
Abstract
Aims/hypothesis
Accumulating evidence has revealed the significant role of glucagon-like peptide-1 (GLP-1) in weight loss. Sirtuin 1 (SIRT1) plays a vital role in the regulation of lipid metabolism. Here, we investigated the contribution of lipolytic and oxidative changes in white adipose tissue (WAT) to the weight-lowering effect induced by the GLP-1 receptor (GLP-1R) agonist exenatide (exendin-4) in mice. We also looked at the role of SIRT1 in this process.
Methods
C57BL/6J mice and Sirt1 +/− mice were treated with exenatide (24 nmol/kg) or an NaCl solution (154 mmol/l) control i.p. for 8 weeks while receiving a high-fat diet (HFD) after a 12 week HFD challenge. Systemic phenotypic evaluations were carried out during and after the intervention. A lentivirus-mediated short hairpin (sh)RNA vector of the Sirt1 gene was transfected into differentiated 3T3-L1 adipocytes. An in vitro model system used adipocytes induced from Sirt1-null mouse embryonic fibroblasts (MEFs).
Results
Exenatide reduced fat mass and enhanced the lipolytic and oxidative capacity of WAT in diet-induced obese C57BL/6J mice. However, these effects were significantly impaired in Sirt1 +/− mice compared with wild-type controls. In vitro, exendin-4 increased lipolysis and fatty acid oxidation by upregulating SIRT1 expression and activity in differentiated 3T3-L1 adipocytes. Conversely, RNA interference (i)-induced knockdown of SIRT1 attenuated the lipolytic and oxidative responses to exendin-4 in differentiated 3T3-L1 adipocytes. Again, these responses were entirely abolished in Sirt1-null MEFs after induction into adipocytes.
Conclusions/interpretation
These data highlight that a GLP-1R agonist promotes brown remodelling of WAT in a SIRT1-dependent manner; this might be one of the mechanisms underlying its effect on weight loss.
Similar content being viewed by others


Introduction
Obesity, which has become a global epidemic, is characterised as excess fat accumulation that predisposes individuals to metabolic disorders such as type 2 diabetes, dyslipidaemia and hypertension [1]. Glucagon-like peptide-1 receptor (GLP-1R) agonists, glucose-lowering drugs well known for their glucose-dependent insulinotropic activity, had a remarkable weight loss effect in type 2 diabetes and obesity [2, 3]. The known mechanisms of its effect on weight loss are inhibition of appetite and suppression of food intake through hypothalamic or parasympathetic pathways [4, 5]. However, little is known about the exact mechanisms underlying its activity in adipose tissue.
White adipose tissue (WAT) is the biggest mammalian triacylglycerol (TG) storage depot. Under fasting conditions, lipolysis is promoted to supply sufficient fatty acids to other organs for oxidative metabolism (reviewed in Nielsen et al [6]). On the other hand, excess fat accumulation could accelerate mitochondrial dysfunction, with subsequent insulin resistance in white fat [7, 8]. Brown fat is morphologically and functionally different from white fat. It is fuelled by mitochondrial oxidation and dissipates chemical energy as heat through uncoupling protein 1 (UCP-1) (reviewed in Cannon and Nedergaard [9]). Recent studies have shown that the browning of adipose tissue, including brown remodelling of WAT and promotion of the brown adipose tissue (BAT) function, could counteract obesity (reviewed in Bartelt and Heeren [10]). Brown remodelling of WAT confers BAT-like features onto WAT and remodels it to exert an energy-disposal capacity rather than just acting as an energy storage site (reviewed in Wu et al [11]).
Sirtuin 1 (SIRT1), an NAD+-dependent deacetylase involved in lipid metabolism, has been proposed as a potential therapeutic target for treating obesity-related metabolic disturbance [12, 13]. Increased SIRT1 activity leads to the activation of adipose triacylglycerol lipase (ATGL), with subsequent triacylglycerol depletion of WAT [14]. Moreover, overexpression of SIRT1 in WAT effected a reduction of fat accumulation in adipocytes, with improved whole-body energy expenditure [15]. Thus, it is possible for a GLP-1 receptor (GLP-1R) agonist to activate SIRT1 to stimulate fat mobilisation and use of WAT.
In this study, we investigated the contribution of brown remodelling of WAT to the weight-lowering-effect induced by the GLP-1R agonist exenatide. We also researched the role of SIRT1 in this process.
