
Abstract
Aims/hypothesis
The mechanisms for diet-induced intramyocellular lipid accumulation and its association with insulin resistance remain contentious. In a detailed time-course study in rats, we examined whether a high-fat diet increased intramyocellular lipid accumulation via alterations in fatty acid translocase (FAT/CD36)-mediated fatty acid transport, selected enzymes and/or fatty acid oxidation, and whether intramyocellular lipid accretion coincided with the onset of insulin resistance.
Methods
We measured, daily (on days 1–7) and/or weekly (for 6 weeks), the diet-induced changes in circulating substrates, insulin, sarcolemmal substrate transporters and transport, selected enzymes, intramyocellular lipids, mitochondrial fatty acid oxidation and basal and insulin-stimulated sarcolemmal GLUT4 and glucose transport. We also examined whether upregulating fatty acid oxidation improved glucose transport in insulin-resistant muscles. Finally, in Cd36-knockout mice, we examined the role of FAT/CD36 in intramyocellular lipid accumulation, insulin sensitivity and diet-induced glucose intolerance.
Results
Within 2–3 days, diet-induced increases occurred in insulin, sarcolemmal FAT/CD36 (but not fatty acid binding protein [FABPpm] or fatty acid transporter [FATP]1 or 4), fatty acid transport and intramyocellular triacylglycerol, diacylglycerol and ceramide, independent of enzymatic changes or muscle fatty acid oxidation. Diet-induced increases in mitochondria and mitochondrial fatty acid oxidation and impairments in insulin-stimulated glucose transport and GLUT4 translocation occurred much later (≥21 days). FAT/CD36 ablation impaired insulin-stimulated fatty acid transport and lipid accumulation, improved insulin sensitivity and prevented diet-induced glucose intolerance. Increasing fatty acid oxidation in insulin-resistant muscles improved glucose transport.
Conclusions/interpretations
High-fat feeding rapidly increases intramyocellular lipids (in 2–3 days) via insulin-mediated upregulation of sarcolemmal FAT/CD36 and fatty acid transport. The 16–19 day delay in the onset of insulin resistance suggests that additional mechanisms besides intramyocellular lipids contribute to this pathology.
Similar content being viewed by others

Introduction
Skeletal muscle insulin resistance has been associated with lifestyle factors, including diet. However, the mechanisms involved have remained the subject of considerable debate. Recent work has focused on insulin resistance induced by changes in intramyocellular lipid metabolism [1–7], possibly due to reductions in peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and mitochondrial content [8–10], and/or incomplete fatty acid oxidation due to mitochondrial fatty acid oversupply [11, 12]. Yet these putative mechanisms remain controversial [13–15]. Alternatively, enhanced transport of fatty acids into muscle could account for a diet-induced increase in intramyocellular lipid accumulation and the induction of skeletal muscle insulin resistance.
Fatty acid entry into skeletal muscle is highly regulated [16]. Among fatty acid transporters, fatty acid translocase (FAT/CD36) is key; it has a high rate of fatty acid transport [17] and, like GLUT4, is insulin sensitive [16]. Yet in insulin-resistant human and rodent muscle, FAT/CD36, unlike GLUT4, is permanently relocated to the sarcolemma via unknown mechanisms [16]. It has been suggested that persistent diet-induced increases in insulin may be key to repositioning FAT/CD36 to the sarcolemma, thereby increasing fatty acid transport and intramyocellular lipid accumulation [18]. Several groups have now begun to question the relationship between intramyocellular lipid accumulation and the onset of insulin resistance in muscle [19–23].
To understand the complex series of molecular and biochemical events that develop in muscle with high-fat feeding we determined the daily and/or weekly changes in variables involved in regulating fatty acid transport and metabolism, as well as glucose transport. Additional studies were performed in Cd36-knockout (KO) mice to identify the role of FAT/CD36, and its insulin dependence, in regulating fatty acid transport and metabolism.
Methods
Male Sprague Dawley rats (Crl:SD) (100–125 g) were purchased (Charles River, St Constant, QC, Canada). Wild-type (WT) and Cd36-KO mice (C57Bl/6: 26.3 ± 0.9 g and 25.9 ± 0.8 g, respectively; a gift from M. Febbraio, Cleveland Clinic, Cleveland, OH, USA) and WT and Akt2-KO mice (C57Bl/6: 20.3 ± 0.6 g and 16.9 ± 0.6 g, respectively; a gift from Pfizer, New York, NY, USA) were bred on site [24]. Mice were age-matched in the experiments. All animals were housed in a temperature-regulated environment (20°C) with a reversed 12:12 h light–dark cycle. The principles of laboratory animal care were followed (as in NIH publication no. 85–23, revised 1985, available at http://grants1.nih.gov/grants/olaw/references/phspol.htm) and the experiments were approved by the Animal Care Committee, University of Guelph.
Experimental design
Upon arrival, animals were assigned alternately to either a chow or high fat diet group. Experimenters were necessarily aware of group assignments, as this information was required for analytical purposes. No animals were excluded from the experiments. Rats were fed isoenergetically for 6 weeks with chow (10% energy from fat, D05092806BM; n = 410) or a high-fat diet (60% energy from fat, D05120801; n = 417) (Research Diets, New Brunswick, NJ, USA). Body weight was monitored every second day. In fasted rats, tail vein blood and muscle samples were obtained for the first 7 days (high fat only) and weekly thereafter (chow and high fat) for 6 weeks.
