
Abstract
Assessing the potential of a new drug to cause drug-induced liver injury (DILI) is a challenge for the pharmaceutical industry. We therefore determined whether cell models currently used in safety assessment (HepG2, HepaRG, Upcyte and primary human hepatocytes in conjunction with basic but commonly used endpoints) are actually able to distinguish between novel chemical entities (NCEs) with respect to their potential to cause DILI. A panel of thirteen compounds (nine DILI implicated and four non-DILI implicated in man) were selected for our study, which was conducted, for the first time, across multiple laboratories. None of the cell models could distinguish faithfully between DILI and non-DILI compounds. Only when nominal in vitro concentrations were adjusted for in vivo exposure levels were primary human hepatocytes (PHH) found to be the most accurate cell model, closely followed by HepG2. From a practical perspective, this study revealed significant inter-laboratory variation in the response of PHH, HepG2 and Upcyte cells, but not HepaRG cells. This variation was also observed to be compound dependent. Interestingly, differences between donors (hepatocytes), clones (HepG2) and the effect of cryopreservation (HepaRG and hepatocytes) were less important than differences between the cell models per se. In summary, these results demonstrate that basic cell health endpoints will not predict hepatotoxic risk in simple hepatic cells in the absence of pharmacokinetic data and that a multicenter assessment of more sophisticated signals of molecular initiating events is required to determine whether these cells can be incorporated in early safety assessment.
Similar content being viewed by others

Introduction
Drug-induced liver injury (DILI) poses a serious issue not only for patients and healthcare professionals, but also for the pharmaceutical industry and regulatory authorities. This is mainly due to the occurrence of human-specific and idiosyncratic adverse reactions at the clinical and post-marketing stages, thus leading to the termination of drug development, black box warnings or even withdrawal of drugs from the market (Bell and Chalasani 2009; Marino et al. 2001).
In vitro models, such as the human hepatocarcinoma cell line HepG2, are widely used across the pharmaceutical and chemical industries, as an initial screen to determine the likely risk of new chemical entities (NCEs) eliciting DILI in man. This is because (1) they can be applied quickly to generate reproducible data, (2) they are amenable to high-content screening and (3) they are relatively cheap and readily available (Gerets et al. 2012; O’Brien et al. 2006). Although not of human origin, rodent liver-derived cell lines and primary hepatocytes (which are more readily available compared with their human counterpart) are also routinely used. However, interspecies differences are known to have a significant impact on predictivity of DILI and non-DILI compounds (Olson et al. 2000). On the other hand, primary human hepatocytes (PHH) are not generally used in such a first screen as they exhibit inter-individual variation, thereby potentially confounding analysis and interpretation of intra-study and/or intra-project results between different NCEs; they are also expensive to acquire, and they undergo significant de-differentiation during culture (Guillouzo et al. 2007; Madan et al. 2003; Richert et al. 2006). However, PHH cultured in vitro are still considered to be useful in the testing paradigm, as the nearest representation of the key metabolically active cell of the liver.
Since DILI continues to pose significant problems in drug development, this suggests that currently used in vitro models are not appropriate for effective screening, but a comprehensive, multicenter, unbiased assessment to test this unequivocally has never been performed. Therefore, the Innovative Medicines Initiative (IMI)-funded consortium ‘Mechanism-based Integrated Systems for the Prediction of Drug-Induced Liver Injury’ (MIP-DILI) has assessed current in vitro cell models, using an evidence-based panel of compounds implicated in DILI in man, in order to determine whether any of these simple cell models per se are actually predictive of human DILI. Furthermore, by using a small panel of DILI- and non-DILI-implicated compounds, and basic measures of cell health, we were able to monitor reproducibility across different sites, thereby ensuring that our data should be more definitive than any currently available.
Materials and methods
PHH, HepG2, HepaRG and Upcyte cells (Table 1) in conjunction with a particular endpoint (referred to as cell models) were evaluated using harmonized protocols which were designed and agreed by all test site participants as detailed below, for their ability to predict DILI liability of NCEs. For the assessment of each cell model, the same protocol was used by all the test sites involved and the supplier and product codes of all materials and reagents included. Each training compound was sourced by all test sites from the same supplier and acquired the same batch/lot number. Plasticware and other reagents such as media and media supplements were sourced by all the test sites from the suppliers and product codes stated in the protocols; however, standardization did not extend to sourcing these items from specific batch/lot numbers. To determine inter-laboratory variation, several of the cell models (cryopreserved PHH, cryopreserved HepaRG, HepG2/ECACC and Upcyte cells) were evaluated by at least two test sites (Table 2). Furthermore, a simple experimental protocol was designed (Fig. 1) and basic endpoints were chosen for our cell models, i.e., resorufin and ATP assays for the assessment of cell viability, to allow generation of rapid data by the test sites involved that could be easily compared.

Cytotoxicity study design. Primary human hepatocytes (cryopreserved and fresh), HepG2 (ECACC and TS clones), HepaRG (cryopreserved and fresh), and Upcyte cells were seeded on the days indicated followed by exposure to the thirteen training compounds (Table 4) as detailed in the method section for 24 or 72 h. After compound treatment, cell viability was assessed by measurement of intracellular ATP and resorufin which is the product when resazurin is reduced (a measure of cellular metabolism)
Cell culture
The different seeding densities used are the optimal for each cell type. Data generated from each cell model was first analyzed relative to the corresponding untreated controls and then comparisons were made across the different cell models. Donor details for the fresh and cryopreserved PHH as well as the Upcyte cells are summarized in Table 3. Allocation of PHH donors for each test site is also summarized in Table 3. Pre-qualification for choosing the five cryopreserved PHH donors was based on their viability post-thawing, ability to adhere to cell culture plates, metabolic profile and material availability to ensure there was sufficient supply for the planned experimental work and any future studies. Donor demographics were collected as summarized in Table 3; however, these were not considered a priority during the pre-qualification stage as the aim was to select a cohort of the population in an unbiased manner.