Methods
Animal models
Seven-week-old male C57BL/6J mice were purchased from the Model Animal Research Center (Nanjing, China). The Sirt1 +/− mice with a C57BL/6J genetic background were from J. Ye’s laboratory at the Pennington Biomedical Research Center (Louisiana State University, Baton Rouge, LA, USA) [16]. The mice were generated by backcrossing of 129/J Sirt1 +/− mice, a gift from F. Alt at the Howard Hughes Medical Institute, Children’s Hospital, Center for Blood Research and Department of Genetics (Harvard University Medical School, Boston, MA, USA) [17], with C57BL/6J mice for more than ten generations. From the age of 8 weeks onwards, the C57BL/6J mice and Sirt1 +/− mice were fed a rodent chow diet (5% fat [wt/wt]; Guangdong Medical Laboratory Animal Center, Guangzhou, China) or a high-fat diet (HFD) (35.8% fat [wt/wt], D12331, Research Diets, New Brunswick, NJ, USA), respectively, for 12 weeks. The mice in the HFD-fed group were randomised according to their body weight, using a random number table generated by SPSS 13.0, to 8 weeks’ treatment with either exenatide (24 nmol/kg; Eli Lilly and Company, Indianapolis, IN, USA) or a normal NaCl solution (154 mmol/l) as control, once daily by i.p. injection [18]. The mice in the control group receiving a normal diet also received i.p. injections of NaCl solution. The body weight and food intake of the mice were measured biweekly. After 8 weeks’ treatment, tolerance tests and metabolic measurements were carried out (see electronic supplementary material (ESM) Methods). All the mice were fasted overnight, anaesthetised and killed for blood and tissue collection. Epididymal fat was subjected to haematoxylin and eosin (H&E) staining, immunohistochemical (IHC) staining, TG and glycerol content per cell, and SIRT1 activity assay (see ESM Methods). The investigators were not blind to group assignment and outcome assessment. The Institutional Animal Care and Use Committees (IACUC) of Sun Yat-sen University approved all animal study procedures.
Western blotting
Total protein was extracted from snap-frozen adipose tissue (∼50 mg) and cell lysate, and then separated by 10% SDS-PAGE. Immunoblotting was performed with specific primary antibodies (see ESM Methods).
Mitochondrial DNA content determination
Mitochondrial DNA (mtDNA) was amplified with quantitative PCR (see ESM Methods). The primer sequences can be found in ESM Table 1.
Quantitative real-time PCR
Quantitative real-time PCR was used to determine the relative expression levels of mRNAs (see ESM Methods). The primer sequences can be found in the ESM Table 2.
3T3-L1 cell culture, treatments and lentiviral transfection
3T3-L1 cell lines (ATCC, Manassas, VA, USA) were induced into mature adipocytes as described previously [19]. The fully differentiated adipocytes were treated with 20 nmol/l exendin-4 (E7144, Sigma-Aldrich, St Louis, MO, USA), 10 μmol/l SRT1720 (S1129, Selleck, Houston, TX, USA), 30 mmol/l nicotinamide (N0636, Sigma-Aldrich, USA), a combination of exendin-4 and SRT1720, or a combination of exendin-4 and nicotinamide, for 24 h, respectively. Intracellular NAD+ quantification, Oil Red O and BODIPY493/503 staining, mitochondria isolation and cytochrome c oxidase assay, and immunoprecipitation were carried out after the treatment (see ESM Methods).
Lentivirus vectors expressing Sirt1 short hairpin (sh)RNA sequence (Genechem, Shanghai, China) and empty vectors as control were transfected into 3T3-L1 pre-adipocytes, and cells were then differentiated into adipocytes and treated with 20 nmol/l exendin-4 for 24 h.
Mouse embryonic fibroblast culture
Mouse embryonic fibroblasts (MEFs) were isolated from 13.5-day-old embryos derived from Sirt1 −/− and wild-type (WT) mice. Embryos were surgically minced and digested. After centrifugation, the digested cells were resuspended, differentiated to adipocytes, and treated with 20 nmol/l exendin-4 for 24 h.
Statistics
There were 5–12 mice in each group of animal study. The outliers with extreme high or low values were excluded in the data analysis. All of the in vitro experiments were repeated at least three times with consistent data. All the values were presented as mean ± SEM. We used unpaired Student’s t test (two-tailed) and ANOVA to evaluate the statistical significance.
Results
Exenatide improves diet-induced obesity and insulin resistance
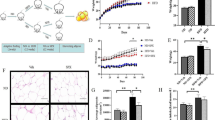
Exenatide treatment decreased the body weight of the HFD-fed mice to a level comparable with that of the normal controls (Fig. 1a). Although exenatide treatment elicited a transient reduction in food intake during the first 2 weeks, no significant differences were found between HFD groups treated with exenatide and NaCl solution from after week 2 to week 8 (Fig. 1b). The results of the i.p. GTT and the insulin tolerance test (ITT) indicated that the impaired glucose tolerance and insulin sensitivity of the mice with HFD-induced obesity were ameliorated by exenatide treatment (Fig. 1c, d).

