alignment of 16S rRNA gene sequences showing the phylogenetic affiliations of anammox bacterial sequences from the different subseafloor sediment of South China Sea. The numbers in parentheses refer to how many clones retrieved by the amplification with the primer sets Brod541F/Amx820R and Amx368/ Amx820R were assigned to an individual phylotype. The consensus phylogenetic tree constructed based on neighbor-joining analysis. The numbers at the nodes are percentages that indicate the levels of bootstrap support based on 500 resampled data sets. Branch lengths correspond to sequence differences as indicated by the scale bar
When all subclusters were compared with each other, zhenghei-I, zhenghei-II, and zhenghei-III (named after He Zheng (also known as Cheng Ho) for his contribution to maritime navigation seven times to the Indian Ocean between 1405–1433 in China) were apparently distant from the others. The zhenghei-I subcluster (Fig. S3-A), containing 17 sequences from all our sampling sites, was most similar to sequences from SCS sediment [42] and Ryukyn Trench deep-sea sediment (AB015552) [25]. The zhenghei-I subcluster showed only 92~96.6% identity with other subclusters within the Candidatus Scalindua genus (Table 2). The zhenghei-II subcluster (Fig. S3-B), which contained 16 sequences from E407, E525, and E401, showed 94.7–96.6% identity with other subclusters within the Candidatus Scalindua genus (Table 2). The zhenghei-II subcluster is the most similar to a clone from Namibian OMZ sea water (EF646011) [57] and an uncultured bacterium in freshwater sediments of the Xinyi River (DQ647431) [59]. Only two clones affiliated with the zhenghei-III subcluster (Fig. S3-C) were related to sequences from Juan de Fuca Ridge sediment (DQ869978) [31] and Candidatus Scalindua sp. enrichment culture (EU142947) [54]. This subcluster has 93.6~97% identity with other subclusters within Candidatus Scalindua genus (Table 2).
Interestingly, nine of the 16S rRNA gene sequences of this study (cluster P) (Fig. S3-D) were grouped into the Planctomycetes but are not closely related to any known anammox bacteria or other described Planctomycetes genera. The sequence identities ranged from 77.7% to 82.2% compared with the known anammox bacteria and from 74.1% to 78.4% with non-anammox Planctomycete genera (Table S1). These nine sequences from the four sampling sites shared 97.3~100% identity to each other.
Rarefaction analysis indicated that site CF5 showed the greatest anammox-16S rRNA gene diversity whereas sites E407 and E525 had the lowest OTUs, which was consistent with the values of Simpson and Shannon indices (Table 2). The OTU numbers in CF5 may have been higher if more clones were sequenced based on non-asymptotic rarefaction curves (Fig. S2).
Phylogenetic Analysis Based on an Alignment of hzo Gene Sequences
Using our new primer pair HZOF1/HZOR1, the PCR amplification products targeting hzo gene were amplified from the deep-sea sediments and subsequently hzo gene libraries were constructed for each sample. A total of 88 clones screened from the libraries showed a high similarity of their deduced protein sequences (Table S2) with those published in previous studies [32, 40, 41]. A phylogenetic analysis of experimental- and GenBank-retrieved hzo gene-deduced protein sequences resulted in a tree, in which the experimental Hzo protein sequences from our study were grouped into hzo cluster 1 defined by previous study (Fig. 2) [40]. Compared to the hzo clusters 1a and 1b, sequences obtained in this study all fell into a distant cluster within hzo cluster 1, for which hzo cluster 1c was proposed. A Candidatus Scalindua sp. enrichment culture (CAQ57909) [40] is also included in the hzo cluster 1c, indicating that the hzo sequences recovered from deep-sea subsurface sediments are closely related to Candidatus Scalindua bacteria. Further cladistic analysis revealed that five distinct subclusters were embedded within the hzo cluster 1c, designated as subclusters 1c-1 to 1c-5. Sequences in subclusters 1c-3 and 1c-5 were obtained from all four sampling sites; sequences in subclusters 1c-2 and 1c-4 were recovered only from the E525 and CF5 sites. To date, no published sequences were found to be closely related to any of the sequences in subclusters 1c-2, 1c-3, 1c-4, and 1c-5. The Hzo protein sequences of the subcluster 1c-1 are all from the E401 site and are most similar to Hzo protein sequence of Candidatus Scalindua sp. enrichment culture (CAQ57909) and two uncultured Planctomycete (CAQ57913 and CAQ 57914) [39]. The sequence identities of subclusters ranged from 83.0% to 94.0% when compared with each other and 77.2% to 83.5% with hzo clusters 1a and 1b, respectively but for other hzo clusters, including clusters 2a, 2b and 3, the identity was less than 50% (Table S2).