Cryopreserved primary human hepatocytes
Cryopreserved PHH from five donors were provided by KaLy-Cell (Plobsheim, France). The human biological samples were sourced ethically and their research use was in accord with the terms of the informed consents. The hepatocytes were thawed in KLC-Thawing Medium (KLC-TM; proprietary medium composition), centrifuged (168×g; 20 min; room temperature), washed in KLC-Washing Medium (KLC-WM; proprietary medium composition; 100×g; 5 min; room temperature) and resuspended in KLC-Seeding Medium (KLC-SM; Williams’ Medium E (Life Technologies, Paisley, UK) supplemented with 10 % heat-inactivated fetal calf serum (FCS, Life Technologies, Paisley, UK), 1 µM dexamethasone (Sigma-Aldrich, St. Louis, MO, USA), 4 µg/mL insulin [(Life Technologies, Paisley, UK) and 10 units penicillin/10 µg streptomycin (Life Technologies, Paisley, UK)]. Cell number and viability were determined by the trypan blue exclusion method, and the cells were plated at a seeding density of 70,000 cells/well of a KaLy-Cell home-coated type I rat tail collagen (10 µg/well) 96-well plate. This seeding density has been optimized and recommended by KaLy-Cell to achieve ≥80 % confluence in a 96-well plate format 24 h after plating. The cells were allowed to attach for 4–6 h (37 °C; 5 % CO2; 95 % air) after which the cells were overlaid with 0.25 mg/mL matrigel in KLC-Seeding Medium and left to incubate overnight (37 °C; 5 % CO2; 95 % air). Cells were used for analysis if the attachment efficiency was ≥80 %. Serum-free KLC-Maintenance Medium (KLC-MM; Hepatocyte Maintenance Medium (Lonza, Basel, Switzerland) supplemented with 1 µM dexamethasone (Sigma-Aldrich, St. Louis, MO, USA), 4 µg/mL insulin (Life Technologies, Paisley, UK) and 10 units penicillin/10 µg streptomycin (Life Technologies, Paisley, UK) was used for compound treatment.
Fresh primary human hepatocytes
Fresh primary human hepatocytes isolated from a different donor (UoL49) to the five cryopreserved PHH donors were also included in our analysis. A liver resection from donor UoL49 was received as surgical waste from Aintree Hospital, Liverpool, United Kingdom, with full patient consent and ethical approval from the relevant authorities (National Research Ethics Service REC reference: 11/NW/0327). Upon collection, the specimen was immediately transported to the University of Liverpool, Liverpool, United Kingdom, on ice in HEPES-buffered saline [HBS; 10 mM Hepes (Sigma-Aldrich, St. Louis, MO, USA), 5 mM KCl (Fisher Scientific, Loughborough, UK), 136 mM NaCl (Fisher Scientific, Loughborough, UK) and 0.5 % glucose (Fisher Scientific, Loughborough, UK)]. Hepatocyte isolation was performed using a 2-step collagenase method as described previously (LeCluyse et al. 2005). Briefly, blood was removed from the liver tissue by blanching through perfusion with HBS pre-warmed to 37 °C and subsequently digested with 0.25 mg/mL collagenase A/IV (Sigma-Aldrich, St. Louis, MO, USA) in HBS containing 0.7 mM CaCl2 (Fisher Scientific, Loughborough, UK), also pre-warmed to 37 °C. The capsule was opened and the hepatocytes released and collected in cold Williams’ Medium E (Sigma-Aldrich, St. Louis, MO, USA). The hepatocytes were separated from the undigested tissue remnants with a 125-μm mesh, washed twice with cold Williams’ Medium E by centrifugation (80×g; 5 min; 4 °C), and the hepatocytes resuspended in cold complete Williams’ Medium E [supplemented with 1 % insulin–transferrin–selenium (Life Technologies, Paisley, UK), 2 mM l-glutamine (Sigma-Aldrich, St. Louis, MO, USA), 100 nM dexamethasone (Sigma-Aldrich, St. Louis, MO, USA), and 1 % penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA)]. Cell number and viability were determined using the trypan blue exclusion method, and the cells were plated at a seeding density of 100,000 cells/well in a BD Biocoat Collagen I 96-well plate (Becton and Dickinson, New Jersey, USA). This seeding density was determined to be optimal for these cells in a 96-well plate format to achieve ≥80 % confluence 24 h after plating. The cells were allowed to attach for 3 h (37 °C; 5 % CO2; 95 % air), the incubation medium removed, and the attached cells overlaid with 0.25 mg/mL matrigel (Becton and Dickinson, New Jersey, USA) in cold complete Williams’ Medium E. The cells were further incubated overnight (37 °C; 5 % CO2; 95 % air) prior to dosing. The cell viability after hepatocyte isolation was 97 %, and the cells were 85 % confluent 16 h post-plating. Complete Williams’ Medium E was serum-free and was also used for compound treatment.
Cryopreserved HepaRG cells
Differentiated cryopreserved HepaRG cells were purchased from Biopredic International (Saint-Gregoire, France). The cells were thawed in Thawing/Plating Medium [Williams’ Medium E supplemented with 1 % penicillin/streptomycin, 1 % l-Glutamine, 10 % FCS and 12.5 % ADD670 cocktail (BioPredic International, Saint-Gregoire, France; proprietary medium composition)], centrifuged (500×g; 5 min; room temperature) and then resuspended in fresh Thawing/Plating Medium. After the addition of the ADD670 cocktail, the final DMSO concentration in the Thawing/Plating Medium was 0.5 %. Cell number and viability were determined by the trypan blue exclusion method, and the cells were plated at a seeding density of 72,000 cells/well in white/clear bottom 96-well plates (Beckton Dickinson, New Jersey, USA). The cells were cultured (37 °C; 5 % CO2; 95 % air) for 24 h after which the Thawing/Plating Medium was replaced with a specified Tox medium (Williams’ Medium E supplemented with 1 % penicillin/streptomycin, 1 % l-Glutamine, and 12.5 % ADD650 cocktail [Biopredic International, Saint-Gregoire, France, proprietary medium composition]). The Tox medium was serum- and DMSO-free. Cells were incubated for another 48 h without additional medium change and subsequently used for experimentation, if the attachment efficiency was at or above 85 %. For compound treatment, the Tox medium was used.
