
Abstract
Aims/hypothesis
The aim of this work was to investigate the prospective relationship between low birthweight (LBW) and type 2 diabetes risk later in life and the mediation effects of type 2 diabetes biomarkers linking LBW to type 2 diabetes risk.
Methods
We measured baseline plasma concentrations of various type 2 diabetes biomarkers in 1,259 incident type 2 diabetes cases and 1,790 controls in the Women’s Health Initiative-Observational Study. Self-report birthweights of the participants were recorded. The total effect of LBW on type 2 diabetes risk was partitioned into effects that were mediated by a specific biomarker and effects that were not mediated by this biomarker, using counterfactual model-based mediation analysis.
Results
LBW was significantly associated with increased risk of type 2 diabetes. Compared with women with birthweight 3.63–4.54 kg, women with LBW (<2.72 kg) had a multivariable-adjusted OR of 2.15 (95% CI, 1.54, 3.00). Insulin resistance (indicated by HOMA-IR) mediated 47% of the total effect. Decreased sex hormone-binding globulin (SHBG) concentration accounted for 24%, elevated E-selectin concentration accounted for 25% and increased systolic blood pressure accounted for 8% of the total effect of LBW on type 2 diabetes risk. (Due to interactions among different mediators, the sum of each individual mediator’s contribution could exceed 100%, without an upper limit.)
Conclusions/interpretation
LBW is directly predictive of higher risk of type 2 diabetes later in life. The effect of LBW on type 2 diabetes risk seems mainly mediated by insulin resistance, which is further explained by circulating levels of SHBG and E-selectin and systolic blood pressure. The study provides potential risk stratification in a population at greater risk of developing type 2 diabetes.
Similar content being viewed by others

Introduction
Previous prospective studies have shown that low birthweight (LBW), an indicator of reduced fetal growth, is predictive of higher risk of type 2 diabetes in adulthood [1]. Impairments in ‘fetal programming’, as reflected by LBW, promote adverse effects on physiology, metabolism and hormonal function during critical phases of fetal development [2, 3]. Insulin resistance has been suggested to be a major factor in mechanistic pathways by which LBW leads to type 2 diabetes [4]. In previous studies, LBW was found to be associated with higher levels of insulin resistance [5–7] and inflammation [8], endothelial dysfunction [9, 10], higher blood pressure [11], greater risk of obesity [12], altered levels of sex steroid hormones and lower levels of sex hormone-binding globulin (SHBG) [13–16], all of which are associated with increased risk of type 2 diabetes. However, how much each of these biomarkers contributes to the LBW–type 2 diabetes relationship remains unknown.
An improved understanding of the mechanisms through which LBW may influence type 2 diabetes risk may improve clinical risk stratification. For instance, an accurate assessment of the specific biomarker mediating the LBW–type 2 diabetes relationship would be helpful in monitoring or preventing the development of type 2 diabetes for those who had suffered from LBW. Herein, we used a newly developed statistical methodology to quantify potential mediators of biological significance, which may link LBW to increased type 2 diabetes risk later in life [17]. Specifically, using mediation modelling, we comprehensively assessed the effect of LBW on type 2 diabetes risks that are explained by potential mediators, including validated biomarkers of insulin resistance, leptin and its receptor, sex steroid hormones and their binding protein, inflammation, endothelial function, cellular ageing and blood pressure.
Methods
Study population
The Women’s Health Initiative (WHI) is a long-term study focused on strategies for preventing chronic diseases in postmenopausal women. The original WHI study included 161,808 postmenopausal women enrolled between 1993 and 1998. The WHI has two major components—Clinical Trial (CT) and Observational Study (OS)—both of which were conducted at 40 clinical centres across the USA. We investigated participants in the OS, which examined the relationship between lifestyle, environmental, medical and molecular risk factors and specific measures of health or disease outcomes. The WHI-OS involves tracking the medical history and health habits of 93,676 women not participating in the CT. The current study was built on a series of nested case–control studies we had completed previously (to investigate the association between different biomarkers and risk of type 2 diabetes in the WHI-OS [18–21]), in which the measurements of blood biomarkers were available. The study was reviewed and approved by human subjects review committees at each participating institution and signed informed consent was obtained from all women enrolled.