During the course of the study, highly oxidative (red tibialis anterior [RTA] and red gastrocnemius [RG]) and highly glycolytic (white tibialis anterior [WTA] and white gastrocnemius [WG]) rat hindlimb muscles were sampled from anaesthetised rats (sodium pentobarbital, 6 mg/100 g body weight i. p.; Ceva Sante Animale, Libourne, France), frozen and stored (−80°C). Muscles were analysed for mitochondrial DNA (mtDNA), proteins and intramyocellular lipids. Substrate transporters were measured in homogenates and sarcolemma. Functional measurements, including fatty acid and glucose transport (basal and insulin-stimulated), mitochondrial fatty acid oxidation and glucose tolerance were also obtained. Additionally, we examined in WT and Cd36-KO mice the effects of insulin-induced FAT/CD36 and GLUT4 translocation, fatty acid and glucose transport, and fatty acid and glucose metabolism. In WT and Akt2-KO mice we examined the effects of insulin on FAT/CD36 translocation and fatty acid transport.
Analytical procedures
Serum samples were analysed for insulin, glucose, triacylglycerol and fatty acids (see electronic supplementary material [ESM] for further details). Basal and adrenaline (epinephrine)-stimulated fatty acid release from adipose tissues was performed as previously described [25].
We determined mtDNA as previously described [26]. Western blotting was used to measure proteins [27, 28] (see ESM for further details). Insulin and radiolabelled substrates were purchased (see ESM for further details).
Fatty acid transport and transporters, and intramyocellular lipids
In overnight-fasted rats sarcolemmal FAT/CD36 (basal and insulin-stimulated) and fatty acid transport were determined. Intramyocellular lipids were determined in freeze-dried muscle [17, 27, 28] (see ESM for further details).
Palmitate oxidation by isolated mitochondria
Palmitate oxidation and carnitine palmitoyltransferase 1 (CPTI) activity were measured in isolated subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria [26, 27] (see ESM for further details).
Glucose tolerance, glucose transport, plasmalemmal GLUT4, Akt2 and AS160
Glucose tolerance was determined in overnight-fasted rats. The insulin sensitivity of glucose transport was determined in basal and insulin-stimulated perfused muscles [26]. An insulin injection was performed to examine phosphorylation of protein kinase B (Akt)-(ser473) and Akt substrate of 160 kDa (AS160) (see ESM for further details).
Effects of improving palmitate oxidation on glucose transport
In insulin-resistant muscle, insulin-stimulated glucose transport was examined when fatty acid oxidation had been increased by Pgc-1α (also known as Ppargc1a) transfection (see ESM for further details).
Studies in Cd36-KO and Akt2-KO mice
We examined insulin-stimulated sarcolemmal FAT/CD36 and fatty acid transport in WT and Cd36-KO mice, and in WT and Akt2-KO mice, and lipid and glucose metabolism in Cd36-KO mice [27–29]. Glucose tolerance was determined in chow- and fat-fed WT and Cd36-KO mice (weeks 0, 3 and 6) (see ESM for further details).
Statistics
Data were analysed using analyses of variance and Fisher’s least significant difference (LSD) post hoc test when appropriate. For some comparisons Student’s t tests were appropriate. Data are reported as mean ± SEM.
Results
In rats, we examined the chronological changes induced by a high-fat diet (ESM Table 1) in molecular and biochemical events involved in regulating: (1) skeletal muscle fatty acid transport and metabolism; and (2) glucose transport. We also examined: (3) the temporal relationship of intramyocellular lipid accretion with the onset of muscle insulin resistance. These studies were supplemented with studies in genetically altered mice. Time-series westerns blots are shown in ESM Fig. 1.
Basal metabolic characteristics in chow- and fat-fed rats
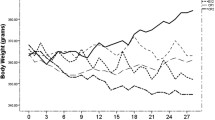
Weight gains were comparable in pair-fed chow- and fat-fed animals (Fig. 1a). In fat-fed animals: insulin (Fig. 1b) and glucose (Fig. 1c) were increased after 2 days and 3 weeks, respectively; circulating levels of triacylglycerol were unaltered (Fig. 1d); but circulating levels of fatty acids (Fig. 1e) were reduced from day 2 onwards. These rapid fatty acid reductions were not attributable to altered adipose tissue fatty acid handling, as no changes were observed in the insulin-mediated suppression of plasma fatty acids (data not shown), hepatic fatty acid transport (data not shown) or fatty acid release from adipose tissue depots (Fig. 1f).

