
Abstract
Soon after the origin of RNA-based life, depletion of prebiotically synthesised ribonucleotides would have driven the evolution of a biosynthetic pathway to these key building blocks. Ribozyme-catalysed nucleosidation—the key biosynthetic step—requires that ribose and the nucleobases are produced by abiotic chemistry and are relatively stable to the conditions of their synthesis. The most plausible prebiotic synthesis of sugars involves photoreduction of cyanohydrins by hydrogen sulphide in the presence of copper(I) cyanide, and we therefore subjected ribose to these conditions whereupon it was partially converted to 2-deoxyribose. Furthermore, a derivative of uracil is reduced under similar conditions to thymine. Thus, DNA biosynthetic precursors can be formed abiotically from those of RNA allowing for an early evolutionary transition to life based on RNA and DNA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
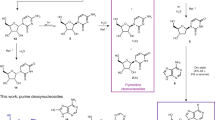
There is a substantial body of evidence that RNA played both catalytic and informational roles at the dawn of life (Joyce 2002). Furthermore, an efficient prebiotic synthesis of the pyrimidine ribonucleotides 1 (Base = Cyt/Ura) has been demonstrated (Scheme 1, black arrows) which suggests that the building blocks of RNA would have been available at the very origin of life (Powner et al. 2009; Szostak 2009; Powner and Sutherland 2010). In this synthesis, 2-aminooxazole 12 reacts with glyceraldehyde 8 to give the aminooxazolines 9, predominantly as the ribo- and arabino-configured stereoisomers (Anastasi et al. 2006). Reaction of 9 with cyanoacetylene 13 and ensuing phosphorylation and irradiation yields 1 (Base = Cyt/Ura), with the free pyrimidine nucleobases cytosine (Cyt–H) 15 and uracil (Ura–H) 16 being major by-products of the process (Powner et al. 2009). A variation of the synthesis of 1 can be conceived (Scheme 1, orange arrows) if 6 and 8 first undergo aldol reaction giving the aldopentoses 11 (Harsch et al. 1984; Kofoed et al. 2005), which could subsequently condense with cyanamide 10 (Sanchez and Orgel 1970; Borsenberger et al. 2004; Springsteen and Joyce 2004) again yielding the aminooxazolines 9. In either case, glycolaldehyde 6 or glyceraldehyde 8 are key, and we recently demonstrated their abiotic syntheses through the reductive homologation of hydrogen cyanide 2 (Scheme 1, blue arrows) (Ritson and Sutherland 2012; Ritson and Sutherland 2013). The reducing power for this process is provided by hydrated electrons and protons from a copper(I)⇔copper(II) photoredox cycle fuelled by hydrogen sulphide 3 (Ritson and Sutherland 2013). It is noteworthy that 2 is also the starting material for prebiotic syntheses of adenine (Ade-H) 17 and guanine (Gua-H) 18 (Sanchez et al. 1967) and these nucleobases have been converted to the corresponding ribonucleotides 1 (Base = Ade/Gua), albeit in low yield (Lohrmann and Orgel 1971; Fuller et al. 1972).

Systems chemistry synthesis of activated pyrimidine ribonucleotides
Hence, the production of ribose and the free nucleobases would appear to be an intrinsic part of the abiotic synthesis of ribonucleotides. Interestingly, ribozymes catalysing the efficient ribosylation of pyrimidine and purine nucleobases have already been isolated from random sequence RNA pools (Unrau and Bartel 1998; Chapple et al. 2003; Lau et al. 2004) suggesting the involvement of this process in early biosynthesis. However, this would require that the nucleobases 15–18 and ribose were stable prebiotic products. Ribose is renowned for it’s relative instability and is unlikely to have accumulated in the free form (Larralde et al. 1995) but chemically stable precursors, such as cyanohydrins 5 and 7, could have accumulated, and then been converted to ribose abiotically in the early biotic era. This possibility would require that ribose be at least partly stable to the conditions under which 5 and 7 are reduced to glycolaldehyde 6 and glyceraldehyde 8, respectively, and we thus set out to investigate this.