Ascertainment of incident diabetes
Participants in the WHI-OS were followed by annual mailed questionnaires and an additional visit to a clinical centre for physical measurements 3 years after enrolment. Of the 93,676 postmenopausal women enrolled into the WHI-OS cohort, 82,069 (87.6%) had no history of diabetes or cardiovascular diseases according to information from baseline questionnaires. Incident cases of diabetes were identified based on post-baseline self-report of first-time use of medication for diabetes (oral glucose-lowering agents or insulin) during a median follow-up period of 6 years. Self-reported diabetes validated against recorded medication histories yielded a positive predictive value of 72% and negative predictive values of > 99.9% [22]. Following the principle of risk-set sampling, for each incident case, appropriate control participants were selected at random from women who remained free from cardiovascular diseases and/or diabetes at the diagnosed time in the case patient. Control participants were matched to the incident cases by age, race/ethnicity, clinical centre, time of blood draw and length of follow-up.
Measurement of birthweight
WHI participants were asked the question: “When you were born, about how much did you weigh?” There were five birthweight categories: < 2.72 kg; 2.72–3.63 kg; 3.63–4.54 kg; ≥ 4.54 kg; and do not know. LBW was defined as a birthweight less than 2.72 kg (6 lb). Given the information available, this definition is slightly different from the standard definition of LBW, which is less than 2.5 kg. We excluded women who reported that they were born pre-term or had formed part of a multiple birth (twin, triplet or quadruplet).
Measurement of biomarkers
Fasting blood samples were collected at study entry into the WHI. Details of the biomarker assays have been described elsewhere [18–21]. Fasting serum specimens collected at baseline from each participant were processed locally, frozen and then shipped to a central repository, where they were stored at −80°C. All biochemical assays were processed in random order by laboratory staff blinded to case status. Samples from cases and their matched controls were handled identically, shipped in the same batch and assayed in the same analytical run to reduce systematic bias and inter-assay variation. Fasting glucose was measured enzymatically on a chemistry analyser (Hitachi 911; Roche Diagnostics, Indianapolis, IN, USA) using reagents from Roche Diagnostics. Fasting insulin concentrations were determined by an ultra-sensitive ELISA from ALPCO Diagnostics (Windham, NH, USA). The CV was 1.7% for fasting glucose and 5.7% for fasting insulin. The HOMA-IR and the HOMA of beta cell function (HOMA-beta) were computed from the mathematical approximation equations originally described by Matthews et al [23].
Circulating leptin and soluble leptin receptor were measured by ELISA (R&D Systems, Minneapolis, MN, USA). To assess the inter-batch variability, high and low control samples of leptin and soluble leptin receptor were run in duplicate. The CV was 9.5% for leptin and 7.4% for soluble leptin receptor.
Plasma concentrations of oestradiol, testosterone and SHBG were measured by electrochemiluminescence immunoassays on the Elecsys 2010 immunoanalyzer (Roche Diagnostics). Competitive immunoassays were used to measure oestradiol and testosterone, whereas a sandwich format was used to measure SHBG. Standardised, quality control serum samples (Liquichek Immunoassay Plus Control; Bio-Rad Laboratories, Hercules, CA, USA) were run with each batch for quality control and evaluation of inter-batch variability. Free oestradiol and free testosterone were calculated using the methods described by Vermuelen et al [24] and Sodergard et al [25], which have been previously validated in postmenopausal women [26, 27]. CVs on quality control samples run on separate days were 5.4% for SHBG, 10.3% for total testosterone and 12.4% for total oestradiol.