In rats, a high-fat diet-induced changes in body mass (a), insulin (b), glucose (c), triacylglycerol (d), fatty acids (e) and basal and adrenaline-stimulated fatty acid release from selected adipose tissue depots (f). Mean ± SEM, n = 5–8 at each time point. The arrows indicate where change (p < 0.05) in a variable first occurred and was then maintained. In (a–e): white circles, chow; and black circles, high fat. In (f) white symbols, basal; black symbols, + adrenaline; squares, chow; circles, high fat. Epid., epidydimal; FA, fatty acids; retrop., retroperitoneal; subcut., subcutaneous
High-fat diet-induced changes in muscle fatty acid transport and lipid metabolism
Diet-induced changes in FAT/CD36 and fatty acid transport in rats
In muscle, no diet-induced changes occurred in the protein expression or sarcolemmal content of selected fatty acid transporters (fatty acid binding protein [FABPpm], fatty acid transporter [FATP]1 or 4; ESM Fig. 2a–f), nor in FAT/CD36 protein expression (Fig. 2a). However, the high-fat diet provoked rapid increases in sarcolemmal FAT/CD36 (Fig. 2b) and palmitate transport (Fig. 2c) after 2 days (red muscle) and 3 days (white muscle). Changes in these two variables were highly correlated (r ≥ 0.92, ESM Fig. 3a), as were the increased rates of fatty acid transport and reductions in circulating fatty acids (r = −0.84, ESM Fig. 3b).

Changes induced by a high-fat diet in rat red and white muscle FAT/CD36 protein (a), sarcolemmal FAT/CD36 (b) and fatty acid transport (c). Mean ± SEM, n = 5–8 at each time point. Western blots are shown in ESM Fig. 1a. Arrows indicate where change (p < 0.05) in a variable first occurred and was maintained. White symbols, chow; black symbols, high fat; circles, red muscle; squares, white muscle. AU, arbitrary units; prot, protein
Insulin sensitivity of FAT/CD36 translocation and its diet-induced impairment in rats
Circulating insulin was highly correlated with sarcolemmal FAT/CD36 content (r ≥ 0.92, Fig. 3a) and fatty acid transport (r ≥ 0.85, ESM Fig. 3c). Insulin administration (bolus) induced the translocation of FAT/CD36 throughout the 6 weeks in chow-fed rats (Fig. 3b), but this was impaired in fat-fed rats. Specifically, after 1 week (red muscle) or 2 weeks (white muscle) of high-fat feeding, insulin failed to increase sarcolemmal FAT/CD36 beyond that induced by the high-fat diet (Fig. 3c).

Correlations in rats between circulating insulin and sarcolemmal FAT/CD36 (a), and the effects of insulin, administered via hindlimb perfusion, on sarcolemmal FAT/CD36 in red and white muscles of chow-fed (b) and high-fat-fed (c) animals. Mean ± SEM, n = 5–8 at each time point. Western blots are shown in ESM Fig. 1b, c. Insulin-stimulated vs basal: * p < 0.05 in red muscle; † p < 0.05 in white muscle. In (a): black triangle, red muscle, r = 0.93; white triangle, white muscle, r = 0.92. In (b, c): circles, red muscle; squares, white muscle; white symbols, basal; and black symbols, insulin. AU, arbitrary units; prot, protein
Diet-induced changes in intramyocellular lipids in rats
To determine the consequences of rapid increases in fatty acid transport we examined intramyocellular lipids. In concert with temporal changes in fatty acid transport, intramyocellular triacylglycerol (Fig. 4a), diacylglycerol (Fig. 4b) and ceramide accumulation (Fig. 4c) was rapidly increased (red muscle, 2 days; white muscle, 3–4 days). These changes were associated with increases in selected fatty acids, including 16:0, 16:1, 18:0, 18:1 and 18:2 (ESM Table 2 and ESM Figs 4–6).

Changes induced by a high-fat diet in rat red and white muscle triacylglycerol (a), diacylglycerol (b), ceramide (c), DGAT2 (d) and GPAT (e), and correlations between fatty acid transport and intramyocellular lipids (f). Mean ± SEM, n = 5–8 at each time point. Western blots are shown in ESM Fig. 1a. White muscle DGAT2 and GPAT are expressed relative to red muscle of chow-fed animals (t = 0). Arrows indicate where change (p < 0.05) in a variable first occurred and was then maintained. Correlation coefficients: triacylglycerol, red muscle, r = 0.87, white muscle, r = 0.87; diacylglycerol, red muscle, r = 0.92, white muscle, r = 0.83; ceramide: red muscle, r = 0.77; white muscle, r = 0.60. Circles, red muscle; squares, white muscle; white symbols, chow; and black symbols, high fat. AU, arbitrary units; CER, ceramide; DAG, diacylglycerol; prot, protein; TAG, triacylglycerol; wt, weight
The rapid diet-induced changes in intramyocellular lipids were not related to changes in selected enzymes, as these remained unaltered (diacylglycerol acyltransferase 2 [DGAT2], Fig. 4d), or were decreased after 1 week (white muscle) and 3 weeks (red muscle) (glycerol-3-phosphate acyltransferase [GPAT], Fig. 4e). In contrast, intramyocellular lipid accumulation in both red and white muscle was correlated with fatty acid transport (r = 0.60–0.92) (Fig. 4f).