Phylogenetic tree constructed from alignments of Hzo protein sequences deduced from deposited hzo gene sequences in the GenBank and experimental sequences obtained by amplifying the community DNA sampled from the subseafloor sediments of the South China Sea at different depths. The numbers in parentheses refer to how many hzo gene clones retrieved using the respective primer set were assigned to an individual phylotype. The numbers at the nodes are percentages that indicate the levels of bootstrap support based on 500 resampled data sets. Branch lengths correspond to sequence differences as indicated by the scale bar. Major clades were named following a proposal of Schmid et al. [40]
Using a 3% cutoff at the amino acids sequence variation to define an OTU, the OTU numbers were almost the same at all four study sites of this study. The values of Simpson and Shannon indices were consistent with OTU richness analysis (Table 2). The non-asymptotic rarefaction curves also showed the OTU numbers in CF5 might have been higher if more clones were sequenced (Fig. S2).
Abundance of the hzo Gene in Deep-Sea Subsurface Sediment
The copy numbers of the hzo gene in all the samples were determined using the fluorescent real-time PCR method with the same primer pair HZOF1/HZOR2 (Fig. 3). The abundance of the hzo gene in CF5 was the highest, up to 7.17 × 104 copies per gram of dry sediments. In E407 and E525, the abundances of the hzo gene were 4.54 × 104 and 5.44 × 104 copies per gram of dry sediments, respectively. The lowest hzo gene copy number of 1.19 × 104 copies per gram of dry sediments was determined at the sampling site E401, where the water column is the deepest (3,300 m below sea surface).

Abundance of hzo gene of anammox bacteria in sediments of selective sampling sites at the South China Sea. The experiment was performed in triplicate. Error bars represent standard deviations of triplicate analysis
Community Structures of Resident Anammox Bacteria
A significant geographical distribution difference of the anammox community structure based on 16S rRNA and hzo genes was determined by the weighted UniFrac PCoA analysis. For 16S rRNA gene, the anammox communities in all four sampling sites of the SCS deep-sea sediment formed a unique group which separated from that of Antarctic sediment, Namibian and Peruvian OMZ (Fig. 4a), indicating a specific niche of SCS for the anammox bacteria, but four sampling sites are indistinguishable (Fig. 4a), The Hzo-based analysis also showed that the anammox communities in all four sampling sites of the SCS were separated from that of deep-sea Tephra deposits, hydrothermal Vent, Jiaozhou Bay, North Carolina groundwater, Upper Cape Fear River sediment and Black River sediment [9, 15]. The weighted UniFrac PCoA analysis for Hzo supported that the diverse distribution pattern is consistent with their actual geographical locations and the community between E401 and E407 as well as between E525 and CF5 was very similar (Fig. 4b).

Weighted UniFrac PCA analyses for (a) 16S rRNA gene sequences and (b) deduced amino acid sequences of Hzo protein in the context of physical and chemical conditions
Relationship Between Nitrogen and the Diversity of Anammox Bacteria
Variations in the concentration of ammonium and nitrate were observed at the four sampling sites. The concentration of ammonium varied from 13.8 mg kg−1 sediment in the E407 sample to 88.6 mg kg−1 sediment at the E401 site. The concentration of nitrate at E401 was as high as 235 mg kg−1 sediment but below detection level at the other three sampling sties (Table 1). However, the concentration of nitrite was fairly consistent at all four sites (Table 1). A Pearson moment correlation analysis for physicochemical parameters and anammox bacteria diversity showed that only the Chao index of Hzo protein sequences at each site significantly correlated with nitrite concentration according to the statistical algorithm (Table 3).
Discussions
In our study, we detected the presence of anammox bacteria with both 16S rRNA gene and hzo functional gene. The results showed that 96 of the 346 screened 16S rRNA gene sequences fell into known anammox species, indicating the presence of anammox bacteria in the deep-sea subsurface sediment of the SCS. In addition, all the 16S rRNA gene sequences from the deep-sea subsurface sediments were closely related to the Candidatus Scalindua cluster. We presumed that the non-selective amplification might have resulted from two possibilities: (1) PCR bias and (2) only Candidatus Scalindua genus anammox bacteria are present predominantly in this environment.