Fresh HepaRG cells
Undifferentiated HepaRG cells were purchased from Biopredic International (Saint-Gregoire, France). For differentiation, one vial of cells was thawed and seeded in a 75-cm2 flask in proliferation medium [Williams’ Medium E supplemented with 2 mM l-Glutamine, 10 % fetal calf serum and ADD710 cocktail (BioPredic International, Saint-Gregoire, France; proprietary medium composition)]. The proliferation medium did not contain DMSO. The cells were allowed to recover and proliferate in culture (37 °C; 5 % CO2; 95 % air) for 2 weeks with a medium change of every 2–3 days. The proliferation medium was then replaced with the differentiation medium [Williams’ Medium E supplemented with 2 mM l-Glutamine, 10 % fetal calf serum, and ADD720 cocktail (Biopredic International, Saint-Gregoire, France; proprietary medium composition)]. After the addition of the ADD720 cocktail, the final DMSO concentration in the differentiation medium was 1.7 %. The cells were maintained in culture for 2 weeks (37 °C; 5 % CO2; 95 % air) after which the cells have reached a hepatocyte-like morphology. Differentiated cells were harvested and seeded in 96-well plates at a density of 72,000 cells/well in differentiation medium. The cells were used for the toxicity study 5 days after seeding. For compound treatment, serum- and DMSO-free Tox medium was used [Williams’ Medium E supplemented with 2 mM l-Glutamine and ADD650 cocktail (Biopredic International, Saint-Gregoire, France; proprietary medium composition)].
HepG2/ECACC and HepG2/TS cells
A specific clone of HepG2 cells was purchased from the European Collection of Cell Cultures (ECACC). The cells were cultivated and low passage frozen stocks were generated and banked by one MIP-DILI partner designated as the cell bank. The cells were distributed to the relevant MIP-DILI partners. The HepG2 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Lonza, Basel, Switzerland) supplemented with 10 % FBS (Lonza, Basel, Switzerland), 1 % penicillin/streptomycin (Lonza, Basel, Switzerland), 1 % l-glutamine (Lonza, Basel, Switzerland) and 1 % non-essential amino acids (NEAA; Sigma-Aldrich, St. Louis, MO, USA) and passaged upon reaching 80 % confluence using Trypsin–EDTA (Lonza, Basel, Switzerland). Cell number and viability were determined using the trypan blue exclusion method, and the cells were plated at a seeding density of 20,000 cells/well in BD Biocoat Collagen I 96-well plates (Beckton Dickinson, New Jersey, USA). The cells were then incubated for 24 h (37 °C; 5 % CO2; 95 % air) prior to compound treatment during which serum-free culture medium was used. To determine whether there are differences in response between two clones of the same cell type, an in-house clone of HepG2 from one test site referred to as the HepG2/TS clone was also assessed for its sensitivity to the compounds and compared with the HepG2/ECACC clone. The same conditions were applied during the culture of the HepG2/TS cells with the exception of using a NucleoCounter (NC-100, Chemometec) for cell counting, TrypLE (Life Technologies, Paisley, UK) for passaging, and the seeding density being 30,000 cells/well.
Upcyte hepatocytes
Preliminary assessment of two Upcyte hepatocyte donors with regards to their sensitivity to a small set of compounds was performed prior to the current study and showed no significant difference (data not presented). Furthermore, despite the infant age of the donor (422A) used in the current study, the basal metabolic profile of this donor including phase I and II enzyme activities which are crucial in the clearance of compounds from the body, as provided by the supplier, was not dissimilar to the adult donors. As such, the use of donor 422A was ultimately due to the large amount of material available to allow for future studies to be carried out should the need arise.
Upcyte hepatocytes from donor 422A (Medicyte, Heidelberg, Germany) were thawed in Upcyte Thawing Medium [Upcyte High Performance Medium (Medicyte, Heidelberg, Germany) and 10 % FCS (Fisher Scientific, Paisley, UK)]. The cells were then centrifuged (90×g; 5 min; room temperature) and resuspended in fresh Upcyte High Performance Medium (Medicyte, Heidelberg, Germany). Cell number and viability were determined via the trypan blue exclusion method, and the cells were seeded at 18,750 cells/well in white/clear bottom 96-well plates (Beckton Dickinson, New Jersey, USA). After seeding, the cells were incubated for 72 h (37 °C; 5 % CO2; 95 % air) without additional medium change prior to compound treatment.
Dosing
For this study, DILI compounds are defined as those that have been reported to cause hepatotoxicity in man while non-DILI compounds have not. Postulated mechanisms of hepatotoxicity of the DILI-implicated compounds and corresponding references can be found in Table 4. Final concentrations used in the cell incubations for all compounds are also detailed in Table 4. The compound concentrations used were chosen as representative for each compound when conducting an in vitro safety assessment study. All stock solutions, except for metformin, were prepared as 200-fold stocks in DMSO (Sigma-Aldrich, St. Louis, MO, USA). Stock aliquots were stored at −20 °C and only thawed once. Metformin stock solutions were prepared as 200-fold stocks in distilled water. Cells were dosed either once and exposed for 24 h or once daily over a 3-day period for a 72 h exposure. For the repeat dose, the old dosing solution was discarded each day and replaced with fresh dosing solution to ensure that the compound and DMSO concentrations in which the cells were exposed to were constant throughout the 72-h period. Dosing solutions were prepared fresh every day. To minimize or prevent non-specific binding to proteins, all media used for dosing did not contain serum. Cells were dosed in technical triplicates with a final concentration of 0.5 % DMSO. Controls were cells treated with 0.5 % DMSO in dosing medium, in the absence of compounds.
Resorufin assay
After incubation with the compounds, cell viability was determined using a 4.5 mM stock solution of resazurin (Sigma-Aldrich, St. Louis, MO, USA) prepared in phosphate buffer. This stock solution was added into the cells at 10 % of the cell culture volume to give a final concentration of 450 µM resazurin. The cells were incubated for 1 h (37 °C; 5 % CO2; 95 % air) after which the medium samples were transferred into black, flat-bottomed 96-well plates. Reduction of the resazurin dye results in the highly fluorescent product, resorufin which is measured at 530–560 nm excitation wavelength and 590 nm emission wavelength.
ATP assay
The CellTiter Glo assay kit (Promega, Madison, WI, USA) was used to determine the ATP content in the cells after exposure to the thirteen compounds for 24 or 72 h. After incubation with the compounds, the cells were washed twice with PBS and added with fresh PBS and CellTiter Glo solution in equal volumes. The cells were placed in a plate shaker for 2 min to induce lysis and left to incubate for 10 min at room temperature. The supernatant samples were transferred into opaque flat-bottomed 96-well plates (Greiner-Bio-One, Frickenhausen, Germany) and the luminescence measured.