TNF-α receptor 2 (TNF-α-R2) was measured by an ELISA (R&D Systems). IL-6 was measured by an ultra-sensitive ELISA (R&D Systems). High-sensitivity C-reactive protein (hsCRP) was measured on a chemistry analyser (Hitachi 911; Roche Diagnostics) using reagents and calibrators from Denka Seiken (Niigata, Japan). Soluble E-selectin, intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) were measured by an ELISA (R&D Systems). The CVs were 3.5% for TNF-α-R2, 7.6% for IL-6, 1.61% for hsCRP, 6.5% for E-selectin, 6.7% for ICAM-1 and 8.9% for VCAM-1.
For measurement of leucocyte telomere length, we applied the method proposed by O’Callaghan et al [28] in a high-throughput 384-well format using Applied Biosystems 7900HT PCR System (Applied Biosystems by Life Technologies, Carlsbad, CA, USA). Average telomere length per chromosome was calculated by the following formula: (telomere length/copies of diploid genome) / (23 × 2). The overall intra-plate and inter-plate CV of the telomere assays was 0.8% and 5.7%, respectively.
Measurement of covariates
Self-administered questionnaires were used to collect information on demographic characteristics and lifestyle factors at study entry into the WHI. Participants were divided into the following categories according to their smoking history: never smokers, former smokers and current smokers. Alcohol and total energy intakes were calculated from food frequency questionnaires. Body weight, height and waist and hip circumferences were measured at baseline entry into the WHI. BMI was calculated as body weight (kg) divided by height (m) squared, and waist-to-hip ratio (WHR) was calculated as waist circumference divided by hip circumference. The level of physical activity (metabolic equivalent hours per week) was estimated on the basis of self-reported duration of different types of exercise, weighted by intensity levels. Blood pressure was measured using standardised procedures and instruments at the WHI enrolment visit.
Statistical analysis
Baseline characteristics were summarised according to type 2 diabetes case and control status. The distributions of all the potential intermediate variables were summarised according to birthweight. Categorical variables were shown as percentage, normal-distributed continuous variables were shown as mean (SD) and non-normal-distributed continuous variables were shown as geometric mean (95% CI).
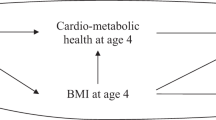
We excluded 167 participants with missing information on family history of diabetes. In all regression models, missing values of other covariates were imputed using median values (95.8% of the participants had no missing covariates besides family history of diabetes; 3.84% had one missing covariate and 0.37% had two to four missing covariates). Logistic regression was used to assess the association between birthweight and type 2 diabetes risk. Our primary goal was to estimate the portion of the total effect of LBW on type 2 diabetes risk mediated by specific biomarkers, after accounting for potential confounding by measured covariates (Fig. 1 shows the potential causal structure in the current study). Two sets of confounders were considered: the first set (C1) was determined before birth, including race/ethnicity (white, black, Hispanic or Asian/Pacific Islander) and family history of diabetes (yes or no); the second set (C2) was determined after birth, including age (six categories), smoking (three categories), alcohol consumption (six categories), physical exercise (five categories), dietary fibre intake (four categories), dietary glycaemic load (four categories) and BMI (six categories). Weighted linear regression was used to model biomarker concentrations in terms of birthweight and potential confounders, and logistic regression was used to model type 2 diabetes status [17]. These two regressions were integrated to obtain the effect mediated by a specific biomarker and the effect that was not mediated by this biomarker (i.e. the effect that came about through all other mechanisms besides this biomarker, including as yet unknown pathways) using the method proposed by VanderWeele et al [17] The proportion of mediating effects was calculated in the risk difference scale. The 95% CIs of the portions of effects were obtained via bootstrapping. The p values for multiplicative interaction were obtained using Wald test of the interaction coefficient in the logistic regression. All the statistical analyses were conducted using SAS 9.3 (SAS Institute, Cary, NC, USA).