Exenatide improves diet-induced obesity and insulin resistance. Mice in the HFD group were randomised to receive exenatide or NaCl solution for 8 weeks after a 12 week HFD challenge. (a) Body weight change in chow-fed mice (white circles), mice receiving NaCl solution on an HFD (black circles) and exenatide-treated mice on an HFD (grey circles). (b) Food intake during treatment. Intraperitoneal GTT (c) and ITT (d) were performed at the end of the intervention. The AUCs were determined. n = 5/group. Values are mean ± SEM. *p < 0.05 and **p < 0.01 compared with normal control group; † p < 0.05 and †† p < 0.01 compared with the HFD group. White bars, normal control; black bars, HFD-fed mice treated with NaCl solution; grey bars, HFD-fed mice receiving exenatide. Exe, exenatide; NC, normal control
Exenatide improves metabolic disorders in serum of HFD-challenged mice
After the 8 week exenatide treatment, we found a dramatic reduction in fasting blood glucose (FBG) and lipid metabolites, including TG, glycerol and NEFA, in the serum of the HFD group with exenatide treatment (Table 1). The GLP-1 level was found to be lowered in the HFD group treated with NaCl solution, and recovered with exenatide treatment (Table 1). The level of the pro-inflammatory cytokine TNF-α was increased in the NaCl-solution-treated mice on HFD and decreased with exenatide treatment (Table 1). This indicates an amelioration of the inflammation. Taken together, these results show that exenatide improves HFD-induced metabolic disorders.
Exenatide reduces adiposity by promoting lipolysis, fatty acid oxidation and mitochondrial biogenesis in the WAT of obese mice
When challenged with HFD, the weight of epididymal fat from C57BL/6J mice increased dramatically compared with normal controls, and decreased remarkably with exenatide treatment (Fig. 2a). H&E staining showed a greater frequency of smaller adipocytes, while IHC staining showed more UCP-1 (Fig. 2b) in the exenatide-treated mice than in the NaCl-treated HFD group. Moreover, the TG content per cell was fourfold higher in HFD-fed mice than in control mice, but this effect was not seen in HFD-fed mice treated with exenatide (Fig. 2c). The amount of glycerol, a product of lipolysis, per adipocyte was lower in the exenatide-treated group (Fig. 2d). We found significantly augmented levels of lipolytic signalling proteins, including SIRT1, and phosphorylated AMP-activated protein kinase (AMPK) and acetyl-CoA carboxylase (ACC) in the exenatide-treated group (Fig. 2e, ESM Fig. 1a). In addition, we observed an increase in ATGL (Fig. 2e, ESM Fig. 1a) and phosphorylated hormone-sensitive lipase (HSL) (ESM Fig. 2), which are hallmarks of lipolysis. The protein levels of peroxisome proliferation activated receptor α (PPARα), peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), UCP-1 (Fig. 2f, ESM Fig. 1b) and carnitine palmitoyltransferase 1 (CPT1) (ESM Fig. 2), representative of fatty acid oxidation, increased significantly after the exenatide treatment. SIRT1 activity was also increased by exenatide (Fig. 2g). Furthermore, the mtDNA copy number in HFD-challenged mice was elevated by exenatide (Fig. 2h), in line with the upregulation of genes involved in mitochondrial biogenesis and function (Fig. 2i). These data indicate that exenatide promotes lipolysis and mitochondrial function in the WAT of diet-induced obese mice.

Exenatide reduces adiposity by promoting lipolysis, fatty acid oxidation and mitochondrial biogenesis in the WAT of HFD-induced C57BL/6J mice. (a) Morphology and weight of epididymal fat. (b) H&E and IHC staining of UCP-1 in epididymal fat and quantification of mean adipocyte size. Magnification ×200. (c) TG and (d) glycerol content per cell in epididymal fat. The relative content values were normalised to the normal control group. (e) Western blotting of SIRT1, lipolytic signals and (f) oxidative signals in epididymal fat. (g) SIRT1 activity in epididymal fat. (h) Relative mtDNA content analysed by quantitative PCR using primers specific for Cox2 and normalised to genomic 18 s rRNA. Values were normalised to the normal control group. (i) Relative expression of genes related to mitochondrial biogenesis and function in epididymal fat analysed by quantitative real-time PCR. Relative expression values were normalised to the normal control group. n = 5/group. Values are mean ± SEM. *p < 0.05 and **p < 0.01 compared with the normal control group; † p < 0.05, and †† p < 0.01 compared with the HFD group. White bars, normal control; black bars, HFD-fed mice treated with NaCl solution; grey bars, HFD-fed mice with exenatide treatment. Exe, exenatide; NC, normal control
Exenatide decreases adiposity through SIRT1
To clarify the role of SIRT1 in WAT during the exenatide treatment, we used the Sirt1 +/− mouse model. The effect of exenatide on reducing body weight and maintaining glucose homeostasis is attenuated in Sirt1 +/− mice (ESM Fig. 3). Exenatide reduced epididymal fat weight to a lesser extent in Sirt1 +/− mice (1.53 ± 0.24 g with NaCl solution vs 1.11 ± 0.24 g with exenatide), compared with that of WT controls (0.92 ± 0.34 g with NaCl solution vs 0.29 ± 0.05 g with exenatide). This represented a 28% decrease in Sirt1 +/− mice vs a 68% decrease in WT controls (Fig. 3a). The adipocyte hypertrophy was sustained in the Sirt1 +/− group after exenatide treatment (Fig. 3b). The increased amount of UCP-1 in the epididymal fat of WT mice after exenatide treatment was diminished in Sirt1 +/− mice (Fig. 3b). Both protein level and activity of SIRT1 were higher in WT mice after exenatide treatment, while no obvious change was observed in Sirt1 +/− mice regardless of exenatide treatment (Fig. 3c, d). The gene expression of Sirt1, Atgl (also known as Pnpla2), Acc, Hsl (also known as Lipe), Cpt1 (also known as Cpt1a), Pgc1α (also known as Ppargc1a) and Pparα (also known as Ppara) in epididymal fat was upregulated in WT mice with exenatide treatment; however, no increase in gene expression was observed in Sirt1 +/− mice with exenatide treatment (Fig. 3e).