Data analysis
Cell viability was determined as the percentage of the fluorescent resorufin after incubation with resazurin in the treated cells compared with vehicle control or the percentage of ATP detected in the treated cells compared with vehicle control. Individual EC50 values from all participants were calculated using GraphPad Prism 6 (version 6.03) at one partner site. Concentration series for each compound and cell type were log-transformed and fitted with a sigmoidal regression function assuming a lower viability plateau with increasing compound concentration of 0 and a Hill coefficient of −1 using Python and Prism (GraphPad Software). Differences between two dilution series were computed using F-tests. Differences were considered significant when p < 0.05 and, to compensate for biological significance, when relative EC50-differences > three-fold. ‘No fit’ indicates no decrease in viability of at least one compound in the respective pairwise comparison. Variability coefficients were calculated by dividing the number of tests with significantly different responses by the number of all pairwise comparisons. Hierarchical clustering was performed using Qlucore Omics Explorer 3.1 on mean-centered sigma-normalized data using maximum linkage. Correlation of ATP and resorufin values was calculated using Pearson correlation. Statistical difference between 24 and 72 h toxicity measurements was computed using a paired Student’s t test. In order to relate compound concentrations leading to loss of viability to therapeutic concentrations, the EC50 values were divided by the corresponding Cmax values (EC50/Cmax). References used to obtain these values (µM) are provided in Table 4: amiodarone—0.807; nefazodone—0.859; paracetamol—139; tolcapone—27.81; diclofenac—7.44; ximelagatran—0.3; troglitazone—6.387; perhexiline—1.525; bosentan—7.4; buspirone—0.005; entacapone—1.5; metformin—7.74; pioglitazone—2.672.
Results
The primary aim of this study was to determine, in an unbiased fashion, if several simple cell models (defined as a cell type in conjunction with a specific endpoint), currently used in industry, can distinguish between NCEs with respect to their potential to cause DILI in man, in the absence of exposure data. Nine compounds that are associated with human DILI and four that are not (Table 4) were investigated using four simple cell types: PHH, HepG2, HepaRG and Upcyte cells. In order for this study to be conducted in a standardized manner across multiple centers in Europe (for study scheme and participants, see Fig. 1 and Table 2, respectively), two simple endpoints, ATP and resorufin, were measured to assess toxicity. Our results demonstrated good correlation between these two endpoints for all compounds, time points and cells (r = 0.94, Supplementary Table 1); therefore, only ATP data are shown in the figures in the main body of the paper, for the sake of clarity.
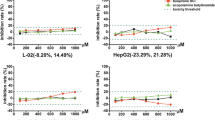
To assess the primary aim of our study, the EC50 values for the ATP response to each of the compounds grouped as DILI and non-DILI, by the HepG2/ECACC, cryopreserved PHH, cryopreserved HepaRG and Upcyte cells after a 24 h and 72 h exposures, are shown in Fig. 2. No clear segregation between the DILI and non-DILI compounds was observed based on the EC50 values for each compound per cell type demonstrating that none of the cell models were able to faithfully distinguish between DILI and non-DILI compounds, at either time point.

Simple-cell models using a basic measure of cell health cannot discriminate between DILI-implicated and non-DILI-implicated compounds. Scattergram of EC50s derived from intracellular ATP content measurements after exposure to the training compounds of each cell type for 24 (a) or 72 h (b), expressed as the mean of multiple determinations carried out across all the test sites involved in assessing each cell type
We then corrected for in vivo exposure by calculating EC50 (ATP)/Cmax for each compound in each cell model at 24 and 72 h (Fig. 3a–f). For clarity, Fig. 3c and d are on a linear scale to aid comparisons between the cell models. Importantly, while no cell model was able to distinguish between DILI- and non-DILI-implicated compounds based on primary EC50 values as obtained in the early stages of drug development, primary human hepatocytes and, to a lesser extent, HepG2 cells can indicate DILI risks when exposure levels are accounted for. As no unequivocal critical EC50/Cmax is defined in the literature, we assessed how many DILI compounds were classified as potentially hepatotoxic using a range of values (Supplementary Fig. 1). With an EC50/Cmax of 20 (Fig. 3e, f), all DILI-implicated compounds are recognized as such in PHH after 72 h (but not after 24 h), with the exception of ximelagatran, while only one of the control compounds falls below the EC50/Cmax = 20 level (entacapone). With the exception of paracetamol, HepG2 cells generated similar profiles to the cryopreserved PHH after exposure to the compounds for 72 h (Fig. 3d).

When Cmax data are available, primary human hepatocytes are the most sensitive of the cell models for the assessment of cellular toxicity. a, b Clustered column plots showing the EC50/Cmax values for all thirteen compounds as detected by all seven forms of the cell models assessed after 24 (a) and 72 h (b). The four compounds not implicated in DILI are indicated. c, d DILI-implicated compounds after 24- (c) and 72-h exposure (d) are shown on a linear axis to aid comparisons between the different cell models. e, f Stacked column plots visualizing the number of compounds classified as toxic by the different cell models with a EC50/Cmax of 20 (red line in panel a) after 24 (e) and 72 h (f). Note that primary human hepatocytes (PHH) are the most sensitive cell type recognizing 8 of the 9 compounds implicated in DILI after 72 h (color figure online)
We then explored inter- and intra-laboratory variation in our data, the effect of inter-donor and clonal variation, as well as differences in sensitivity to the compounds between fresh and cryopreserved cells. As part of our experimental design, cryopreserved PHH, HepG2/ECACC, cryopreserved HepaRG, and Upcyte cells were evaluated by at least two test sites to allow pairwise comparison of the cytotoxicity profiles generated (Table 2). For each of the cell models evaluated, the statistical significance of all pairwise comparisons between cytotoxicity curves was determined. We detected statistically significant inter-laboratory variation in the cytotoxicity profiles generated by each test site that assessed cryopreserved PHH, HepG2/ECACC, and Upcyte cells for their response to all the training compounds. These differences appeared to be of varying degrees, while the Upcytes (12.3 %) showed minor variability between the laboratories, and the HepG2/ECACC (23.4 %) and primary human hepatocytes (48.5 %) elicited clear variable responses. On the other hand, the HepaRG cells showed no difference in all pairwise comparisons performed. Furthermore, the degree in which each compound varied per cell type across the relevant test sites also differed with the exception of ximelagatran, for which no toxicity was detected even at the highest doses despite the use of harmonized protocols (Fig. 4). As such, the inter-laboratory differences appeared to be cell type- as well as compound-dependent.