Materials and Methods
General Experimental
Reagents and solvents were bought from Sigma-Aldrich, Alfa Aesar and Santa Cruz Biotechnology and were used without further purification. Photochemical reactions were carried out using a Rayonet RPR-200 photochemical reactor chamber (Hg bulbs with a principle emission of 254 nm) in Spectrosil quartz cuvettes. A Mettler Toledo SevenEasy pH Meter S20 was used to monitor pH, and deoxygenation of solutions was achieved by sparging argon through the solution for 20 min. 1H and 13C NMR spectra were acquired using a Bruker Ultrashield 400 Plus machine (at 400.1 and 100.6 MHz, respectively). Samples consisting of H2O/D2O mixtures were analysed using HOD suppression to collect 1H NMR data. The notations s, d and br s represent singlet, doublet and broad signal and chemical shifts (δ) are given in ppm. Low resolution mass spectrometry was performed using an Agilent Technologies 1200 series/6130 Quadrupole LC/MS.
Procedure for Photochemical Reduction of Ribose/Arabinose (Ribo-/Arabino-11) to 2-Deoxyribose 19
H2O/D2O (2.5/0.4 mL) was degassed and NaH2PO4.2H2O (0.100 mmol, 16 mg), KSCN (0.030 mmol, 3 mg), ribose/arabinose (0.030 mmol, 4.5 mg) and NaSH.xH2O (60 %, 0.030 mmol, 3 mg) were added and the solution was adjusted to pH 7 using degassed NaOH/HCl. The mixture was then transferred to a quartz cuvette containing CuCN (ca. 0.5 mg, 20 mol%) which was sealed immediately. The cuvette was placed in the Rayonet reactor and the cooling fan was turned on (temperature inside reactor ca. 37 °C). In the absence of mechanical stirring the reaction was sonicated for 10 s after 3 h of irradiation.
Procedure for Photochemical Reduction of Uracil 16 to 5,6-Dihydrouracil 22
H2O/D2O (2.5/0.4 mL) was degassed and NaH2PO4.2H2O (0.100 mmol, 16 mg), KSCN (0.030 mmol, 3 mg) and NaSH.xH2O (60 %, 0.100 mmol, 9 mg) were added and the solution was adjusted to pH 7 using degassed NaOH/HCl. Uracil (0.030 mmol, 3.3 mg) was then added and the tube was sealed and agitated until the uracil had dissolved. The mixture was transferred to a quartz cuvette which was then sealed immediately. The cuvette was placed in the Rayonet reactor and the cooling fan was turned on (temperature inside reactor ca. 37 °C). The mixture was irradiated for the desired amount of time at which point an aliquot of the reaction mixture was removed and examined by NMR spectrometry.
Procedure for Photochemical Reduction of 5-Mercaptomethyluracil 21 to Thymine 24
H2O/D2O (2.5/0.4 mL) was degassed then 15 % (0.44 mL) of the solution was removed. NaH2PO4.2H2O (0.200 mmol, 32 mg), KSCN (0.030 mmol, 3 mg) and NaSH.xH2O (60 %, 0.030 mmol, 3 mg) were added and the solution was adjusted to pH 6 using degassed NaOH/HCl. Degassed formamide (0.44 mL) and 5-mercaptomethyluracil 21 (0.030 mmol, 4.7 mg) were added and the tube was sealed. The tube was agitated/sonicated until the solid had dissolved and the resultant solution was transferred to a quartz cuvette which was then sealed immediately. The cuvette was placed in the Rayonet reactor and the cooling fan was turned on (temperature inside reactor ca. 37 °C). In the absence of mechanical stirring the reaction was sonicated for 10 s after 2 h of irradiation. The mixture was irradiated for the desired amount of time at which point an aliquot of the reaction was removed and examined by NMR spectrometry.
Results and Discussion
The system we employed for the reduction of 5 and 7 was made by mixing a phosphate-buffered solution of the cyanohydrin and hydrogen sulphide 3 with solid catalytic copper(I) cyanide (Ritson and Sutherland 2013). In addition, it appeared that thiocyanate made the copper catalyst a more competent reductant, and so we examined it’s effect here also. Optimal reduction of the cyanohydrins to the aldehydic products 6 and 8 was observed after 2–6 h of UV irradiation of the system. We thus investigated the stability of ribose in the same system and time frame.