Diet-induced changes in fatty acid oxidation and mitochondrial biogenesis in rats
Increases in intramyocellular lipids were not attributable to mitochondrial dysfunction, as 2 weeks of high-fat feeding increased fatty oxidation by red muscle IMF (Fig. 5a) and SS mitochondria (Fig. 5b), independent of changes in CPTI activity (Fig. 5c, d). Diet-induced increases in PGC-1α protein occurred after 2 weeks (red muscle only) (Fig. 5e), while DNA copy number was increased after 3 weeks (red muscle) and 6 weeks (white) (Fig. 5f). Based on the relative muscle content of SS and IMF mitochondria [30], and the increase in muscle mitochondria (Fig. 5f), we calculated that there was a diet-induced increase in whole-muscle fatty acid oxidation after 3 weeks (red muscle) and 5 weeks (white muscle) (Fig. 5g).

Changes induced by a high-fat diet in rat red and white muscle: mitochondrial fatty acid oxidation in IMF (a) and SS (b) mitochondria; CPTI activity in IMF (c) and SS (d) mitochondria; muscle PGC-1α (e); mtDNA (f); and muscle fatty acid oxidation (g). Arrows indicate where change (p < 0.05) in a variable first occurred and was maintained. Western blots are shown in ESM Fig. 1a. In (f) white muscle is set to 1 at week 0. In (g), muscle palmitate oxidation index = mitochondrial content (mtDNA) × the relative contributions of IMF and SS mitochondria [30]. Mean ± SEM, n = 5–8 at each time point. In (a–g): white symbols, chow; black symbols, high fat; circles, red muscle; and squares, white muscle. AU, arbitrary units; prot, protein
Cd36-KO and Akt2-KO mice demonstrate that fatty acid transport and intramyocellular lipid accumulation are insulin-, FAT/CD36- and Akt2-dependent
To establish the role of insulin on FAT/CD36, we examined the effect of insulin in the muscle of Cd36-KO and Akt2-KO mice. Insulin stimulated fatty acid transport in WT mice by inducing the translocation of FAT/CD36 to the sarcolemma (Fig. 6a), via Akt2 activation (Fig. 6b). In Cd36-KO mice, reduced fatty acid transport (Fig. 6a) was associated with reductions in intramyocellular lipids (Fig. 6c), basal and insulin-stimulated fatty acid esterification (Fig. 6d), and basal and caffeine-stimulated fatty acid oxidation (Fig. 6e).

Insulin-stimulated fatty acid transport and sarcolemmal FAT/CD36 in WT and Cd36-KO mice (a) and in WT and Akt2-KO mice (b). In Cd36-KO mice, intramyocellular lipid content (c) insulin-stimulated palmitate esterification and caffeine-stimulated palmitate oxidation (d) and oxidation (e). Mean ± SEM, n = 4–7 at each point. In (a, b): white, basal; black, insulin. * p < 0.05, insulin vs basal. In (c–e): white bars, WT; and black bars, Cd36-KO. * p < 0.05, KO vs WT; † p < 0.05 vs basal. AU, arbitrary units; B, basal; Caf, caffeine; CER, ceramide; DAG, diacylglycerol; Ins, insulin; ND, not determined; R, red; TAG, triacylglycerol; W, white
Diet-induced skeletal muscle insulin resistance
Diet-induced changes in insulin-stimulated GLUT4 translocation and glucose transport in rats
After 3 weeks of high-fat feeding, whole-body glucose tolerance (Fig. 7a), insulin-stimulated glucose transport (Fig. 7b, c) and GLUT4 translocation, but not GLUT4 expression (Fig. 7d), were impaired (Fig. 7e, f). Diet-induced impairments in GLUT4 translocation did not coincide with concurrent impairments in insulin signalling (Fig. 7g–l), as in white muscle only, there were diet-induced increases in Akt2 (week 4) and AS160 proteins (week 1) (Fig. 7g, j). Concurrently, insulin-stimulated Akt phosphorylation (ser473), although somewhat reduced, was not significantly altered (p > 0.05, Fig. 7h, i). Insulin-stimulated AS160 phosphorylation was reduced under basal (red muscle, week 1) and insulin-stimulating conditions (red muscle, week 2; white muscle, week 1) (Fig. 7k, l), 2 weeks prior to the onset of insulin resistance.

Changes induced by a high-fat diet in rats: glucose tolerance (a); and in red and white muscle, basal and insulin-stimulated glucose transport (b, c), muscle GLUT4 (d), sarcolemmal GLUT4 (e, f), muscle Akt2 (g), pAkt (ser473) (h, i), AS160 (j) and pAS160 (k, l). In (a–l), arrows denote the point where a change (p < 0.05) in a given variable first occurred and was maintained; mean ± SEM, n = 5–8 at each time point. Western blots are shown in ESM Fig. 1a, b. White symbols, chow; black symbols, high fat. In (d, g, j): circles, red muscle; squares, white muscle. In (b, c, e, f, h, i, k, l): circles, basal; squares, insulin. AU, arbitrary units
Cd36-KO mice display improved insulin sensitivity and resistance to diet-induced impairment in glucose tolerance
We examined sarcolemmal GLUT4, glucose transport and glucose tolerance in Cd36-KO mice in which intramyocellular lipids are reduced (see Fig. 4c). In Cd36-KO mice, insulin-stimulated sarcolemmal GLUT4 content (Fig. 8a), and glucose transport (Fig. 8b) and oxidation (Fig. 8c) were increased. With high-fat feeding, the impaired glucose tolerance observed from week 3 onwards in WT mice was not observed in Cd36-KO mice (Fig. 8d).