Causal diagram hypothesised for mediation and confounding, characterising the relationship between LBW and type 2 diabetes risk later in life. C1 represents the potential confounders that occurred before birth (race/ethnicity and family history of diabetes) and C2 indicates the potential confounders that occurred after birth (age, smoking, alcohol consumption, physical exercise, total fibre intake, dietary glycaemic load and BMI)
Results
During a median follow-up period of 6 years, a total of 1,259 type 2 diabetes cases and 1,790 controls were identified and included in the current study, although the actual number of participants entered in the analyses differed by specific biomarkers. Type 2 diabetes cases had a higher prevalence of traditional diabetes risk factors at baseline than controls (Table 1): they tended to be older, smokers, less physically active, overweight or obese and had a family history of diabetes. Women with incident type 2 diabetes had significantly higher levels of HOMA-IR, leptin, free oestradiol, total and free testosterone, TNF-α-R2, IL-6, hsCRP, E-selectin, ICAM-1 and VCAM-1 and significantly higher systolic blood pressure at baseline, whereas the controls had significantly higher levels of HOMA-beta, soluble leptin receptor and SHBG and significantly longer leucocyte telomere length. The proportion of women with LBW was significantly higher in incident type 2 diabetes cases than in controls (p = 0.01).
LBW women had significantly higher E-selectin levels and systolic blood pressure than women with normal birthweight (p < 0.05, Table 2). LBW was associated with an increased risk of type 2 diabetes (p-trend < 0.001, Fig. 2a). Compared with women with a birthweight of 2.72–3.63 kg and 3.63–4.54 kg, LBW women had a multivariable-adjusted OR for type 2 diabetes of 1.27 (95% CI, 1.02, 1.58) and 2.15 (95% CI, 1.54, 3.00), respectively. We did not observe significant interaction between any of the mediators and LBW. Thus, models assuming no interaction between exposure and mediators were adopted. Four of the biomarkers (HOMA-IR, SHBG, E-selectin and systolic blood pressure) in our study mediated a statistically significant proportion of the total effect (Table 3 and Fig. 2b).

Association between birthweight and type 2 diabetes risk later in life (a) and the potential mediating factors (b). In (a), women with a birthweight of 3.63–4.54 kg served as the reference group. Logistic regression model was adjusted for cigarette smoking, alcohol consumption, physical activity, dietary fibre intake, dietary glycaemic load, BMI and family history of diabetes. Error bars indicate 95% CIs. In (b), the area of each circle is proportional to the percentage of the total effect that was mediated by that particular biomarker. Overlapping among circles represents potential interactions between these mediating factors. SBP, systolic blood pressure
Insulin resistance (indicated by HOMA-IR), being a necessary cause of type 2 diabetes, mediated 47% of the total effect. As the indicator of abdominal obesity, WHR is a direct risk factor for insulin resistance, mediating 14% of the total effect. In contrast, the effect of LBW on type 2 diabetes risk mediated by beta cell function seemed to be in the opposite direction, although it did not reach statistically significance (OR of mediated effect 0.93; 95% CI 0.84, 1.03).