Since the 16S rRNA gene is not specific enough in detecting the relevant anammox community in the environmental samples, the hzo gene was used for further more specific detection. HZO, a member of the octaheme cytochrome c hydroxylamine oxidoreductase protein family and a key step in the anammox biochemical process, dehydrogenates the unique anammox intermediate, hydrazine, to N2 [17, 35]. So far, the hzo gene has been isolated from several anammox enrichment cultures [35, 41] including a new genus of anammox bacteria Candidatus Jettenia sp. enrichment culture [32] and corresponding sequences were identified in the genome of Candidatus Kuenenia stuttgartiensis [43]. Recently, Klotz and Stein [20] proposed that genes encoding hydrazine oxidoreductase (Hzo) can be used as a functional phylogenetic marker. Schmid et al. [40] employed several degenerate primer sets for detecting hzo genes in various ecosystems and suggested that the hzo gene sequences in cluster 1 (targeted by two primer sets hzocl1F1-hzocl1R2 and hzocl1F1l-hzoclR2) were most appropriate and feasible for phylogenetic analysis because these genes appeared to be functional and present in all anammox bacteria studied. In addition, Quan et al. [32] amplified hzo genes from anammox granular biomass using another degenerate primer set (Ana-hzo1F/Ana-hzo2R). The primer pair HZOF1/HZOR1, which has been successfully used to amplify hzo gene sequences of multitype anammox genera in a previous study [26], was used to further detect anammox bacteria in the deep-sea subsurface sediment. Surprisingly, the sequences obtained with primer pair HZOF1/HZOR1 were also closely related to the Candidatus Scalindua cluster the closest. Thus, the results of 16S rRNA and hzo gene analyses support that the Candidatus Scalindua genus bacteria are the only anammox species present in the deep-sea subsurface sediments of the SCS.
Previous studies showed that anammox bacteria in the marine environments mainly belonged to the Candidatus Scalindua genus. However, in some marine environments strongly affected by the anthropogenic input of wastewater, such as the Cape Fear River Estuary [6, 15], Jiaozhou Bay [9], Japan coastal marine sediments [2], and in the intertidal wetland of Hong Kong [26], genera other than Candidatus Scalindua, including Candidatus Kuenenia sp., and Candidatus Brocadia sp. and Candidatus Jettenia sp. were also found. Several anammox species existed concurrently in the deep-sea hydrothermal vent [3]. Based on the available information and our analyses, the anammox bacteria of Candidatus Scalindua genus were found in the low-temperature environment of the sea water column, within the surface and subsurface of the ocean sediment. The diversity of the anammox bacteria is much higher in the marine habitat of a hydrothermal vent or anthropogenic impacted environments. The Candidatus Scalindua species may dominate the anammox bacterial community because most marine environments are low in temperature.
An analysis of both 16S rRNA and hzo genes revealed a high diversity of Candidatus Scalindua bacteria in the deep-sea subsurface sediment of the SCS. As the Candidatus Scalindua sorokinii has high identity (98.6%) with Candidatus Scalindua brodae, the two species generally fell into one subcluster in the phylogenic analysis. In this study, the 16S rRNA gene sequences from SCS deep-sea subsurface sediment were distributed into five distant subclusters in the phylogenetic tree, which included three new subclusters. Moreover, the new subclusters showed less than 97.4% sequence identities with other identified Candidatus Scalindua phylotypes. The results in this study (Fig. 1 and Table S1) suggest that the subclusters “zhenghei-I,” “zhenghei-II,” and “zhenghei-III” comprise three individual phylotypes within the Candidatus Scalindua genus. Thus, the three specific clusters of the anammox bacteria found in the SCS deep-sea sediment provide new information about anammox diversity within a natural ecosystem. An analysis of the clone library composition showed that the Candidatus Scalindua brodae/sorokinii species was still the dominant group present and the Candidatus “Scalindua zhenghei-I” phylotypes were the second most abundant group in the sediment.