In WT and Cd36-KO mice, insulin-stimulated sarcolemmal GLUT4 (KO and WT order is reversed from plotted graph order) (a), glucose transport (b) and oxidation (c), and glucose tolerance in chow- and fat-fed mice at 3 and 6 weeks (d). Mean ± SEM, n = 4–7 at each time point. In (a–c): white bars, WT; black bars, Cd36-KO. In (d): white bars, week 0; grey bars, week 3; black bars, week 6. * p < 0.05, KO vs WT (a–c); and high fat vs respective weeks in chow (d). † p < 0.05, insulin-stimulated vs basal in respective groups (a–c); and WT high fat vs KO high fat in respective weeks (d). AU, arbitrary units; Ch, chow; HF, high fat; HQ, hindquarter; Ins, insulin
Marked delay between alterations in intramyocellular lipids and insulin resistance
In both muscle types there was a striking 16–19 day delay between the rapid (2–3 days), diet-induced increases in selected aspects of lipid metabolism and the onset of insulin resistance (3 weeks) (Fig. 9). In red muscle compared with white muscle, the rapid increases in fatty acid transport and intramyocellular lipid accumulation occurred 1–2 days earlier and were greater. In addition, fatty acid oxidation upregulation occurred 2 weeks earlier (red muscle, week 3; white muscle, week 5). Nevertheless, muscle insulin resistance occurred at the same time (week 3) in both muscle types (see Fig. 7b, c, e, f). Interestingly, in diet-induced insulin-resistant muscle at 4 weeks, a PGC-1α-induced upregulation of fatty acid oxidation improved insulin-stimulated glucose transport (ESM Fig. 7).

Schematic summary of the temporal relationships in rat red muscle for changes induced by a high-fat diet in selected variables of fatty acid transport and metabolism, mitochondrial biogenesis and insulin sensitivity. White bars, no difference between chow- and fat-fed animals; black bars, onset and continuation of increases (upwards arrow) or decreases (downwards arrow) in fat-fed animals. FA, fatty acid; CER, ceramide; DAG, diacylglycerol; Ox, oxidation; SL, sarcolemmal; TAG, triacylglycerol
Discussion
We have shown that, independent of changes in either fatty acid oxidation or selected lipid metabolism enzymes, a high-fat diet very rapidly increases intramyocellular lipids (2–3 days), concurrent with rapid increases, within 2–3 days, in insulin, sarcolemmal FAT/CD36 and fatty acid transport. These effects in lipid metabolism preceded the onset of muscle insulin resistance by 16–19 days.
High-fat-diet-induced increase in insulin, plasmalemmal FAT/CD36, fatty acid transport and intramyocellular lipids
Our present study and others have observed rapid (≤7 days) increases in insulin (+60 to +100%) induced by a high-fat diet [18, 31, 32]. The dietary-associated mechanism(s) for these rapid insulin increases remain undefined. It is possible that enhanced insulin secretion is induced by dietary fats via their stimulation of gastric inhibitory polypeptide (GIP), which has potent insulinotropic effects on beta cells [33]. Alternatively, enhanced insulin secretion may be induced by dietary fatty acids via a FAT/CD36-dependent process in pancreatic beta cells (MIN6) [34]. Finally, the rapid development of insulin resistance in liver and adipose tissues, which occurs several weeks prior to muscle insulin resistance [21, 32, 35], may also contribute to the early insulin increase. In view of the high correlations between circulating insulin and sarcolemmal FAT/CD36 (r ≥ 0.92), and the insulin sensitivity of FAT/CD36-mediated fatty acid transport, it appears that the diet-induced increase in insulin regulates the permanent redistribution of FAT/CD36 to the sarcolemma. The concurrent upregulation of fatty acid transport may account for the concomitant reduction in circulating fatty acids (r ≥ −0.84), as hepatic and adipose tissue fatty acid handling were not altered.
Diet-induced increases in intramyocellular triacylglycerol, diacylglycerol and ceramide occur much more rapidly (2–3 days) (present study, [18]) than has previously been recognised. Yet we and others have shown that neither changes in selected lipid metabolism enzymes nor fatty acid oxidation can account for rapid intramyocellular lipid accumulation (present study, [19, 36–38]). Instead, we found that, just as in human obesity and type 2 diabetes [36, 39, 40], fatty acid transport is highly correlated with the accumulation of intramyocellular lipids (r = 0.60–0.92). Moreover, our present studies in Cd36-KO mice demonstrate a central role for FAT/CD36 in intramyocellular lipid accumulation.
Diet-induced insulin resistance of FAT/CD36 and GLUT4
With high-fat feeding, FAT/CD36 insulin sensitivity became impaired several weeks before the onset of GLUT4 insulin resistance. This might suggest that FAT/CD36 is more insulin sensitive than GLUT4. Nevertheless, after ≥3 weeks the GLUT4 and FAT/CD36 subcellular localisations were juxtaposed, with GLUT4 being compartmentalised intracellularly and FAT/CD36 being retained at the sarcolemma. Insulin signalling via the phosphoinositide 3-kinase (PI3)–kinase–Akt2 pathway is required to increase sarcolemmal FAT/CD36 (present study, [41]), just as this is required for GLUT4 translocation. However, given the juxtaposed subcellular repositioning of FAT/CD36 and GLUT4 in insulin-resistant muscle, their signalling and/or trafficking diverge at some point. AS160 is discounted as impaired FAT/CD36 insulin insensitivity was temporally dissociated from impaired insulin-stimulated AS160 phosphorylation. Substrate transporter selectivity may not necessarily be modulated at the level of signalling, but may occur at the level of trafficking (i.e. via soluble NSF attachment protein receptor [SNARE] proteins), particularly as vesicle associated membrane protein (VAMP)4 and 7 have been found to discriminate between FAT/CD36 and GLUT4 traffic, respectively [42].