SHBG levels significantly mediated the LBW–type 2 diabetes relationship (OR 1.16; 95 CI 1.03, 1.33), accounting for 24% of the total effect of LBW on type 2 diabetes. In contrast, neither oestradiol nor testosterone mediated any significant amount of the total effect. E-selectin accounted for 25% of the total effect of LBW on type 2 diabetes (OR of mediated effect 1.12; 95% CI 1.02, 1.23), but other markers of endothelial function (ICAM-1 and VCAM-1) did not quantitatively account for the effect of LBW on type 2 diabetes. The effect that was mediated by systolic blood pressure was significant (OR 1.03; 95% CI 1.01, 1.07), although it only mediated 8% of the total effect. Diastolic blood pressure did not contribute to the total effect. Of note, due to interactions between different mediators, the sum of each individual mediator’s contribution could exceed 100%, without an upper limit. In total, SHBG, E-selectin and systolic blood pressure mediated 32% of the total effect of LBW on type 2 diabetes risk. Leptin, soluble leptin receptor, inflammation markers (TNF-α-R2, IL-6 and hsCRP) and leucocyte telomere length did not mediate any significant proportion of the total effect of LBW on type 2 diabetes. When we used HOMA-IR and HOMA-beta as the outcomes (Table 4), SHBG, E-selectin and systolic blood pressure accounted for 33%, 28%, and 13% of the total effect on insulin resistance due to LBW, respectively. In contrast, among these biomarkers, only systolic blood pressure contributed moderately to the total effect on beta cell function. In a sensitivity analysis using traditional ‘difference’ method comparing the regression coefficients of the models with and without the specific biomarkers as mediators, the proportions appeared in the same directions although the magnitudes varied (20% for SHBG, 26% for E-selection and 11% for systolic blood pressure).
Discussion
In this prospective study of women in the USA, we confirmed that LBW was significantly associated with increased type 2 diabetes risk later in life. We found that insulin resistance mediated a considerable amount of the total effect on type 2 diabetes risk due to LBW. This effect was further mediated by low SHBG concentration, elevated blood E-selectin level and increased systolic blood pressure.
Significant associations between LBW—a result and surrogate of early nutrition inadequacy—and higher type 2 diabetes risk have been observed consistently in both animal and human studies. A systematic review of previous population studies investigating this relationship found that, in most populations studied, birthweight was inversely associated with type 2 diabetes risk, and the pooled OR for type 2 diabetes was 0.75 (95% CI 0.70, 0.81) per kg birthweight [1]. Although findings from an appreciable number of studies have supported this inverse association, the potential mechanisms remain unknown [29]. To examine the potential influence of excluding prevalent cases at baseline on the estimates of the association between birthweight and type 2 diabetes risk, we also modelled the relationship of birthweight to prevalent type 2 diabetes cases among the whole WHI-OS cohort [30]. We observed similar inverse associations as with the incident cases (p for trend < 0.001).
Previous studies indicate that increased susceptibility to type 2 diabetes may be predominantly determined by epigenetic variations in early life [31]. Nonetheless, proximal mediators of the effect (i.e. changes in type 2 diabetes-related biomarkers before onset of the disease) are more relevant to targeted type 2 diabetes prevention among LBW persons yet this topic remains understudied. Statistical analysis of mediation was first proposed in psychological and social sciences [32, 33] and has been further developed in the framework of causal analysis [17, 34]. Under appropriate causal structures justified by substantive scientific knowledge, mediation analysis measures the extent to which specific intermediate factors contribute to the total effect of an exposure on an outcome of interest, thus addressing directly the questions of how and why the specific exposures and outcomes of interest are related [17, 33]. Our study evaluated the potential mediation effects of validated biomarkers of type 2 diabetes and resulted in the observation that several biomarkers may be partially determined by birthweight and also predict type 2 diabetes risk later in life.
Insulin resistance underpins type 2 diabetes pathogenesis and is a defining feature of the metabolic syndrome. In previous studies, several surrogate markers of insulin resistance have been found to be associated with LBW [5]. Animal work has shown that insufficient intrauterine nutrition may lead to growth retardation characterised by diminished skeletal muscle mass and fewer insulin-producing pancreatic islets—highly correlated, respectively, with insulin resistance and beta cell dysfunction in adulthood [35]. Our data appear to indicate that insulin resistance, rather than beta cell function, may have a significant mediating effect on type 2 diabetes risk due to LBW, though our use of the HOMA-beta model may be less than ideal in assessing beta cell function.