The Hzo protein sequences were used for a further differentiation of closely related clones and to measure the diversity of anammox bacteria in the deep-sea sediment. A total of 88 Hzo sequences from SCS formed five distant subclusters in Hzo cluster 1c, which generally confirms the corresponding 16S rRNA gene phylogeny. However, because there are limited number of hzo sequences available at present, including only two Candidatus Scalindua enrichment cultures [40], these hzo subclusters cannot be further classified. Under this condition, it is difficult to match the subcluster in hzo cluster 1c to that in the 16S rRNA gene Scalindua cluster one to one. Further investigation is needed for identifying the hzo sequences more clearly.
The sequences in cluster P formed a separated branch in the 16S rRNA gene phylogenic tree, showing clearly low identities with known anammox bacteria though they have a closer relationship with the known anammox clade than other non-anammox Planctomycetes, which might comprise a new potential Planctomycetes genus which remains to be elucidated further.
The PCoA analysis showed that anammox bacteria have obvious geographical distribution characteristics. It can be concluded that the special geographical conditions of subsurface sediment in the SCS could be a factor that influenced the evolution of diverse Candidatus Scalindua anammox bacteria different from other marine environment.
A series of Integrated Ocean Drilling Programs demonstrated that the subsurface deep-sea biosphere harbors a huge biomass and complex physical–ecological processes [49]. As the most important elements for life, the nitrogen biogeochemical cycle is an essential process in this system. According to our data, inorganic nitrogen is not limited for microbial metabolism in the deep-sea sediment of the SCS. The removal of inorganic N is an important step in maintaining N cycling in this ecosystem. With the amplification of 16S rRNA and hzo genes, anammox bacteria were detected in the subsurface sediments of the SCS and hzo gene abundance was 1.19~7.17 × 104 copies per gram of dry sediments, suggesting that anammox could be involved in the nitrogen removal process. Furthermore, the correlations of anammox bacterial diversity and abundance with the different nitrogen substrates provided further evidence to the potential activity of anammox bacteria in deep-sea biosphere. However, the exact roles of anammox in the subsurface ecosystem of the SCS remain unknown and require further investigation in the future.
References
American Public Health Association (1995) Vol, 19th edn. APHA, Washington DC
Amano T, Yoshinaga I, Okada K, Yamagishi T, Ueda S, Obuchi A, Sakoand Y, Suwa Y (2007) Detection of anammox activity and diversity of anammox bacteria-related 16S rRNA genes in coastal marine sediment in Japan. Microbes Environ 22:232–242
Byrne N, Strous M, Crepeau V, Kartal B, Birrien J-L, Schmid M, Lesongeur F, Schouten S, Jaeschke A, Jetten MSM, Prieur D, Godfroy A (2009) Presence and activity of anaerobic ammonium-oxidizing bacteria at deep-sea hydrothermal vents. ISME J 3:117–123
Chen YLL, Chen HY, Karl DM, Takahashi M (2004) Nitrogen modulates phytoplankton growth in spring in the South China Sea. Cont Shelf Res 24:527–541
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM (2005) The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 33:294–296
Dale OR, Tobias CR, Song B (2009) Biogeographical distribution of diverse anaerobic ammonium oxidizing (anammox) bacteria in Cape Fear River Estuary. Environ Microbiol 11:1194–1207
Dalsgaard T, Canfield DE, Petersen J, Thamdrup B, Acuna-Gonzalez J (2003) N2 production by the anammox reaction in the anoxic water column of Golfo Dulce, Costa Rica. Nature 422:606–608
Dang H, Luan X, Zhao J, Li J (2009) Diverse and novel nifH and nifH-Like gene sequences in the deep-sea methane seep sediments of the Okhotsk Sea. Appl Environ Microbiol 75:2238–2245