Exenatide decreases adiposity through SIRT1. After 12 weeks of chow diet or HFD challenge, mice were randomised to receive i.p. injections of exenatide or NaCl solution for 8 weeks. (a) Morphology and weight of epididymal fat. It represented a 28% decrease in Sirt1 +/− mice vs a 68% decrease in WT controls. (b) H&E and IHC staining for UCP-1, and quantification of mean adipocyte size in epididymal fat. Magnification ×200. (c) Protein level and the quantification of SIRT1 and (d) SIRT1 activity in epididymal fat. (e) Relative mRNA expression of genes involved in lipolysis and oxidation. Values were normalised to the WT mice fed an HFD. n = 5–12/group. Values are mean ± SEM. *p < 0.05 and **p < 0.01 compared with WT + HFD mice; † p < 0.05 and †† p < 0.01 compared with Sirt1 heterozygous knockout mice (SHK) + HFD mice; ‡ p < 0.05 and ‡‡ p < 0.01 compared with WT + HFD + Exe mice. White bars, WT; dark grey bars, exenatide-treated WT mice; light grey bars, SHK; black bars, exenatide-treated SHK mice on an HFD. Exe, exenatide
Exendin-4 enhances lipolytic and oxidative pathways with SIRT1 activation in vitro
We treated differentiated 3T3-L1 adipocytes with 1 nmol/l, 20 nmol/l or 100 nmol/l of exendin-4 for 24 h. Exendin-4 was used as an exenatide substitute to exclude interference from auxiliary material. In agreement with in vivo findings, exendin-4 treatment led to upregulation of SIRT1 protein production, as well as activation of the AMPK/ACC pathway and ATGL (Fig. 4a, b). Moreover, the protein levels of PPARα, PGC-1α and UCP-1 were amplified by exendin-4 in 3T3-L1 adipocytes (Fig. 4c, d). Notably, a build-up of cellular NAD+ levels, which SIRT1 activity is dependent on, provided further support for exendin-4 activation of SIRT1 not only by increasing the SIRT1 protein level but also by upregulating its activity (Fig. 4e, f). As 20 nmol/l exendin-4 had been found to significantly upregulate lipolytic and oxidative pathways, we used 20 nmol/l exendin-4 in the in vitro study. Overall, the results show that exendin-4 enhances lipolysis and lipid oxidation in 3T3-L1 adipocytes with the upregulation of SIRT1 production and activity.

Exendin-4 enhances lipolytic and oxidative pathways with SIRT1 activation in vitro. Differentiated 3T3-L1 adipocytes were exposed to exendin-4 (1 nmol/l, 20 nmol/l and 100 nmol/l) for 24 h. (a) Immunoblotting analysis of SIRT1 and signalling proteins involved in lipid metabolism. (b) The expression, relative to β-tubulin, of SIRT1 and ATGL, p-AMPK to AMPK, and p-ACC to ACC. (c) Immunoblotting analysis of oxidative signals and (d) the expression of PPARα, PGC-1α and UCP-1 relative to β-tubulin. (e) Intracellular NAD+ level and (f) NAD+/NADH ratio. Values are mean ± SEM. *p < 0.05 and **p < 0.01 compared with the controls; †† p < 0.01 compared with mice receiving exendin-4, 1 nmol/l. Ctrl, control; Exe-4, exendin-4; NC, normal control
Exendin-4 promotes lipolysis and mitochondrial function through SIRT1 in vitro
Exendin-4 and SRT1720, a SIRT1 activator, both led to a reduction of lipid droplets compared with control group as shown by Oil Red O staining of differentiated 3T3-L1 adipocytes. This effect was blunted by nicotinamide, an inhibitor of SIRT1 (Fig. 5a). Further analysis of BODIPY fluorescence staining for lipids was conducted and normalised to numbers of nuclei. The result was the same as had been observed with Oil Red O staining (Fig. 5a). The TG content per adipocyte was in line with the morphologic analysis (Fig. 5b). The levels of proteins involved in lipolytic signalling were increased in differentiated 3T3-L1 adipocytes after treatment with exendin-4 and SRT1720. These salient effects were inhibited by nicotinamide (Fig. 5c and ESM Fig. 4a).