Hepatokines (proteins that are exclusively or predominantly secreted by the liver) are known to directly affect glucose metabolism [36, 37]. SHBG may be yet another important hepatokine [38] in the regulation of glucose homeostasis [13, 39]. Recent prospective studies have identified SHBG as an independent risk factor for type 2 diabetes [13, 18]. In three prospective cohorts of men and women, we have consistently observed that higher concentration of plasma SHBG was predictive of considerably lower risk of type 2 diabetes [13, 18]. Several germ-line mutations in the SHBG gene were also linked to type 2 diabetes susceptibility [13]. Individuals born with LBW tend to have lower levels of plasma SHBG [15, 16]. In the current study, we confirmed that decreased SHBG concentration explained approximately one-quarter of the total effect due to LBW.
Endothelial dysfunction, assessed by circulating E-selectin and ICAM-1 concentrations, has been associated with insulin resistance [40, 41] and also elevated risk of type 2 diabetes [20]. LBW was associated with endothelial dysfunction measured by an ultrasound-based approach in different populations [9, 10]. Compared with other biomarkers of endothelial function, E-selectin is arguably a more sensitive and robust biomarker of early endothelial dysfunction, given its exclusive expression in endothelial cells [40]. However, no previous studies have reported the association between birthweight and E-selectin concentration. In the current study, E-selectin concentration was significantly higher in LBW women than in their normal-birthweight counterparts. In addition, we observed that increased E-selectin concentration accounted for 25% of the total effect of LBW on type 2 diabetes risk. High blood pressure was also shown to be associated with endothelial dysfunction [41]. In addition, high blood pressure is regarded as an important component of the metabolic syndrome and is often concomitant with the presence of type 2 diabetes. However, high blood pressure was also found to be a strong predictor of type 2 diabetes, independent of traditional risk factors for type 2 diabetes and antihypertensive medication [42, 43]. In the current study, we found that systolic blood pressure was significantly elevated in LBW compared with normal-birthweight women. Moreover, increased systolic blood pressure explains a moderate but significant amount of the total effect of LBW on type 2 diabetes risk.
Several aspects of our study are worthy of further discussion. First, self-report of LBW may result in measurement errors. However, previous work has shown that self-reported birthweight was correlated reliably with birthweights recorded on birth certificates (r = 0.64–0.83) [44, 45]. Moreover, characterising birthweight data into an LBW vs normal-birthweight group may further reduce the probability of misclassification (as opposed to treating birthweight as a continuous variable as in previous reliability studies). Nonetheless, future studies that link to birth registry data will be helpful in providing precise information and thus avoid the loss of information in categorising birthweight. Second, the validity of our study is based on the assumption that we have sufficiently controlled the confounding of the exposure–mediator, exposure–outcome and mediator–outcome relationships. As far as possible, we included all the covariates that may confound these relationships. Also, we grouped all the covariates into multiple categories to avoid residual confounding due to potential non-linear relationships. Some researchers proposed another hypothesis that LBW and type 2 diabetes may all be phenotypes of the same insulin-resistance genotype (the fetal insulin hypothesis) [46], which may confound the association of interest. In the current analysis, we used family history of diabetes as a surrogate of this potential insulin-resistance genotype, but residual confounding is still possible. Moreover, if some variables in the C2 set are caused by LBW, they will bias the mediation analysis. We conducted a sensitivity analysis excluding BMI (which is possibly affected by LBW) from C2, and the results did not differ significantly. Third, we used the novel counterfactual model-based mediation analysis rather than the traditional method that compares differences of the regression coefficients between the models with and without the mediators. The traditional ‘difference method’ cannot address the issues due to non-collapsibility when logistic regression models are used [47] whereas the method we chose models the association between exposure and mediator separately to circumvent the problem of non-collapsibility. Finally, due to the strong effect of LBW on type 2 diabetes, we observed significant mediation by several biomarkers with a relatively limited sample size for the LBW group. However, the sample size may still not be large enough to provide more precise estimates or to detect mediation by other potential factors with smaller mediation effects. Future longitudinal studies with larger sample size of LBW participants are warranted.