Monitoring the reaction by 1H NMR spectroscopy (Fig. 1) revealed that ribose was not fully stable to the reducing conditions, and after 6 h partial conversion to another sugar product or products was immediately suggested by three new anomeric proton signals downfield of the HOD signal (Fig. 1a, b). That the product or products were the result of reduction was suggested by the appearance of new signals which were upfield from any due to ribose. Suspecting that ribose might have been reduced to 2-deoxyribose 19 (Scheme 2), we compared the spectrum of the reaction products (Fig. 1b) with a spectrum of a commercial sample of 19 (Fig. 1c), whereupon this suspicion was confirmed—the three new anomeric proton signals corresponded to the α- and β-furanose and β-pyranose forms of 19. The anomeric signal for the α-pyranose form of 19 was obscured by the HOD signal both in the spectrum of the reaction products and in that of the standard. Further support for the assignment of 19 as the reaction product was provided by comparison of the midfield region of spectra (Supplementary Fig. S1a) and sample spiking with a commercial standard. Integration of signals for ribose and 2-deoxyribose 19 relative to those of an added standard enabled quantification of the conversion after 6 h of irradiation. It was found that there was 42 % residual ribose and that 19 had been produced in 52 % yield leaving only 6 % of the starting material unaccounted for presumably as minor unidentified products. Thus, a continuous reduction and aldolisation process for the abiotic formation of ribose from the cyanohydrins 5 and 7 can be envisaged, but 2-deoxyribose 19 would appear to be an inevitable by-product.

Partial 1H NMR analysis of the effect of exposing ribose (ribo-11) to the photoreduction conditions that generate the C2 and C3 sugars 6 and 8 from their cyanohydrin precursors 5 and 7. a Spectrum of ribose in D2O. b Spectrum of the photoreduction products of ribose in H2O-D2O (with HOD signal suppression). c Spectrum of 2-deoxyribose 19 in D2O. Full 1H NMR spectra are shown in Supplementary Figs. S1a, b. The various tautomers of ribo-11 and 19 are indicated in parentheses: α and β refer to anomeric hydroxyl group stereochemistry; f and p refer to furanose and pyranose forms

Prebiotic syntheses of 2-deoxyribose 19
We considered that α-deoxygenation of the aldehydic form of ribose to be the likely mechanism and, therefore, that arabinose, arabino-11, another major product of the aldolisation of 6 and 8 (Harsch et al. 1984; Kofoed et al. 2005), would also be photoreduced to the same product. In the event, arabino-11 was reduced to 19 in 43 % yield (Scheme 2, Supplementary Figs. S4a, b). Investigating the system further we found that CuCN and H2S 3 were essential for the α-deoxygenation of ribo-11 or arabino-11 to proceed, whereas thiocyanate could be omitted, although the efficiency of the reaction decreased by a factor of ca. 3. This is in accordance with our previous findings and suggests that thiocyanate binds to CuCN making a more efficient catalyst for the photochemical production of hydrated electrons (Ritson and Sutherland 2013). The prebiotic synthesis of 19 has been claimed before via an aldol reaction of acetaldehyde and glyceraldehyde 8 to give 2-deoxyribose 19 in about 3 % yield (Scheme 2) (Oró and Cox 1962). In contrast, aldol reaction of glycolaldehyde 6 and 8 gives ribose in 20 % yield and arabinose in 14 % yield (Kofoed et al. 2005) and we have demonstrated photoreduction of ribose and arabinose to 19 in 52 and 43 % yield, respectively.
With a developing model for the abiogenesis of the sugars needed for early nucleic acid biosynthesis, we next turned our attention back to the nucleobases. Although 15–18 are intrinsically relatively stable, we recognised that they might undergo photochemical reduction or reaction with aldehyde intermediates during prebiotic ribonucleotide synthesis. In particular, we were concerned about uracil 16 as the 5,6-double bond of this nucleobase struck us as prone to saturation under the photoreduction conditions. Furthermore, 16 is known to undergo reaction with formaldehyde 4 giving rise in moderate yield to 5-hydroxymethyluracil 20 which, in turn, is converted essentially quantitatively to 5-mercaptomethyluracil 21 by reaction with hydrogen sulphide 3 (Scheme 3) (Robertson and Miller 1995).