Exendin-4 promotes lipolysis and mitochondrial function through SIRT1 in vitro. The differentiated 3T3-L1 adipocytes were treated with 20 nmol/l exendin-4, 10 μmol/l SRT1720, 30 mmol/l nicotinamide, a combination of 20 nmol/l exendin-4 and 10 μmol/l SRT1720, or a combination of 20 nmol/l exendin-4 and 30 mmol/l nicotinamide, for 24 h, respectively. (a) Oil Red O and BODIPY staining. DAPI dye shows the nuclei. Magnification ×200. (b) Relative TG content per cell. Protein levels of (c) SIRT1 and lipid-metabolism-related signals and (d) oxidative signals. (e) The relative mtDNA content. (f) Cytochrome c oxidase activity. (g) Intracellular NAD+ level and (h) NAD+/NADH ratio. (i) The protein acetylation of PPARα and PGC-1α was measured by an anti-acetyl lysine primary antibody and total protein was also evaluated by immunoprecipitation (IP). Values are mean ± SEM. *p < 0.05 and **p < 0.01 compared with the control group; † p < 0.05 and †† p < 0.01 compared with the exenatide group; ‡ p < 0.05 compared with the group receiving nicotinamide. Ctrl, control; Exe-4, exendin-4; SRT, SRT1720; Nico, nicotinamide
Both exendin-4 and SRT1720 upregulated the levels of oxidative signalling proteins, including PPARα, PGC-1α and UCP-1 in differentiated 3T3-L1 adipocytes. This effect was partly reversed by the SIRT1 inhibitor nicotinamide (Fig. 5d and ESM Fig. 4b). We detected changes in mitochondria biogenesis, and a role for SIRT1. The mtDNA copy number was increased by exendin-4 and SRT172 and reduced by nicotinamide, compared with control (Fig. 5e). We also examined the cytochrome c oxidase activity. Similarly, the activity was enhanced by exendin-4 and SRT1720, and inhibited by nicotinamide (Fig. 5f). These results suggest that SIRT1 plays a central role in facilitating mitochondrial biogenesis and function with exendin-4 treatment.
SIRT1 is an NAD+-dependent deacetylase. We determined the cellular NAD+ level and the NAD+/NADH ratio in vitro. Both were upregulated with exendin-4 and SRT1720 treatment and downregulated when treated with nicotinamide (Fig. 5g, h). We also detected the deacetylation by checking the acetylation status of SIRT1 target proteins involved in fatty acid oxidation. The acetylation of PPARα and PGC-1α was decreased with exendin-4 and SRT1720 treatment but increased significantly after nicotinamide treatment (Fig. 5i and ESM Fig. 4c).
SIRT1 is required for exendin-4-induced lipolysis and fatty acid oxidation in vitro
To verify that the beneficial effects of exendin-4 are SIRT1 dependent, we used two other in vitro models. First, we knocked down SIRT1 expression by Sirt1 RNA interference (i) using a lentivirus shRNA vector transfected into differentiated 3T3-L1 adipocytes. Oil Red O staining showed fewer lipid droplets with exendin-4 treatment and more lipid droplets after the SIRT1 RNAi treatment, compared with control (Fig. 6a). The effects of exendin-4 on lipolysis and fatty acid oxidation dramatically faded after SIRT1 knockdown as shown by relative protein levels (Fig. 6b and ESM Fig. 5).

SIRT1 is required for exendin-4-induced lipolysis and fatty acid oxidation in vitro. (a) Oil Red O staining of differentiated 3T3-L1 adipocytes infected with lentivirus to cause SIRT1 knockdown, or empty vectors, and treated with exendin-4 for 24 h. Magnification ×200. (b) Protein levels of SIRT1, lipid-metabolism-related signals and oxidative signals in the 3T3-L1 adipocytes prepared as described in (a). (c) Oil Red O staining of the differentiated MEFs after 24 h of exendin-4 treatment. Magnification ×200. (d) Protein levels of SIRT1, lipid-metabolism-related signals and oxidative signals in the cells prepared in as described in (c). Ctrl, control; Exe-4, exendin-4
Second, we collected MEFs and differentiated them into adipocytes. Oil Red O staining revealed that exendin-4 treatment could reduce the number of lipid droplets in the WT MEFs, but not in the SIRT1-null MEFs (Sirt1 −/−) (Fig. 6c). The higher levels of lipolytic and oxidative signalling proteins that occurred in response to exendin-4 in the WT MEFs were completely absent in the SIRT1-null MEFs (Fig. 6d and ESM Fig. 6). Overall, the increased lipolytic and oxidative capacity induced by exendin-4 requires SIRT1 in adipocytes.
Discussion
GLP-1 and the incretin mimetics have been found to play important roles in weight loss that occurs primarily as a result of a reduction in fat tissue [2, 20]. In the current work, we demonstrate that a GLP-1 mimetic, exenatide, significantly improves diet-induced obesity through increased lipolysis and fatty acid oxidation of WAT in a SIRT1-dependent manner.