FAT/CD36 contributes to the regulation of insulin-stimulated glucose transport
In Cd36-KO mice, reductions in intramyocellular lipids were associated with increased insulin-stimulated glucose transport (present study, [28, 43]). In addition, a high-fat diet did not impair glucose tolerance (present study) or glucose use in Cd36-KO mice [44], and FAT/CD36 ablation mitigated against diet-induced selected metabolic changes [45]. Thus, by limiting FAT/CD36-associated fatty acid transport and/or intramyocellular lipid accumulation, insulin sensitivity is preserved.
Diet-induced changes in insulin resistance, lipid metabolism and mitochondrial variables
It is well known that fat quality can influence insulin sensitivity. However, it is unclear how much dietary monounsaturated fat (%) is required to counterbalance the reduction in muscle insulin sensitivity induced by dietary saturated fatty acids. In our study, the high-fat diet did induce insulin resistance, although its magnitude may have been attenuated by dietary monounsaturated fats.
Diet-induced increases in intramyocellular triacylglycerol, diacylglycerol and ceramide (present study, [21, 23]), were not attributable to reductions in fatty acid oxidation, as this was unaltered for at least 3–5 weeks. Thereafter, it was upregulated because of increases in mitochondrial content and mitochondrial fatty acid oxidation. When we increased muscle fatty acid oxidation by overexpressing PGC-1α in insulin-resistant muscle from either Zucker obese rats [27] or high-fat-fed rats (present study), insulin-stimulated glucose transport was increased. This suggests that the upregulation of fatty acid oxidation observed in fat-fed rats (present study, [21, 23]) may be adaptive to limit further impairment in glucose transport.
Long delay between intramyocellular lipid accumulation and insulin resistance
Although intramyocellular lipids have frequently been linked with the onset of insulin resistance, this relationship has recently been questioned [20–23]. In our study, diet-induced impairments in glucose tolerance and insulin-stimulated GLUT4 translocation and glucose transport occurred 16–19 days after increases in intramyocellular diacylglycerol and ceramide. Moreover, the intramyocellular diacylglycerol and ceramide content observed at the onset of insulin resistance (week 3) had been attained 16–19 days earlier. Clearly, muscle insulin resistance, if it is induced via intramyocellular lipid accumulation, requires considerable time to develop, and likely involves additional mechanisms that remain to be identified.
Are studies of insulin resistance in rodents relevant to humans?
Direct comparison between so-called high-fat studies in rodents and/or humans are difficult as high-fat diets can differ with respect to the content of fat, their fatty acid composition (saturated vs unsaturated) and the use of various supplements, all of which may have separate and independent effects [46, 47]. Thus, whether the present observations in fat-fed rats can be applied to humans is perhaps uncertain. Although there is disagreement about whether the increase in muscle fatty acid oxidation observed in insulin-resistant rat muscle (present study, [15, 48]) is also observed in human insulin resistance (obesity and type 2 diabetes) [13, 14], a recent analysis by Holloszy [13] concluded that ‘most of the studies comparing fat oxidation in insulin-sensitive and insulin-resistant individuals have shown that fat oxidation is higher in T2DM patients and obese insulin-resistant individuals than in insulin-sensitive control subjects’ [13].
The effects of a high-fat diet on FAT/CD36 in humans are equivocal, as muscle FAT/CD36 protein expression and/or its sarcolemmal content may or may not be increased [31, 49, 50]. Nevertheless, there are very similar relationships between circulating insulin and sarcolemmal FAT/CD36 in fat-fed rats (Fig. 3a, r ≥ 0.92) and insulin-resistant humans (r = 0.80, unpublished data), and between fatty acid transport and intramyocellular triacylglycerol in fat-fed rats (Fig. 4f, r ≥ 0.87) and insulin-resistant humans (r = 0.98, [39]). This indicates that in fat-fed rodent models of insulin resistance and human insulin resistance there is considerable consistency in: (1) the insulin-associated increases in sarcolemmal FAT/CD36; and (2) the positive relationship between FAT/CD36-mediated fatty acid transport and intramyocellular lipid accumulation.
Experimental caveats
We controlled body weights in the two groups via pair-feeding. We acknowledge that this does not imply that body compositions between the groups were comparable. Also, in the present study we supplemented our studies with ex vivo studies. We recognise that these cannot fully mimic the in vivo situation. Nevertheless, ex vivo studies are commonly used to derive possible mechanistic explanations for in vivo observations.