Degree of inter-laboratory variability depends on the cell model and the compound. Stacked column plots demonstrating the degree of inter-laboratory variability for HepG2/ecacc (a), Upcyte (b), cryopreserved HepaRG cells (c), and cryopreserved PHH (d). On the y-axis, the number of different (shades of red) and not different (shades of blue) pairwise comparisons between the participating partner laboratories is depicted. While HepaRG cells and Upcytes (12.3 %) show no or minor variability between laboratories, HepG2/ecacc (23.4 %) and primary human hepatocytes (48.5 %) elicit variable responses (color figure online)
Similar to our findings on inter-laboratory variability, differing degrees of intra-laboratory variability were observed between cell types. In particular, HepG2 (15.4 %) and Upcyte (11.5 %) cells differed markedly between replicate experiments in the same laboratory while HepaRG (5.7 %) and PHH (2 %) varied considerably less. In addition, intra-laboratory analysis was also observed to vary between compounds ranging from no significant variation between replicate experiments observed with paracetamol and diclofenac compared with pioglitazone demonstrating differences in 21.1 % of comparisons. Based on these results, we conclude that intra-laboratory variation and thus result reproducibility can also differ between cell types and compounds.
In order to examine differences in compound sensitivity between cryopreserved and fresh cells, cryopreserved and fresh HepaRG cells were compared in their response to the compounds tested (Fig. 5). We observed a biologically significant difference between cryopreserved and fresh HepaRG cells for only three compounds. Similarly, we also compared the cytotoxicity profiles generated for each of the training compounds from one fresh PHH donor with the five cryopreserved donors and found a biologically significant difference for two compounds (Supplementary Fig. 2).

Fresh and cryopreserved cells elicit overall similar responses to the compounds. Stacked column plots showing the responses of fresh and cryopreserved HepaRG cells. On the y-axis, the number of different (shades of red) and not different (shades of blue) pairwise comparisons between fresh and cryopreserved cells are plotted. Differences in responses are detected in 18.4 % (HepaRG) of all pairwise comparisons (color figure online)
Differences in basal activities of phases I and II enzymes have been reported in HepG2 cells that have been obtained from different sources (Hewitt and Hewitt 2004). Therefore, we determined whether different clones of the same cell line vary in their response to chemical insult by comparing two clones of HepG2 cells (Fig. 6). The ECACC clone is a commercially available HepG2 clone that is used by the MIP-DILI consortium for all HepG2-related studies for consistency. This allows direct comparison of results between studies conducted within the consortium. The results indicated differences between the two clones for only four of the compounds.

Comparison of the responses of two commonly used HepG2 clones indicates only minor differences between clones. Stacked column plot showing the differences in response to the compounds between HepG2/ecacc and HepG2/TS clones. On the y-axis, the number of different (shades of red) and non-different (shades of blue) pairwise comparisons is depicted. While responses to amiodarone and nefazodone are dissimilar, only few differences in the response to the other compounds are detected (overall 16.2 % differences) (color figure online)
Five cryopreserved hepatocyte donors were also assessed for inter-donor variability, and the influence of inter-donor variation was found to be only minor (8.1 %, Fig. 7). Interestingly, hierarchical clustering of the four cell types based on the EC50 values derived from the ATP measurements demonstrated close clustering between all the primary human hepatocyte donors while the HepG2, HepaRG and Upcyte cells constitute a second cluster (Fig. 8). This analysis further demonstrates the minor influence of donor variation between the five donors used for cryopreserved PHH in this study, as well as cryopreserved versus fresh cells (HepaRG and PHH) and clonal differences (HepG2).

Inter-donor variability in response to the compounds is negligible. Stacked column plot visualizing the differences in responses of cryopreserved PHH isolated from five different donors. In total, differences are only detected in 8.1 % of pairwise comparisons suggesting that responses to chemical insult by the compounds are only marginally affected by the genetic background of the donor

Hierarchical clustering separates primay human hepatocytes from the assessed cell line culture systems. Cell types were clustered using maximum distance measure based on their mean-centered sigma-normalized EC50 values as obtained from ATP measurements. Note that while the different primary human hepatocyte donors cluster closely together, HepG2s, HepaRGs, and Upcyte cells constitute a second cluster. Coloring indicates deviation from mean
Discussion
The prediction of drug-induced liver injury (DILI) remains a challenge for medical professionals, the pharmaceutical industry, and regulatory authorities (Chen et al. 2014; Marino et al. 2001). We therefore conducted a comprehensive and unbiased acute cytotoxicity assessment of four cell models (PHH, HepG2, HepaRG and Upcyte cells, in conjunction with basic measures of cell health, namely ATP and resorufin) that are currently used in industry as part of a testing strategy to determine DILI risk. Furthermore, we used a multinational multicenter approach, involving six industry partners and one academic partner, with the majority of the cell models tested in two or more independent sites (Table 2). To our knowledge, this is the first time such a study has been done for the types of basic cell models that are commonly used in hepatotoxicity studies. Cytotoxicity profiles were generated in response to nine human DILI-implicated compounds, which were chosen according to their proposed mechanisms of hepatotoxicity in human liver, and four compounds without known human DILI liability (Table 4). Furthermore, other factors relevant in safety assessment including clonal differences between cell lines, the use of cryopreserved and fresh cells, as well as inter-donor variation, were also addressed.
We found that based on the EC50 values obtained from this study, none of the four cell models assessed could distinguish between DILI and non-DILI compounds. This is important as it clearly demonstrates, particularly when simple endpoints are used—a common approach during drug screening—that for the medicinal chemist, none of these cell models is suitable for the purpose of predicting the likely risk of an NCE to cause DILI in man. Thus, current in vitro models must be further developed with respect to cell composition as well as endpoints measured in order to increase predictability for the various forms of DILI that can occur in man. For the simple cell types described here, which still retain some significant advantages, e.g., cost, availability, and ease-of-use, their utility in safety assessment may be specialized to specific forms of cellular stress, e.g., mitochondrial damage, functional changes in endoplasmic reticulum, nuclear damage, and lysosomal perturbation, many of which are currently used in high-content screens, combined with appropriate endpoints. We have recently determined the basal hepatic proteome of each of the cell types assessed here, data which could be valuable in deciding how these cells are used appropriately (Sison-Young et al. 2015). For the current study, this proteomic analysis is particularly useful when interpreting the cytotoxicity data as it gives an indication of each cell type’s hepatic phenotype at the protein level which could be indicative of their functional capability. For example, we have demonstrated varying protein levels of Phase I and Phase II enzymes in each cell type which is an important consideration for the current study as several of the compounds tested are known to be hepatotoxic through the generation of reactive intermediates (Gustafsson et al. 2014; McGill et al. 2012; Tang et al. 1999; Zahno et al. 2011). However, to fully characterize each of these cell types, functional measurements would have to be performed as one cannot assume that the level of abundance of a protein directly correlates with its activity. Nevertheless, once we try to go beyond modeling the initial chemical insult in the hepatocyte and take account of DILI mechanisms including the action of non-parenchymal cells as well as blood cells, it is anticipated that either the complexity of in vitro models will have to be increased in order to gain higher sensitivity for DILI prediction or more relevant readouts will be needed in order to capture the early molecular events and pathways within the hepatocyte which eventually lead to DILI as it occurs in a patient. This is particularly important for compounds such as ximelagatran, in which inflammation and the immune system are known to play an important role (Edling et al. 2008), and our results clearly show that the cell models tested in the current study are inadequate for modeling this.