Dang H, Chen R, Wang L, Guo L, Chen P, Tang Z, Tian F, Li S, Klotz MG (2010) Environmental factors shape sediment anammox bacterial communities in hypernutrified Jiaozhou Bay, China. Appl Environ Microbiol 76:7036–7047
D’Hondt S, Rutherford S, Spivack AJ (2002) Metabolic activity of subsurface life in deep-sea sediments. Science 295:2067–2070
D’Hondt S, Jørgensen BB, Miller DJ, Batzke A, Blake R, Cragg BA, Cypionka H, Dickens GR, Ferdelman T, Hinrichs K-U, Holm NG, Mitterer R, Spivack A, Wang G, Bekins B, Engelen B, Ford K, Gettemy G, Rutherford SD, Sass H, Skilbeck CG, Aiello IW, Guèrin G, House CH, Inagaki F, Meister P, Naehr T, Niitsuma S, Parkes RJ, Schippers A, Smith DC, Teske A, Wiegel J, Padilla CN, Acosta JLS (2004) Distributions of microbial activities in deep subseafloor sediments. Science 306:2216–2221
Francis CA, Beman JM, Kuypers MMM (2007) New processes and players in the nitrogen cycle: the microbial ecology of anaerobic and archaeal ammonia oxidation. ISME J 1:19–27
Galan A, Ulloa O, Molina V, Thamdrup B, Woebken D, Lavik D, Kuypers MM (2009) Anammox bacteria and the anaerobic oxidation of ammonium in the oxygen minimum zone off northern Chile. Deep Sea Res II 56:1021–1031
Hamersley MR, Lavik G, Woebken D, Rattray JE, Lam P, Hopmans EC, Sinninghe Damsté JS, Krúger S, Graco M, Gutirréz D, Kuypers MMM (2007) Anaerobic ammonium oxidation in the Peruvian oxygen minimum zone. Limnol Oceanogr 52:923–933
Hirsh MD, Long ZT, Song B (2011) Anammox bacterial diversity in various aquatic ecosystems based on the detection of hydrazine oxidase gene (hzoA/hzoB). Microb Ecol. 61:264–267
Jetten MSM, Sliekers O, Kuypers M, Dalsgaard T, van Niftrik L, Cirpus I, van de Pas-Schoonenl K, Lavik G, Thamdrup B, Le Paslier D, Op den Camp HJM, Hulth S, Nielsen LP, Abma W, Third K, Engström P, Kuenen JG, Jørgensen BB, Canfield DE, Sinninghe Damsté JS, Revsbech NP, Fuerst J, Weissenbach J, Wagner M, Schmidt I, Schmid M, Strous M (2003) Anaerobic ammonium oxidation by marine and freshwater Planctomycete-like bacteria. Appl Microbiol Biotechnol 63:107–114
Jetten MSM, van Niftrik L, Marc S, Kartal B, Keltjens JT, Op den Camp HJM (2009) Biochemistry and molecular biology of anammox bacteria. Crit Rev Biochem Mol Biol 44:65–84
Kartal B, Rattray J, van Niftrik LA, van de Vossenberg J, Schmid MC, Webb RI, Schouten S, Fuerst JA, Sinninghe Damsté J, Jetten MSM, Strous M (2007) Candidatus “Anammoxoglobus propionicus” gen. nov., sp. nov., a new propionate oxidizing species of anaerobic ammonium oxidizing bacteria. Syst Appl Microbiol 30:39–49
Kartal B, van Niftrik L, Rattray J, van de Vossenberg JLCM, Schmid MC, Sinninghe Damsté J, Jetten MSM, Strous M (2008) Candidatus ‘Brocadia fulgida’: an autofluorescent anaerobic ammoniumoxidizing bacterium. FEMS Microbiol Ecol 63:46–55
Klotz MG, Stein LY (2008) Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol Lett 278:146–156
Kuenen JG (2008) Anammox bacteria: from discovery to application. Nature Rev Microbiol 6:320–326
Kuypers MMM, Sliekers AO, Lavik G, Schmid M, Jergensen BB, Kuenen JG, Sinninghe Damsté JS, Strous M, Jetten MSM (2003) Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. Nature 422:608–611
Kuypers MMM, Lavik G, Woebken D, Schmid M, Fuchs BM, Amann RB, Jørgensen B, Jetten MSM (2005) Massive nitrogen loss from the Benguela upwelling system through anaerobic ammonium oxidation. Proc Natl Acad Sci USA 102:6478–6483
Lam P, Lavik Jensen G, van de Vossenberg M, Schmid JM, Woebken D, Gutiérrez D, Amanna R, Jetten MSM, Kuypers MMM (2009) Revising the nitrogen cycle in the Peruvian oxygen minimum zone. Proc Natl Acad Sci USA 106:4752–4757
Li L, Kato C, Horikoshi K (1999) Bacterial diversity in deep-sea sediments from different depths. Biodivers Conserv 8:659–677