Summary
We found that, independent of changes in fatty acid oxidation and selected enzymes, a high-fat diet induced a very rapid, insulin-associated increase in sarcolemmal FAT/CD36 and fatty acid transport (2–3 days), which accounted for the very rapid intramyocellular accumulations of triacylglycerol, diacylglycerol and ceramide. However, it appears that other mechanisms besides intramyocellular accumulations contribute to the onset of insulin resistance, as rapid intramyocellular lipid accumulations (2–3 days) preceded the onset of diet-induced insulin resistance by 16–19 days.
Abbreviations
- Akt:
-
Protein kinase B
- AS160:
-
Akt substrate of 160 kDa
- CPTI:
-
Carnitine palmitoyltransferase 1
- DGAT2:
-
Diacylglycerol acyltransferase 2
- GPAT:
-
Glycerol-3-phosphate acyltransferase
- FAT/CD36:
-
Fatty acid translocase
- FATP:
-
Fatty acid transporter
- FABPpm:
-
Fatty acid binding protein
- IMF:
-
Intermyofibrillar
- KO:
-
Knockout
- mtDNA:
-
Mitochondrial DNA
- PGC-1α:
-
Peroxisome proliferator-activated receptor γ co-activator 1α
- SS:
-
Subsarcolemmal
- WT:
-
Wild type
References
Cantley JL, Yoshimura T, Camporez JP et al (2013) CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A 110:1869–1874
Erion DM, Shulman GI (2010) Diacylglycerol-mediated insulin resistance. Nat Med 16:400–402
Choi CS, Savage DB, Kulkarni A et al (2007) Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 282:22678–22688
Jornayvaz FR, Birkenfeld AL, Jurczak MJ et al (2011) Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc Natl Acad Sci U S A 108:5748–5752
Jornayvaz FR, Shulman GI (2012) Diacylglycerol activation of protein kinase cepsilon and hepatic insulin resistance. Cell Metab 15:574–584
Holland WL, Brozinick JT, Wang LP et al (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Ussher JR, Koves TR, Cadete VJ et al (2010) Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 59:2453–2464
Mootha VK, Lindgren CM, Eriksson KF et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Patti ME, Butte AJ, Crunkhorn S et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100:8466–8471
Holloway GP, Thrush AB, Heigenhauser GJ et al (2007) Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am J Physiol Endocrinol Metab 292:E1782–E1789
Koves TR, Ussher JR, Noland RC et al (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7:45–56
Bell JA, Reed MA, Consitt LA et al (2010) Lipid partitioning, incomplete fatty acid oxidation, and insulin signal transduction in primary human muscle cells: effects of severe obesity, fatty acid incubation, and fatty acid translocase/CD36 overexpression. J Clin Endocrinol Metab 95:3400–3410
Holloszy JO (2013) “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62:1036–1040
Goodpaster BH (2013) Mitochondrial deficiency is associated with insulin resistance. Diabetes 62:1032–1035
Hancock CR, Han DH, Chen M et al (2008) High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A 105:7815–7820
Glatz JFC, Luiken JJFP, Bonen A (2010) Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev 90:367–417
Nickerson JG, Alkhateeb H, Benton CR et al (2009) Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1, and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J Biol Chem 284:16522–16530
Lee YS, Li P, Huh JY et al (2011) Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60:2474–2483
Amati F, Dube JJ, Alvarez-Carnero E et al (2011) Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60:2588–2597
Sitnick MT, Basantani MK, Cai L et al (2013) Skeletal muscle triacylglycerol hydrolysis does not influence metabolic complications of obesity. Diabetes 62:3350–3361
Turner N, Kowalski GM, Leslie SJ et al (2013) Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56:1638–1648
Bosma M, Kersten S, Hesselink MK, Schrauwen P (2012) Re-evaluating lipotoxic triggers in skeletal muscle: relating intramyocellular lipid metabolism to insulin sensitivity. Prog Lipid Res 51:36–49
Mullen KL, Pritchard J, Ritchie I et al (2009) Adiponectin resistance precedes the accumulation of skeletal muscle lipids and insulin resistance in high-fat-fed rats. Am J Physiol Regul Integr Comp Physiol 296:R243–R251
Garofalo RS, Orena SJ, Rafidi K et al (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest 112:197–208
Wan Z, Ritchie I, Beaudoin MS, Castellani L, Chan CB, Wright DC (2012) IL-6 indirectly modulates the induction of glyceroneogenic enzymes in adipose tissue during exercise. PLoS One 7, e41719
Benton CR, Nickerson J, Lally J et al (2008) Modest PGC-1α overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in SS, not IMF, mitochondria. J Biol Chem 283:4228–4240
Benton CR, Holloway GP, Han XX et al (2010) Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 53:2008–2019
McFarlan JT, Yuko Yoshida Y, Jain SS et al (2012) In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance and training-induced adaptation of fatty acid oxidation. J Biol Chem 287:23502–23516
Bonen A, Han X-X, Habets DDJ, Febbraio M, Glatz JFC, Luiken JJFP (2007) A null mutation in skeletal muscle FAT/CD36 reveals its essential role in insulin-, and AICAR-stimulated fatty acid metabolism. Am J Physiol Endocrinol Metab 292:E1740–E1749