We acknowledge the importance of pharmacokinetic data in the development of better models and assessment strategies given the central role of the liver in drug disposition (Smith et al. 2005). As such, Cmax values were factored into our analysis, thereby deriving some human exposure relevance. Based on this, we found that PHH provided the most accurate cell model in distinguishing compounds implicated in DILI, reaching detection rates of 89 % after 72 h (Fig. 3). It is worth pointing out that while the medicinal chemist would not generally have Cmax data at an early stage in drug development, a predicted therapeutic exposure can be calculated early on in development, and theoretically this could be used as a prediction. In the absence of known pharmacological Cmax and AUC data, basic cell models, as described here, are limited to simple and rapid early discovery screens for the ranking of chemical series within projects. Therefore, no distinction can be made between these cell models regarding sensitivity, selectivity, or relevant EC50/Cmax values to predict liver safety, without a priori knowledge of the human pharmacokinetic characteristics of the chemical entities per se. We have illustrated this by the use of an unbiased panel of DILI and non-DILI reference compounds commonly used in industry, and the subsequent analysis of cytotoxicity readouts both with and without clinical pharmacokinetic data.
The importance of chronic exposure, which is more representative of the clinical setting, was clearly evident in the current study, as a clear segregation between DILI and non-DILI compounds in PHH was only observed after a 72-h exposure to the training compounds, a pattern that was not observed at the 24-h time point. This has also been demonstrated by Holmgren and colleagues (Holmgren et al. 2014) in human stem cell-derived hepatocyte-like cells (HLCs) wherein a shift in the cytotoxicity curves of the HLCs after a 7 or 14 day exposure to drugs was observed compared with HLCs as well as HepG2 cells that have been exposed to the same drugs for 48 h. It would therefore be worthwhile in future studies to perform a chronic study in order to assess whether each of the cell models tested here can better distinguish between DILI and non-DILI compounds under chronic rather than acute exposure. In addition to the cell models tested in the present study, HLCs are also under assessment for their potential as an in vitro tool for predicting DILI risk by several groups in the field (Pradip et al. 2016; Szkolnicka et al. 2014).
Another aim of this study was to ensure that the data set generated was sufficiently robust for use as a reference tool for the evaluation of initial markers of chemical insult in simple cell models, and to benchmark more complex models of DILI. Importantly, we detected different degrees of inter-laboratory variation across the cell types assessed (Fig. 4). The largest inter-laboratory variation was observed in the PHH (48.5 %), followed by the HepG2/ECACC cells (23.4 %). It is not unexpected that the most complex and sophisticated cell in our study, the hepatocyte, shows the greatest inter-laboratory variation among the different cell models, despite the use of common reagents, plasticware and protocols. However, the variation in the HepG2 data was surprising particularly as these cells also demonstrated the highest level of intra-laboratory variation (11.54 %) among all the cell types assessed. Although identical harmonized protocols for each cell model were used by all test sites, our results show differences between laboratories that require even more stringent protocols. With the HepG2 cells, our intra-laboratory data suggests an inherent level of variability even within one type of clone. Alternatively, it is conceivable that early markers of the initial chemical insult would be less variable compared with basic cell health endpoints measured over several days in culture, and would be more appropriate (Xu et al. 2004). However, despite these variations observed and although none of the cell models could faithfully predict DILI risk, the PHH and HepG2 cells were still relatively accurate in discriminating DILI risk when exposure levels are taken into account and that this accuracy was not diminished by inter- and intra-laboratory variation.
We also assessed differences between fresh and cryopreserved HepaRG cells as well as the effect of clonal variation in the HepG2 cell line. Interestingly, cryopreservation had only minor effects on response to chemical insult in the HepaRG cells (Fig. 5). A similar observation was made between one donor of fresh and five of cryopreserved PHH (Supplementary Fig. 2). As only one donor of fresh PHH was used for comparison, additional donors would need to be tested in order to make this observation conclusive. Nonetheless, our current finding is in agreement with the minimal effects of cryopreservation in PHH that has been previously reported by other groups (Richert et al. 2006; Smith et al. 2012). Also differences between HepG2 clones when assessed by one laboratory were found to be of less importance (16.3 %, Fig. 6) than inter-laboratory variations of the same clone (23.4 %, Fig. 4a). The TS HepG2 clone used to compare with the HepG2/ECACC clone is an established HepG2 clone at one of the participating test sites and is a cell line that is routinely used for their drug screening studies. While this is not a widescale test of all HepG2 clones that are available, this does begin to address the frequently discussed, but rarely tested, issue of inter-clonal variation in response to compound exposure.
When inter-donor variation among the PHH was examined, only 8.1 % of all pairwise comparisons was observed to be different (Fig. 7). Furthermore, all five cryopreserved PHH donors and one fresh hepatocyte donor were shown to cluster together (Fig. 8), suggesting that although donor variations were observed in the response of the hepatocytes to the compounds, these variations were much less important than differences from the other cell models.
In summary, when used with simple endpoints such as the measurement of ATP and resorufin, none of the cell models currently used in industry (HepG2, HepaRG, Upcyte and PHH) can completely distinguish between established drugs with respect to their propensity to cause DILI in man, and are therefore unlikely to be able to predict the DILI hazard and risk for NCEs. However, when in vivo exposure levels are taken into account, PHH are the most accurate cell model, identifying 8 out of 9 DILI compounds as such after 72 h, but not after 24 h of exposure. Our study revealed significant inter-laboratory variation for EC50 values obtained in PHH, HepG2, and Upcyte cells, but not in HepaRG cells. Inter-donor and inter-clonal differences and the effect of cryopreservation were found to be of less importance than differences between the cell models. Therefore, while PHH can provide an expedient model to aid in the indication of possible DILI risks in later stages of drug development, e.g., when in vivo exposure data is already available, none of the cell models are suitable to indicate the risk of DILI early in drug development. Hence, a multicenter assessment of more reliable cell models, i.e., simple cell types such as used here, or more complex 3D or co-culture models, in conjunction with more sophisticated endpoints of molecular initiating events that report on the chemical insult in the hepatocyte is needed.