Chemistry of uracil 16 under the conditions of sugar synthesis
We first investigated the stability of the nucleobases 15–17 towards the photoreduction conditions (guanine 18 was not investigated because it is insufficiently soluble for NMR analysis). Cytosine 15 and adenine 17 proved stable, but as we had suspected uracil 16 was not and was efficiently converted into two new products according to 1H NMR analysis (Supplementary Fig. S2a). One of these, dihydrouracil 22, was indeed the product of reduction of the 5,6-double bond, and the other was the photohydrate 23 (Scheme 3) (Moore 1958). After 16 h of irradiation, 9 % of 16 remained, while 22 was produced in 62 % yield and the photohydrate 23 in 29 % yield. However, it is known that 23 undergoes elimination of water to regenerate uracil 16, thus the partial production of 23 under the photoreduction conditions effectively protects a sizeable portion of 16 from reduction. The water elimination reaction to regenerate 16 is extremely slow at room temperature, but can be accelerated by heating (Supplementary Fig. S2b). Unlike the reduction of ribose to 2-deoxyribose, the photoreduction of 16 neither requires copper nor thiocyanate, although their presence does not affect the reduction.
We next considered the consequences of some of the prebiotically synthesised uracil 16 having reacted with formaldehyde 4. As thiolysis of the initial hydroxymethylation product 20 to 5-mercaptomethyluracil 21 is so efficient (Robertson and Miller 1995), we reasoned that 20 would not accumulate in the presence of hydrogen sulphide 3 in the dark, but that 21 would. We then wondered what would happen if 21 was subsequently subjected to the photoreduction conditions. Although the 5,6-double bond of 21, like that of uracil 16, is potentially prone to saturation, the C–S bond is also a potential site of reduction. In the event, the C–S bond proved more susceptible to the reduction conditions, and the thiomethyl group of 21 was converted to a methyl group giving thymine 24 in 36 % yield after 6 h (Supplementary Figs. S3a, b). 5-Hydroxymethyluracil 20 has been reduced directly to 24 in 3 % yield by heating in 2 M formic acid (Choughuley et al. 1977), conditions which are prebiotically questionable. In contrast, the conversion of 20 to 5-mercaptomethyluracil 21 occurs by heating at neutral pH and proceeds in 99 % yield (Robertson and Miller 1995), and we have demonstrated photoreduction of 21 to 24 in 36 % yield. As with the photoreduction of uracil 16, we found that neither copper nor thiocyanate were required for the reaction, although their inclusion in the system did not inhibit the reduction of 21 to thymine.
We have not investigated in detail the mechanisms by which the various photoreductions described herein proceed. However, we have ascertained whether all the components of the system are necessary for reduction to occur and this provides certain mechanistic clues (for a full discussion see Supplementary Table S1 and Scheme S1). Whatever the mechanisms of the reactions we have discovered, the fact that a common set of reactants and conditions converts biosynthetic precursors of RNA to those of DNA is noteworthy in the context of the earliest biology. If the ribozymes responsible for the biosynthesis of ribonucleotides had relaxed substrate specificity, they could also have been able to synthesise deoxyribonucleotides including thymidine. Furthermore, if the earliest nucleic acid polymerase also had relaxed substrate specificity, a mixed nucleic acid R/DNA, or MNA, might have been produced (Sutherland and Whitfield 1997; Trevino et al. 2011). Such a polymer offers a potential advantage over a single pure polymer in that those variants of a particular genetic sequence that is deoxyribose rich, R/DNA, would be more easily copied, whereas those which were ribose rich, R/DNA, would have been more structured, and thus more suited to catalytic roles. By polymerase gene duplication and specificity refinement, biology could evolve along a smooth trajectory from being based on R/DNA to being based on RNA plus DNA. Finally, the presence of environmental dihydrouracil 22—from the partial reduction of uracil 16 as described herein—would have allowed its incorporation into tRNA where it contributes to essential structural flexibility (Yu et al. 2011).
In summary, it appears that RNA monomers and amino acids could have emerged simultaneously during prebiotic synthesis (Ritson and Sutherland 2013) and concurrently components of DNA would have been produced (vide supra). If a suitable abiotic coupling/activation chemistry was found, polymers of amino acids and ribonucleotides could form at a similar time and would be present before the earliest forms of life. MNA or DNA, however, would have to wait for biology to develop the requisite N-glycosidation ribozyme before deoxyribonucleotides could be biosynthesised.