Many studies have suggested that the effect of GLP-1 on weight loss is attributable to the suppression of food intake. However, most of these studies observed the short-term effect on food intake, such as over 24 h or 1 week [4, 21, 22]. In a study with 28 days of exendin-4 treatment in mice with diet-induced obesity, the initial transient reduction in food intake disappeared over time [23]. Consistent with this observation, exenatide-treated mice in our study showed a decrease in food intake during the first 2 weeks, and then recovered to a level comparable with that of the control group from after week 2 to week 8. This indicates that the effect of exenatide on weight loss is independent of food intake during chronic administration.
Our study found that exenatide (exendin-4) led to pronounced attenuation of fat reserves by enhanced lipolysis in adipocytes both in vivo and in vitro. This finding is supported by a previous study which showed that GLP-1 stimulated lipolysis in both 3T3-L1 cells and primary adipocytes isolated from the visceral fat of human [24]. In contrast, another study, using in situ microdialysis of GLP-1, observed that GLP-1 did not alter the lipolysis rate in abdominal subcutaneous adipose tissue from non-obese individuals [25]. This inconsistency reflects the different fat locations and individuals used in the studies. We studied epididymal fat, as exenatide has been revealed to reduce visceral rather than subcutaneous fat content [26]. The effect of GLP-1 on lipolysis might only exist in pathological conditions, and pharmacologically GLP-1 is likely to act in visceral rather than subcutaneous fat.
Our results show that the lipolytic effect of exenatide on WAT is SIRT1 dependent. SIRT1 promotes lipolysis in adipocytes via activation of the rate-limiting lipolytic enzyme ATGL [14]. Here, we found that exenatide (exendin-4) reduced adiposity by activating SIRT1 and consequently upregulating ATGL. In addition, AMPK, another SIRT1-interacting factor with a vital role in the regulation of hydrolysis and energy dissipation [27, 28], was activated by exenatide in WAT. Our findings indicate that exenatide induces the phosphorylation of AMPK which, in turn, activates SIRT1 by regulating NAD+ concentration [29], triggering a lipolytic cycle.
Despite the remarkable increase in lipolysis in adipocytes exposed to exenatide (exendin-4), levels of the products of lipolysis, such as glycerol and fatty acids, were strikingly decreased in serum. While WAT glycerol content was also significantly decreased in exenatide-treated mice, it is reasonable to speculate that exenatide not only induces lipolysis in adipocytes but also promotes glycerol and lipid consumption. A growing number of studies have demonstrated the capacity of WAT to burn off excessive energy [30–32]; thus, the reduced adiposity is potentially connected with the increased oxidative metabolism in white adipocytes. In our study, a set of oxidative factors normally silent in white adipocytes, including UCP-1, PPARα and PGC-1α, were increased by exenatide both in vitro and in vivo. Moreover, mtDNA copy number and mitochondrial-related gene expression were enhanced in the WAT of exenatide-treated mice. Exenatide also promoted fat oxidation in other sites, such as liver, muscle and BAT, as indicated by the increase in expression of oxidative genes (ESM Fig. 7).
SIRT1 has emerged as a key factor to enhance thermogenesis in BAT via activation of UCP-1 [33]. Resveratrol, a SIRT1 agonist, promotes the remodelling of white adipocytes by increasing oxidative gene expression [34]. Our data suggest that exenatide promotes oxidative metabolism in WAT by activating SIRT1. The upregulation of UCP-1, PPARα and PGC-1α, together with the increased mtDNA content and cytochrome c oxidase activity, occurred in a SIRT1-dependent manner with exenatide treatment.
A clinical study revealed that a dipeptidyl peptidase-4 (DPP-4) inhibitor that decreases endogenous GLP-1 degradation augmented adipose tissue lipolysis and the rate of systemic lipid oxidation in type 2 diabetic patients [35]. Moreover, others have demonstrated that acute activation of central nervous system (CNS) GLP-1Rs promotes BAT thermogenesis and WAT browning [36–38]. However, browning of WAT induced by CNS infusion of GLP-1R agonist was abolished in mice with diet-induced obesity [36, 38]. We observed that chronic peripheral treatment with GLP-1R agonist promoted the browning of WAT in diet-induced obese mice. This finding indicates that central and peripheral GLP-1R activation has different roles in the regulation of WAT metabolism. GLP-1R agonists may have a direct effect on WAT. The poor access of CNS-injected GLP-1R agonists to peripheral tissues, and the short periods of treatment (<7 days) in the central infusion studies, may both contribute to the discrepancy in the results of central vs peripheral administration of GLP-1R agonists on WAT browning.