References
Amoah AG, Gould BJ, Parke DV, Lockhart JD (1986) Further studies on the pharmacokinetics of perhexiline maleate in humans. Xenobiotica 16(1):63–68. doi:10.3109/00498258609043506
Antherieu S, Chesne C, Li R et al (2010) Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos 38(3):516–525. doi:10.1124/dmd.109.030197
Bandyopadhyay S, Klaunig JE, Somani P (1990) Cytotoxic interactions of cardioactive cationic amphiphilic compounds in primary rat hepatocytes in culture. Hepatology 12(1):48–58
Barbhaiya RH, Buch AB, Greene DS (1996) A study of the effect of age and gender on the pharmacokinetics of nefazodone after single and multiple doses. J Clin Psychopharmacol 16(1):19–25
Bell LN, Chalasani N (2009) Epidemiology of idiosyncratic drug-induced liver injury. Semin Liver Dis 29(4):337–347. doi:10.1055/s-0029-1240002
Burkard A, Dahn C, Heinz S et al (2012) Generation of proliferating human hepatocytes using Upcyte(R) technology: characterisation and applications in induction and cytotoxicity assays. Xenobiotica 42(10):939–956. doi:10.3109/00498254.2012.675093
Chen M, Bisgin H, Tong L et al (2014) Toward predictive models for drug-induced liver injury in humans: are we there yet? Biomark Med 8(2):201–213. doi:10.2217/bmm.13.146
Dahlin DC, Miwa GT, Lu AY, Nelson SD (1984) N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci USA 81(5):1327–1331
Dake MD, Madison JM, Montgomery CK et al (1985) Electron microscopic demonstration of lysosomal inclusion bodies in lung, liver, lymph nodes, and blood leukocytes of patients with amiodarone pulmonary toxicity. Am J Med 78(3):506–512
Edling Y, Sivertsson L, Andersson TB, Porsmyr-Palmertz M, Ingelman-Sundberg M (2008) Pro-inflammatory response and adverse drug reactions: mechanisms of action of ximelagatran on chemokine and cytokine activation in a monocyte in vitro model. Toxicol In vitro Int J Publ Assoc BIBRA 22(6):1588–1594. doi:10.1016/j.tiv.2008.06.011
Fattinger K, Funk C, Pantze M et al (2001) The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 69(4):223–231. doi:10.1067/mcp.2001.114667
Fromenty B, Pessayre D (1997) Impaired mitochondrial function in microvesicular steatosis. Effects of drugs, ethanol, hormones and cytokines. J Hepatol 26(Suppl 2):43–53
Gerets HH, Tilmant K, Gerin B et al (2012) Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol Toxicol 28(2):69–87. doi:10.1007/s10565-011-9208-4
Gripon P, Rumin S, Urban S et al (2002) Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA 99(24):15655–15660. doi:10.1073/pnas.232137699
Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F, Guguen-Guillouzo C (2007) The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem Biol Interact 168(1):66–73. doi:10.1016/j.cbi.2006.12.003
Gustafsson F, Foster AJ, Sarda S, Bridgland-Taylor MH, Kenna JG (2014) A correlation between the in vitro drug toxicity of drugs to cell lines that express human P450 s and their propensity to cause liver injury in humans. Toxicol Sci Off J Soc Toxicol 137(1):189–211. doi:10.1093/toxsci/kft223
Gutierrez MM, Nicolas LB, Donazzolo Y, Dingemanse J (2013) Relative bioavailability of a newly developed pediatric formulation of bosentan vs. the adult formulation. Int J Clin Pharmacol Ther 51(6):529–536. doi:10.5414/CP201806
Heikkinen H, Saraheimo M, Antila S, Ottoila P, Pentikainen PJ (2001) Pharmacokinetics of entacapone, a peripherally acting catechol-O-methyltransferase inhibitor, in man. A study using a stable isotope techique. Eur J Clin Pharmacol 56(11):821–826
Hewitt NJ, Hewitt P (2004) Phase I and II enzyme characterization of two sources of HepG2 cell lines. Xenobiotica; Fate Foreign Compd Biol Syst 34(3):243–256. doi:10.1080/00498250310001657568
Holmgren G, Sjogren AK, Barragan I et al (2014) Long-term chronic toxicity testing using human pluripotent stem cell-derived hepatocytes. Drug Metab Dispos Biol Fate Chem 42(9):1401–1406. doi:10.1124/dmd.114.059154
Jorga K, Fotteler B, Heizmann P, Gasser R (1999) Metabolism and excretion of tolcapone, a novel inhibitor of catechol-O-methyltransferase. Br J Clin Pharmacol 48(4):513–520
Kalgutkar AS, Vaz AD, Lame ME et al (2005) Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metab Dispos 33(2):243–253. doi:10.1124/dmd.104.001735
Kaplowitz N (2005) Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov 4(6):489–499. doi:10.1038/nrd1750
Keisu M, Andersson TB (2010) Drug-induced liver injury in humans: the case of ximelagatran. Handb Exp Pharmacol (196):407–418. doi:10.1007/978-3-642-00663-0_13
Kostrubsky SE, Strom SC, Kalgutkar AS et al (2006) Inhibition of hepatobiliary transport as a predictive method for clinical hepatotoxicity of nefazodone. Toxicol Sci 90(2):451–459. doi:10.1093/toxsci/kfj095
Lambert CB, Spire C, Renaud MP, Claude N, Guillouzo A (2009) Reproducible chemical-induced changes in gene expression profiles in human hepatoma HepaRG cells under various experimental conditions. Toxicol In Vitro 23(3):466–475. doi:10.1016/j.tiv.2008.12.018
Lautala P, Ethell BT, Taskinen J, Burchell B (2000) The specificity of glucuronidation of entacapone and tolcapone by recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos 28(11):1385–1389
LeCluyse EL, Alexandre E, Hamilton GA et al (2005) Isolation and culture of primary human hepatocytes. Methods Mol Biol 290:207–229
Loi CM, Randinitis EJ, Vassos AB, Kazierad DJ, Koup JR, Sedman AJ (1997) Lack of effect of type II diabetes on the pharmacokinetics of troglitazone in a multiple-dose study. J Clin Pharmacol 37(12):1114–1120
Loi CM, Alvey CW, Vassos AB, Randinitis EJ, Sedman AJ, Koup JR (1999) Steady-state pharmacokinetics and dose proportionality of troglitazone and its metabolites. J Clin Pharmacol 39(9):920–926