Nielsen J, Mogensen M, Vind BF et al (2009) Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am J Physiol Endocrinol Metab 298:E706–E713
Jordy AB, Serup AK, Karstoft K, Pilegaard H, Kiens B, Jeppesen J (2014) Insulin sensitivity is independent of lipid binding protein trafficking at the plasma membrane in human skeletal muscle: effect of a 3-day, high-fat diet. Am J Physiol Regul Integr Comp Physiol 307:R1136–R1145
Paglialunga S, Ludzki A, Root-McCaig J, Holloway GP (2015) In adipose tissue, increased mitochondrial emission of reactive oxygen species is important for short-term high-fat diet-induced insulin resistance in mice. Diabetologia 58:1071–1080
Nasteska D, Harada N, Suzuki K et al (2014) Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes 63:2332–2343
Noushmehr H, D’Amico E, Farilla L et al (2005) Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 54:472–481
Lee YS, Kim JW, Osborne O et al (2014) Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 157:1339–1352
Li M, Paran C, Wolins NE, Horowitz JF (2011) High muscle lipid content in obesity is not due to enhanced activation of key triglyceride esterification enzymes or the suppression of lipolytic proteins. Am J Physiol Endocrinol Metab 300:E699–E707
Lally JS, Snook LA, Han XX, Chabowski A, Bonen A, Holloway GP (2012) Subcellular lipid droplet distribution in red and white muscles in the obese Zucker rat. Diabetologia 55:479–488
Holloway GP, Benton CR, Mullen KL et al (2009) In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am J Physiol Endocrinol Metab 296:E738–E747
Bonen A, Parolin ML, Steinberg GR et al (2004) Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J 18:1144–1146
Aguer C, Mercier J, Yong Wai Man C et al (2010) Intramyocellular lipid accumulation is associated with permanent relocation ex vivo and in vitro of fatty acid translocase (FAT)/CD36 in obese patients. Diabetologia 53:1151–1163
Luiken JJFP, Dyck DJ, Han X-X et al (2002) Insulin induces the translocation of the fatty acid transporter FAT/CD36 to the plasma membrane. Am J Physiol Endocrinol Metab 282:E491–E495
Schwenk RW, Dirkx E, Coumans WA et al (2010) Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 53:2209–2219
Goudriaan JR, Dahlmans VE, Teusink B et al (2003) CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J Lipid Res 44:2270–2277
Hajri T, Han XX, Bonen A, Abumrad NA (2002) Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest 109:1381–1389
Koonen DP, Sung MM, Kao CK et al (2010) Alterations in skeletal muscle fatty acid handling predisposes middle-aged mice to diet-induced insulin resistance. Diabetes 59:1366–1375
Buettner R, Scholmerich J, Bollheimer LC (2007) High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring) 15:798–808
Warden CH, Fisler JS (2008) Comparisons of diets used in animal models of high fat feeding. Cell Metab 7:277
Turner N, Bruce CR, Beale SM et al (2007) Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 56:2085–2092
Cameron-Smith D, Burke LM, Angus DJ et al (2003) A short-term, high-fat diet up-regulates lipid metabolism and gene expression in human skeletal muscle. Am J Clin Nutr 77:313–318
Roepstorff C, Helge JW, Vistisen B, Kiens B (2004) Studies of plasma membrane fatty acid-binding protein and other lipid-binding proteins in human skeletal muscle. Proc Nutr Soc 63:239–244
Acknowledgements
We thank A. Piatelli (Department of Human Health and Nutritional Sciences, University of Guelph, Guelph, ON, Canada) for his animal care work.
Funding
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (AB, DCW, GPH), the Canadian Institutes of Health Research (AB, DCW), the Heart and Stroke Foundation of Ontario (AB) and the Canada Research Chair program (AB, DCW). M-SB was a recipient of a doctoral scholarship from the Natural Sciences and Engineering Research Council of Canada. SSJ and LAS were recipients of doctoral Canada Graduate Scholarships from the Natural Sciences and Engineering Research Council of Canada. DCW was the Canada Research Chair (Tier II) in Lipids, Metabolism and Health. AB was the Canada Research Chair (Tier I) in Metabolism and Health.
Duality of interest
The authors declare there is no duality of interest associated with this manuscript.
Contribution statement
AB conceived, designed and supervised the study, analysed the data and wrote the manuscript. SSJ, LAS, X-XH, YY, KHB, JSL, EDP, SP, M-SB and EH performed experiments, interpreted data and revised the manuscript. AC, DCW, JFCG, JJFPL and GPH contributed intellectual expertise, interpreted data and reviewed and edited the manuscript. AB is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed to reviewing and editing the manuscript, and approved its final version.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 90 kb)
ESM Table 1
(PDF 23 kb)
ESM Table 2
(PDF 30 kb)
ESM Fig. 1
(PDF 0.99 mb)
ESM Fig. 2
(PDF 148 kb)
ESM Fig. 3
(PDF 104 kb)
ESM Fig. 4
(PDF 155 kb)
ESM Fig. 5
(PDF 155 kb)
ESM Fig. 6
(PDF 155 kb)
ESM Fig. 7
(PDF 94 kb)
Rights and permissions
About this article

Cite this article
Bonen, A., Jain, S.S., Snook, L.A. et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 58, 2381–2391 (2015). https://doi.org/10.1007/s00125-015-3691-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3691-8