Another GLP-1R agonist, liraglutide, has recently been approved as an anti-obesity drug because of abundant evidence of its prominent effect on weight loss (reviewed in van Bloemendaal et al [39]). A recent paper described how liraglutide did not affect BAT thermogenesis, but significantly reduced body weight and adiposity in obese mice [40]. This indicates that there is another physiological mechanism underlying the effect of GLP-1R agonists on weight. In the present study, we demonstrated that the GLP-1R agonist exenatide triggers a process of brown remodelling of WAT by promoting lipolysis, fatty acid oxidation and mitochondrial biogenesis in a SIRT1-dependent manner. This reveals a new mechanism by which GLP-1R agonism counters obesity, and provides us with new insights into novel therapeutic targets to treat obesity and its associated metabolic disorders.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- AMPK:
-
AMP-activated protein kinase
- ATGL:
-
Adipose triacylglycerol lipase
- BAT:
-
Brown adipose tissue
- CPT1:
-
Carnitine palmitoyltransferase 1
- FBG:
-
Fasting blood glucose
- GLP-1:
-
Glucagon-like peptide-1
- GLP-1R:
-
GLP-1 receptor
- H&E:
-
Haematoxylin and eosin
- HFD:
-
High-fat diet
- HSL:
-
Hormone-sensitive lipase
- IHC:
-
Immunohistochemical
- ITT:
-
Insulin tolerance test
- MEF:
-
Mouse embryonic fibroblast
- PGC-1α:
-
Peroxisome proliferator-activated receptor γ coactivator-1α
- mtDNA:
-
Mitochondrial DNA
- PPARα:
-
Peroxisome proliferation activated receptor α
- RNAi:
-
RNA interference
- sh:
-
Short hairpin
- SIRT1:
-
Sirtuin 1
- TG:
-
Triacylglycerol
- UCP-1:
-
Uncoupling protein-1
- WAT:
-
White adipose tissue
References
Twig G, Afek A, Derazne E et al (2014) Diabetes risk among overweight and obese metabolically healthy young adults. Diabetes Care 37:2989–2995
Astrup A, Rossner S, Van Gaal L et al (2009) Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 374:1606–1616
DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD (2005) Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 28:1092–1100
Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR (2011) Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 152:3103–3112
Wettergren A, Wojdemann M, Holst JJ (1998) Glucagon-like peptide-1 inhibits gastropancreatic function by inhibiting central parasympathetic outflow. Am J Physiol 275:G984–G992
Nielsen TS, Jessen N, Jorgensen JO, Moller N, Lund S (2014) Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 52:R199–R222
Wilson-Fritch L, Nicoloro S, Chouinard M et al (2004) Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114:1281–1289
Yin X, Lanza IR, Swain JM, Sarr MG, Nair KS, Jensen MD (2014) Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrinol Metab 99:E209–E216
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Bartelt A, Heeren J (2014) Adipose tissue browning and metabolic health. Nat Rev Endocrinol 10:24–36
Wu J, Cohen P, Spiegelman BM (2013) Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 27:234–250
Feige JN, Lagouge M, Canto C et al (2008) Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab 8:347–358
Milne JC, Lambert PD, Schenk S et al (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450:712–716
Chakrabarti P, English T, Karki S et al (2011) SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J Lipid Res 52:1693–1701
Xu C, Bai B, Fan P et al (2013) Selective overexpression of human SIRT1 in adipose tissue enhances energy homeostasis and prevents the deterioration of insulin sensitivity with ageing in mice. Am J Transl Res 5:412–426
Xu F, Gao Z, Zhang J et al (2010) Lack of SIRT1 (mammalian sirtuin 1) activity leads to liver steatosis in the SIRT1 +/- mice: a role of lipid mobilization and inflammation. Endocrinology 151(6):2504–2514
Cheng HL, Mostoslavsky R, Saito S et al (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A 100(19):10794–10799
Xu F, Li Z, Zheng X et al (2014) SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes 63:3637–3646
Anil KK, Marita AR (2000) Troglitazone prevents and reverses dexamethasone induced insulin resistance on glycogen synthesis in 3T3 adipocytes. Br J Pharmacol 130:351–358
Jendle J, Nauck MA, Matthews DR et al (2009) Weight loss with liraglutide, a once-daily human glucagon-like peptide-1 analogue for type 2 diabetes treatment as monotherapy or added to metformin, is primarily as a result of a reduction in fat tissue. Diabetes Obes Metab 11:1163–1172
Richard JE, Anderberg RH, Goteson A, Gribble FM, Reimann F, Skibicka KP (2015) Activation of the GLP-1 receptors in the nucleus of the solitary tract reduces food reward behavior and targets the mesolimbic system. PLoS One 10, e119034
van Bloemendaal L, IJzerman RG, Ten KJ et al (2014) GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes 63:4186–4196
Wei Q, Li L, Chen JA, Wang SH, Sun ZL (2015) Exendin-4 improves thermogenic capacity by regulating fat metabolism on brown adipose tissue in mice with diet-induced obesity. Ann Clin Lab Sci 45:158–165
Vendrell J, El BR, Peral B et al (2011) Study of the potential association of adipose tissue GLP-1 receptor with obesity and insulin resistance. Endocrinology 152:4072–4079