Madan A, Graham RA, Carroll KM et al (2003) Effects of prototypical microsomal enzyme inducers on cytochrome P450 expression in cultured human hepatocytes. Drug Metab Dispos Biol Fate Chem 31(4):421–431
Marino G, Zimmerman HJ, Lewis JH (2001) Management of drug-induced liver disease. Curr Gastroenterol Rep 3(1):38–48
McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H (2012) The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Investig 122(4):1574–1583. doi:10.1172/JCI59755
O’Brien PJ, Irwin W, Diaz D et al (2006) High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol 80(9):580–604. doi:10.1007/s00204-006-0091-3
Olson H, Betton G, Robinson D et al (2000) Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol RTP 32(1):56–67. doi:10.1006/rtph.2000.1399
Pourbaix S, Berger Y, Desager JP, Pacco M, Harvengt C (1985) Absolute bioavailability of amiodarone in normal subjects. Clin Pharmacol Ther 37(2):118–123
Pradip A, Steel D, Jacobsson S et al (2016) High content analysis of human pluripotent stem cell derived hepatocytes reveals drug induced steatosis and phospholipidosis. Stem cells Int 2016:2475631. doi:10.1155/2016/2475631
Rajagopalan R, Iyer S, Perez A (2005) Comparison of pioglitazone with other antidiabetic drugs for associated incidence of liver failure: no evidence of increased risk of liver failure with pioglitazone. Diabetes Obes Metab 7(2):161–169. doi:10.1111/j.1463-1326.2004.00382.x
Richert L, Liguori MJ, Abadie C et al (2006) Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab Dispos Biol Fate Chem 34(5):870–879. doi:10.1124/dmd.105.007708
Sakr A, Andheria M (2001) Pharmacokinetics of buspirone extended-release tablets: a single-dose study. J Clin Pharmacol 41(7):783–789
Schutzer KM, Wall U, Lonnerstedt C et al (2004) Bioequivalence of ximelagatran, an oral direct thrombin inhibitor, as whole or crushed tablets or dissolved formulation. Curr Med Res Opin 20(3):325–331. doi:10.1185/030079903125003035
Sevilla-Tirado FJ, Gonzalez-Vallejo EB, Leary AC, Breedt HJ, Hyde VJ, Fernandez-Hernando N (2003) Bioavailability of two new formulations of paracetamol, compared with three marketed formulations, in healthy volunteers. Methods Find Exp Clin Pharmacol 25(7):531–535
Sison-Young R, Mitsa D, Jenkins RE et al (2015) Comparative proteomic characterisation of four human liver-derived single cell culture models reveals significant variation in the capacity for drug disposition, bioactivation and detoxication. Toxicol Sci Off J Soc Toxicol. doi:10.1093/toxsci/kfv136
Smith KS, Smith PL, Heady TN, Trugman JM, Harman WD, Macdonald TL (2003) In vitro metabolism of tolcapone to reactive intermediates: relevance to tolcapone liver toxicity. Chem Res Toxicol 16(2):123–128. doi:10.1021/tx025569n
Smith NF, Figg WD, Sparreboom A (2005) Role of the liver-specific transporters OATP1B1 and OATP1B3 in governing drug elimination. Expert Opin Drug Metab Toxicol 1(3):429–445. doi:10.1517/17425255.1.3.429
Smith CM, Nolan CK, Edwards MA et al (2012) A comprehensive evaluation of metabolic activity and intrinsic clearance in suspensions and monolayer cultures of cryopreserved primary human hepatocytes. J Pharm Sci 101(10):3989–4002. doi:10.1002/jps.23262
Szkolnicka D, Farnworth SL, Lucendo-Villarin B et al (2014) Accurate prediction of drug-induced liver injury using stem cell-derived populations. Stem Cells transl Med 3(2):141–148. doi:10.5966/sctm.2013-0146
Tang W, Stearns RA, Bandiera SM et al (1999) Studies on cytochrome P-450-mediated bioactivation of diclofenac in rats and in human hepatocytes: identification of glutathione conjugated metabolites. Drug Metab Dispos Biol Fate Chem 27(3):365–372
Tucker GT, Casey C, Phillips PJ, Connor H, Ward JD, Woods HF (1981) Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol 12(2):235–246
Tujios S, Fontana RJ (2011) Mechanisms of drug-induced liver injury: from bedside to bench. Nat Rev Gastroenterol Hepatol 8(4):202–211. doi:10.1038/nrgastro.2011.22
Tzvetkov MV, Vormfelde SV, Balen D et al (2009) The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther 86(3):299–306. doi:10.1038/clpt.2009.92
Wong H, Ozalp Y, Lainesse A, Alpan RS (2004) In vivo bioequivalence of oral antidiabetic agents: pioglitazone tablets. Arzneimittelforschung 54(9A):618–624. doi:10.1055/s-0031-1297059
Xu JJ, Diaz D, O’Brien PJ (2004) Applications of cytotoxicity assays and pre-lethal mechanistic assays for assessment of human hepatotoxicity potential. Chem Biol Interact 150(1):115–128. doi:10.1016/j.cbi.2004.09.011
Zahno A, Brecht K, Morand R et al (2011) The role of CYP3A4 in amiodarone-associated toxicity on HepG2 cells. Biochem Pharmacol 81(3):432–441. doi:10.1016/j.bcp.2010.11.002
Zhu M, Zhao W, Jimenez H et al (2005) Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes. Drug Metab Dispos Biol Fate Chem 33(4):500–507. doi:10.1124/dmd.104.000836
Funding
This work was supported by funding from the Mechanism-based Integrated Systems for the Prediction of Drug-Induced Liver Injury (MIP-DILI) project, a European Community grant under the Innovative Medicines Initiative (IMI) Programme (Grant Agreement Number 115336).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
The human biological samples were sourced ethically.
Informed consent
The research use of the samples was in accord with the terms of the informed consents.
Additional information
1–12. Members of the Innovative Medicines Initiative MIP-DILI (Mechanism-based Integrated systems for the Prediction of Drug-Induced Liver Injury) Consortium.
Rowena Sison-Young, Volker M. Lauschke and Esther Johann have contributed equally to this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sison-Young, R.L., Lauschke, V.M., Johann, E. et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Arch Toxicol 91, 1385–1400 (2017). https://doi.org/10.1007/s00204-016-1745-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-016-1745-4