Li M, Hong Y-G, Klotz MG, Gu J-D (2010) A comparison of primer sets for detecting 16S rRNA and hydrazine oxidoreductase genes of anaerobic ammonium-oxidizing bacteria in marine sediments. Appl Microbiol Biotechnol 86:781–790
Lozupone C, Hamady M, Knight R (2006) UniFrac-an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform 7:371–384
Mehta MP, Butterfield DA, Baross JA (2003) Phylogenetic diversity of nitrogenase(nifH) genes in deep-sea and hydrothermal vent environments of the Juan de Fuca Ridge. Appl Environ Microbiol 69:960–970
Mulder A, van de Graaf AA, Robertson LA, Kuenen JG (1995) Anaerobic ammonium oxidation discovered in a denitrifying fluidized bed reactor. FEMS Microbiol Ecol 16:177–183
Parkes RJ, Webster G, Cragg BA, Weightman AJ, Newberry CJ, Ferdelman TG, Kallmeyer J, Jørgensen BB, Aiello IW, Fry JC (2005) Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436:390–394
Penton CR, Devol AH, Tiedje JM (2006) Molecular evidence for the broad distribution of anaerobic ammonium-oxidizing bacteria in freshwater and marine sediments. Appl Environ Microbiol 72:6829–6832
Quan ZX, Rhee SK, Zuo JE, Yang Y, Bae JW, Park JR, Lee S-T, Park Y-H (2008) Diversity of ammonium-oxidizing bacteria in a granular sludge anaerobic ammonium-oxidizing (anammox) reactor. Environ Microbiol 10:3130–3139
Rich JJ, Dale OR, Song B, Ward BB (2007) Anaerobic ammonium oxidation (Anammox) in Chesapeake Bay sediments. Microb Ecol 55:311–320
Risgaard-Petersen N, Meyer RL, Schmid M, Jetten MSM, Enrich-Prast A (2004) Anaerobic ammonium oxidation in an estuarine sediment. Aquat Microb Ecol 36:293–304
Schalk J, de Vries S, Kuenen JG, Jetten MSM (2000) Involvement of a novel hydroxylamine oxidoreductase in anaerobic ammonium oxidation. Biochemistry 39:5405–5412
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schmid M, Twachtmann U, Klein M, Strous M, Juretschko S, Jetten MSM, Metzger JW, Schleifer KH, Wagner M (2000) Molecular evidence for genus level diversity of bacteria capable of catalyzing anaerobic ammonium oxidation. Syst Appl Microbiol 23:93–106
Schmid M, Walsh K, Webb R, Rijpstra WI, van de Pas-Schoonen K, Verbruggen MJ, Hill T, Moffett B, Fuerst J, Schouten S, Damsté JS, Harris J, Shaw P, Jetten MSM, Strous M (2003) Candidatus "Scalindua brodae", sp. nov., Candidatus "Scalindua wagneri", sp. nov., two new species of anaerobic ammonium oxidizing bacteria. Syst Appl Microbiol 26:529–538
Schmid MC, Risgaard-Petersen N, van de Vossenberg J, Kuypers MMM, Lavik G, Petersen J, Hulth S, Thamdrup B, Canfield D (2007) Anaerobic ammonium-oxidizing bacteria in marine environments: widespread occurrence but low diversity. Environ Microbiol 9:1476–1484
Schmid MC, Hooper AB, Klotz MG, Woebken D, Lam P, Kuypers MM, Pommerening-Roeser A, op den Camp HJM, Jetten MSM (2008) Environmental detection of octahaem cytochrome c hydroxylamine/hydrazine oxidoreductase genes of aerobic and anaerobic ammonium-oxidizing bacteria. Environ Microbiol 10:3140–3149
Shimamura M, Nishiyama T, Shigetomo H, Toyomoto T, Kawahara Y, Furukawa K, Fujii T (2007) Isolation of a multiheme protein with features of a hydrazine-oxidizing enzyme from an anaerobic ammonium-oxidizing enrichment culture. Appl Environ Microbiol 73:1065–1072
Shu Q, Jiao N (2008) Profiling Planctomycetales diversity with reference to anammox-related bacteria in a South China Sea, deep-sea sediment. Mar Ecol 29:413–420
Strous M, Pelletier E, Mangeno S, Rattei T, Lehner A, Taylor MW, Horn M, Daims H, Bartol-Mavel D, Wincker P, Barbe V, Fonknechten N, Vallenet D, Segurens B, Schenowitz-Truong C, Médigue C, Collingro A, Sne B, Dutilh BE, Op den Camp HJM, van der Drift C, Cirpus I, van de Pas-Schoonen KT, Harhangi HR, van Niftrik L, Schmid M, Keltjens J, van de Vossenberg J, Kartal B, Meier H, Frishman D, Huynen MA, Mewes H-W, Weissenbach J, Jetten MSM, Wagner M, Le Paslier D (2006) Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440:790–794