References
Anastasi C, Crowe MA, Powner MW, Sutherland JD (2006) Direct assembly of nucleotide precursors from two- and three-carbon units. Angew Chem Int Ed 45:6176–6179
Borsenberger V, Crowe MA et al (2004) Exploratory studies to investigate a linked prebiotic origin of RNA and coded peptides. Chem Biodivers 1:203–246
Chapple KE, Bartel DP, Unrau PJ (2003) Combinatorial minimization and secondary structure determination of a nucleotide synthase ribozyme. RNA 9:1208–1220
Choughuley ASU, Subbaraman AS, Kazi ZA, Chadha MS (1977) A possible prebiotic synthesis of thymine: uracil-formaldehyde-formic acid reaction. BioSystems 9:73–80
Fuller WD, Sanchez RA, Orgel LE (1972) Studies in prebiotic synthesis: VI. Synthesis of purine nucleosides. J Mol Biol 67:25–33
Harsch G, Bauer H, Voelter W (1984) Kinetik, Katalyse und mechanismus der sekundärreaktion in der schlußphase der formose-reaktion. Liebigs Ann Chem 1984:623–635
Joyce GF (2002) The antiquity of RNA-based evolution. Nature 418:214–221
Kofoed J, Reymond J-L, Darbre T (2005) Prebiotic carbohydrate synthesis: zinc-proline catalyses direct aqueous aldol reactions of α-hydroxy aldehydes and ketones. Org Biomol Chem 3:1850–1855
Larralde R, Robertson MP, Miller SL (1995) Rates of decomposition of ribose and other sugars: implications for chemical evolution. Proc Natl Acad Sci USA 92:8158–8160
Lau MWL, Cadieux KE, Unrau PJ (2004) Isolation of fast purine nucleotide synthase ribozymes. J Am Chem Soc 126:15686–15693
Lohrmann R, Orgel LE (1971) Urea-inorganic phosphate mixtures as prebiotic phosphorylating agents. Science 171:490–494
Moore AM (1958) Ultraviolet irradiation of pyrimidine derivatives: II. Note on the synthesis of the product of reversible photolysis of uracil. Can J Chem 36:281–283
Oró J, Cox AC (1962) Non-enzymic synthesis of 2-deoxyribose. Fed Proc 21:80
Powner MW, Sutherland JD (2010) Phosphate-mediated interconversion of ribo- and arabino-configured prebiotic nucleotide intermediates. Angew Chem Int Ed 49:4641–4643
Powner MW, Gerland B, Sutherland JD (2009) Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459:239–242
Ritson D, Sutherland JD (2012) Prebiotic synthesis of simple sugars by photoredox systems chemistry. Nature Chem. 4:895–899
Ritson DJ, Sutherland JD (2013) Synthesis of aldehydic ribonucleotide and amino acid precursors by photoredox chemistry. Angew Chem Int Ed 52:5845–5847
Robertson MP, Miller SL (1995) Prebiotic synthesis of 5-substituted uracils: a bridge between the RNA world and the DNA-protein world. Science 268:702–705
Sanchez RA, Orgel LE (1970) Studies in prebiotic synthesis: V. Synthesis and photoanomerization of pyrimidine nucleosides. J Mol Biol 47:531–543
Sanchez RA, Ferris JP, Orgel LE (1967) Studies in prebiotic synthesis: II. Synthesis of purine precursors and amino acids from aqueous hydrogen cyanide. J Mol Biol 30:223–253
Springsteen G, Joyce GF (2004) Selective derivatization and sequestration of ribose from a prebiotic mix. J Am Chem Soc 126:9578–9583
Sutherland JD, Whitfield JN (1997) Prebiotic chemistry: A bioorganic perspective. Tetrahedron 53:11493–11527
Szostak JW (2009) Origins of life: Systems chemistry on early Earth. Nature 459:171–172
Trevino SG et al (2011) Evolution of functional nucleic acids in the presence of nonheritable backbone heterogeneity. Proc Natl Acad Sci USA 108:13492–13497
Unrau PJ, Bartel DP (1998) RNA-catalysed nucleotide synthesis. Nature 395:260–263
Yu F et al (2011) Molecular basis of dihydrouridine formation on tRNA. Proc Natl Acad Sci USA 108:19593–19598
Acknowledgments
This work was supported by the Medical Research Council (project number: MC_UP_A024_1009), a grant from the Simons Foundation (Award Number: 290362 to JDS), and an award from the Origin of Life Challenge—we thank Harry Lonsdale for the latter. We also thank Drs. Stefan Freund and Trevor Rutherford for assistance with NMR spectroscopy.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Ritson, D.J., Sutherland, J.D. Conversion of Biosynthetic Precursors of RNA to Those of DNA by Photoredox Chemistry. J Mol Evol 78, 245–250 (2014). https://doi.org/10.1007/s00239-014-9617-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-014-9617-0