Conclusions
In the current study, we confirmed that LBW was consistently associated with increased risk of type 2 diabetes later in life in a multi-ethnic population of women. In addition, we found that the total effect of LBW on risk of type 2 diabetes is mainly mediated by insulin resistance, which is further explained by circulating levels of SHBG and E-selectin and systolic blood pressure. These prospective data provide quantifiable mechanistic evidence linking LBW to increased risk of type 2 diabetes while presenting risk stratification in a population at greater risk of developing type 2 diabetes later in life.
Abbreviations
- CT:
-
Clinical trial
- HOMA-beta:
-
HOMA of beta cell function
- hsCRP:
-
High-sensitivity C-reactive protein
- ICAM-1:
-
Intercellular adhesion molecule 1
- LBW:
-
Low birthweight
- OS:
-
Observational study
- SHBG:
-
Sex hormone-binding globulin
- TNF-α-R2:
-
Tumour necrosis factor α receptor 2
- VCAM-1:
-
Vascular cell adhesion molecule 1
- WHI:
-
Women’s Health Initiative
- WHR:
-
Waist-to-hip ratio
References
Whincup PH, Kaye SJ, Owen CG et al (2008) Birth weight and risk of type 2 diabetes: a systematic review. JAMA 300:2886–2897
Hales CN, Barker DJ, Clark PM et al (1991) Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303:1019–1022
Hales CN, Barker DJ (1992) Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35:595–601
Mericq V (2006) Low birth weight and endocrine dysfunction in postnatal life. Pediatr Endocrinol Rev 4:3–14
Phillips DI, Barker DJ, Hales CN, Hirst S, Osmond C (1994) Thinness at birth and insulin resistance in adult life. Diabetologia 37:150–154
Dabelea D, Pettitt DJ, Hanson RL, Imperatore G, Bennett PH, Knowler WC (1999) Birth weight, type 2 diabetes, and insulin resistance in Pima Indian children and young adults. Diabetes Care 22:944–950
Li C, Johnson MS, Goran MI (2001) Effects of low birth weight on insulin resistance syndrome in Caucasian and African-American children. Diabetes Care 24:2035–2042
Tzoulaki I, Jarvelin MR, Hartikainen AL et al (2008) Size at birth, weight gain over the life course, and low-grade inflammation in young adulthood: northern Finland 1966 Birth Cohort study. Eur Heart J 29:1049–1056
Leeson CP, Kattenhorn M, Morley R, Lucas A, Deanfield JE (2001) Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation 103:1264–1268
Goodfellow J, Bellamy MF, Gorman ST et al (1998) Endothelial function is impaired in fit young adults of low birth weight. Cardiovasc Res 40:600–606
Law CM, Shiell AW (1996) Is blood pressure inversely related to birth weight? The strength of evidence from a systematic review of the literature. J Hypertens 14:935–941
Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ (1996) Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation 94:3246–3250
Ding EL, Song Y, Manson JE et al (2009) Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med 361:1152–1163
Vanbillemont G, Lapauw B, Bogaert V, et al Birth weight in relation to sex steroid status and body composition in young healthy male siblings. J Clin Endocrinol Metab 95:1587–1594
Pearce MS, Groom A, Relton CL, Peaston RT, Pollard TM, Francis RM (2011) Birth weight and early socio-economic disadvantage as predictors of sex hormones and sex hormone binding globulin in men at age 49-51 years. Am J Hum Biol 23:185–189
Ibanez L, Lopez-Bermejo A, Diaz M, Suarez L, de Zegher F (2009) Low-birth weight children develop lower sex hormone binding globulin and higher dehydroepiandrosterone sulfate levels and aggravate their visceral adiposity and hypoadiponectinemia between six and eight years of age. J Clin Endocrinol Metab 94:3696–3699
VanderWeele TJ, Vansteelandt S (2010) Odds ratios for mediation analysis for a dichotomous outcome. Am J Epidemiol 172:1339–1348