Bertin E, Arner P, Bolinder J, Hagstrom-Toft E (2001) Action of glucagon and glucagon-like peptide-1-(7-36) amide on lipolysis in human subcutaneous adipose tissue and skeletal muscle in vivo. J Clin Endocrinol Metab 86:1229–1234
Bi Y, Zhang B, Xu W et al (2014) Effects of exenatide, insulin, and pioglitazone on liver fat content and body fat distributions in drug-naive subjects with type 2 diabetes. Acta Diabetol 51:865–873
Gaidhu MP, Fediuc S, Anthony NM et al (2009) Prolonged AICAR-induced AMP-kinase activation promotes energy dissipation in white adipocytes: novel mechanisms integrating HSL and ATGL. J Lipid Res 50:704–715
Ceddia RB (2013) The role of AMP-activated protein kinase in regulating white adipose tissue metabolism. Mol Cell Endocrinol 366:194–203
Um JH, Park SJ, Kang H et al (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59:554–563
Jun HJ, Joshi Y, Patil Y, Noland RC, Chang JS (2014) NT-PGC-1alpha activation attenuates high-fat diet-induced obesity by enhancing brown fat thermogenesis and adipose tissue oxidative metabolism. Diabetes 63:3615–3625
Qiang L, Wang L, Kon N et al (2012) Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 150:620–632
Barbatelli G, Murano I, Madsen L et al (2010) The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab 298:E1244–E1253
Andrade JM, Frade AC, Guimaraes JB et al (2014) Resveratrol increases brown adipose tissue thermogenesis markers by increasing SIRT1 and energy expenditure and decreasing fat accumulation in adipose tissue of mice fed a standard diet. Eur J Nutr 53:1503–1510
Mercader J, Palou A, Bonet ML (2011) Resveratrol enhances fatty acid oxidation capacity and reduces resistin and retinol-binding protein 4 expression in white adipocytes. J Nutr Biochem 22:828–834
Boschmann M, Engeli S, Dobberstein K et al (2009) Dipeptidyl-peptidase-IV inhibition augments postprandial lipid mobilization and oxidation in type 2 diabetic patients. J Clin Endocrinol Metab 94:846–852
Nogueiras R, Perez-Tilve D, Veyrat-Durebex C et al (2009) Direct control of peripheral lipid deposition by CNS GLP-1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet-induced obesity. J Neurosci 29:5916–5925
Beiroa D, Imbernon M, Gallego R et al (2014) GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 63:3346–3358
Kooijman S, Wang Y, Parlevliet ET et al (2015) Central GLP-1 receptor signalling accelerates plasma clearance of triacylglycerol and glucose by activating brown adipose tissue in mice. Diabetologia 58:2637–2646
van Bloemendaal L, Ten Kulve JS, la Fleur SE et al (2014) Effects of glucagon-like peptide 1 on appetite and body weight: focus on the CNS. J Endocrinol 221(1):T1–T16
Heppner KM, Marks S, Holland J et al (2015) Contribution of brown adipose tissue activity to the control of energy balance by GLP-1 receptor signalling in mice. Diabetologia 58:2124–2132
Acknowledgements
We gratefully acknowledge T. Tang from the University of California (Berkeley, CA, USA) and M. Li from Yale University (New Haven, CT, USA) for their valuable suggestions about the writing of this article, and N. Weng from University of New South Wales (Sydney, NSW, Australia) for proofreading. We thank Z. Li and K. Zeng for their assistance with the animal experiments.
Parts of this study were presented in Poster 710 at the 50th EASD Annual Meeting in 2014, Vienna, 15–19 September 2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by grants from the NSFC-CIHR (81261120565 to JW), the Program for Changjiang Scholars and Innovative Research Team in University (82000-18811100 to JW), the Program for ‘973’ project (2012CB517506 to JW), the National Natural Science Foundation of China Grant Award (81300705 to FX) and the Fundamental Research Funds for the Central Universities (12ykpy41 to FX).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
FX contributed to the study design, acquisition and interpretation of data, and drafting and revising of the article. BL contributed to the acquisition and analysis of data, and drafting of the article. XZ, ZC and HC contributed to the acquisition of data and revising of the article. HX and HL contributed to the analysis of data and revising of the article. JW contributed to the study design, interpretation of data and revising of the article. All the authors approved the final version to be published. FX is the guarantor of this work.
Additional information
Fen Xu and Beisi Lin contributed equally to this study
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 209 kb)
ESM Table 1
(PDF 84.8 kb)
ESM Table 2
(PDF 95 kb)
ESM Fig. 1
(PDF 147 kb)
ESM Fig. 2
(PDF 132 kb)
ESM Fig. 3
(PDF 260 kb)
ESM Fig. 4
(PDF 295 kb)
ESM Fig. 5
(PDF 192 kb)
ESM Fig. 6
(PDF 154 kb)
ESM Fig. 7
(PDF 170 kb)
Rights and permissions
About this article

Cite this article
Xu, F., Lin, B., Zheng, X. et al. GLP-1 receptor agonist promotes brown remodelling in mouse white adipose tissue through SIRT1. Diabetologia 59, 1059–1069 (2016). https://doi.org/10.1007/s00125-016-3896-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3896-5