Strous M, Fuerst J, Kramer E, Logemann S, Muyzer G, van de Pas-Schoonen KT, Webb R, Kuenen JG, Jetten MSM (1999) Missing lithotroph identified as new planctomycete. Nature 400:446–449
Tamegai H, Aoki R, Arakawa S, Kato C (2007) Molecular analysis of the nitrogen cycle in deep-sea microorganisms from the Nankai Trough: genes for nitrification and denitrification from deep-sea environmental DNA. Extremophiles 11:269–275
Tal Y, Watts JE, Schreier HJ (2005) Anaerobic ammonia-oxidizing bacteria and related activity in Baltimore inner harbor sediment. Appl Environ Microbiol 71:1816–1821
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–15969
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Teske A, Sørensen K (2008) Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J 2:3–18
Thamdrup B, Dalsgaard T (2002) Production of N2 through anaerobic ammonium oxidation coupled to nitrate reduction in marine sediments. Appl Environ Microbiol 68:1312–1318
Thamdrup B, Dalsgaard T, Jensen MM, Ulloa O, Farias L, Escribano R (2006) Anaerobic ammonium oxidation in the oxygen-deficient waters off northern Chile. Limnol Oceanogr 51:2145–2156
Trimmer M, Nicholls JC, Deflandre B (2003) Anaerobic ammonium oxidation measured in sediments along the Thames estuary, United Kingdom. Appl Environ Microbiol 69:6447–6454
Trimmer M, Nicholls JC, Morley N, Davies CA, Aldridge J (2005) Biphasic behavior of anammox regulated by nitrite and nitrate in estuarine sediment. Appl Environ Microbiol 71:930
van de Vossenberg J, Rattray JE, Geerts W, Kartal B, van Niftrik L, van Donselaar EG, Sinninghe Damsté JS, Strous M, Jetten MSM (2008) Enrichment and characterization of marine anammox bacteria associated with global nitrogen gas production. Environ Microbiol 10:120–312
Whitman WB (1998) Perspective: prokaryotes: the unseen majority. Proc Natl Acad Sci 95:6578–6583
Woebken D, Lam P, Kuypers MM, Naqvi SW, Kartal B, Strous M, Jetten MSM, Fuchs BM, Amann R (2008) A microdiversity study of anammox bacteria reveals a novel Candidatus Scalindua phylotype in marine oxygen minimum zones. Environ Microbiol 10:3106–3119
Woebken D, Fuchs BM, Kuypers MM, Amann R (2007) Potential interactions of particle-associated anammox bacteria with bacterial and archaeal partners in the Namibian upwelling system. Appl Environ Microbiol 73:4648–4657
Wu JF, Chung SW, Wen LS, Liu KK, Chen YLL, Chen HY, Karl DM (2003) Dissolved inorganic phosphorus, dissolved iron, and Trichodesmium in the oligotrophic South China Sea. Glob Biogeochem Cycles 17:1008–1010
Zhang Y, Ruan X-H, Op den Camp HJM, Smits TJM, Jetten MSM, Schmid MC (2007) Diversity and abundance of aerobic and anaerobic ammonium-oxidizing bacteria in freshwater sediments of the Xinyi River (China). Environ Microbiol 9:2375–2382
Acknowledgments
This research was supported by the National Natural Science Foundation of China (30800032, 41076095); PhD studentship (ML) and in part by the Agriculture, Fisheries and Conservation Department of the Hong Kong SAR Government (J-DG); the Knowledge Innovation Key Project of The Chinese Academy of Sciences (KZCX2-YW-QN207); and South China Sea Open Cruise by R/V Shiyan 3, South China Sea Institute of Oceanography, CAS. We thank Jessie Lai for general laboratory assistance during this study at The University of Hong Kong.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yi-Guo Hong and Meng Li contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM
(DOC 324 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hong, YG., Li, M., Cao, H. et al. Residence of Habitat-Specific Anammox Bacteria in the Deep-Sea Subsurface Sediments of the South China Sea: Analyses of Marker Gene Abundance with Physical Chemical Parameters. Microb Ecol 62, 36–47 (2011). https://doi.org/10.1007/s00248-011-9849-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-011-9849-0