Chen BH, Brennan K, Goto A et al (2012) Sex hormone-binding globulin and risk of clinical diabetes in American black, Hispanic, and Asian/Pacific Islander postmenopausal women. Clin Chem 58:1457–1466
You NC, Chen BH, Song Y et al (2012) A prospective study of leukocyte telomere length and risk of type 2 diabetes in postmenopausal women. Diabetes 61:2998–3004
Song Y, Manson JE, Tinker L et al (2007) Circulating levels of endothelial adhesion molecules and risk of diabetes in an ethnically diverse cohort of women. Diabetes 56:1898–1904
Liu S, Tinker L, Song Y et al (2007) A prospective study of inflammatory cytokines and diabetes mellitus in a multiethnic cohort of postmenopausal women. Arch Intern Med 167:1676–1685
Margolis KL, Lihong Q, Brzyski R et al (2008) Validity of diabetes self-reports in the Women's Health Initiative: comparison with medication inventories and fasting glucose measurements. Clin Trials 5:240–247
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Vermeulen A, Verdonck L, Kaufman JM (1999) A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab 84:3666–3672
Sodergard R, Backstrom T, Shanbhag V, Carstensen H (1982) Calculation of free and bound fractions of testosterone and estradiol-17 beta to human plasma proteins at body temperature. J Steroid Biochem 16:801–810
Miller KK, Rosner W, Lee H et al (2004) Measurement of free testosterone in normal women and women with androgen deficiency: comparison of methods. J Clin Endocrinol Metab 89:525–533
Rinaldi S, Geay A, Dechaud H et al (2002) Validity of free testosterone and free estradiol determinations in serum samples from postmenopausal women by theoretical calculations. Cancer Epidemiol Biomarkers Prev 11:1065–1071
O’Callaghan N, Dhillon V, Thomas P, Fenech M (2008) A quantitative real-time PCR method for absolute telomere length. Biotechniques 44:807–809
Couzin-Frankel J (2013) Mysteries of development. How does fetal environment influence later health? Science 340:1160–1161
Ryckman KK, Rillamas-Sun E, Spracklen CN et al (2014) Ethnic differences in the relationship between birth weight and type 2 diabetes mellitus in postmenopausal women. Diabetes Metab 40:379–385
Stoger R (2008) The thrifty epigenotype: an acquired and heritable predisposition for obesity and diabetes? Bioessays 30:156–166
Baron RM, Kenny DA (1986) The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol 51:1173–1182
MacKinnon DP (2008) Introduction to statistical mediation analysis. Taylor & Francis Group, New York
Imai K, Keele L, Tingley D (2010) A general approach to causal mediation analysis. Psychol Methods 15:309–334
Gluckman PD, Hanson MA, Cooper C, Thornburg KL (2008) Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359:61–73
Stefan N, Haring HU (2013) The role of hepatokines in metabolism. Nat Rev Endocrinol 9:144–152
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867
Khan MS, Knowles BB, Aden DP, Rosner W (1981) Secretion of testosterone-estradiol-binding globulin by a human hepatoma-derived cell line. J Clin Endocrinol Metab 53:448–449
Perry JR, Weedon MN, Langenberg C et al (2010) Genetic evidence that raised sex hormone binding globulin (SHBG) levels reduce the risk of type 2 diabetes. Hum Mol Genet 19:535–544
Price DT, Loscalzo J (1999) Cellular adhesion molecules and atherogenesis. Am J Med 107:85–97
Bautista LE (2003) Inflammation, endothelial dysfunction, and the risk of high blood pressure: epidemiologic and biological evidence. J Hum Hypertens 17:223–230
Stolk RP, van Splunder IP, Schouten JS, Witteman JC, Hofman A, Grobbee DE (1993) High blood pressure and the incidence of non-insulin dependent diabetes mellitus: findings in a 11.5 year follow-up study in the Netherlands. Eur J Epidemiol 9:134–139
Gress TW, Nieto FJ, Shahar E, Wofford MR, Brancati FL (2000) Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus. Atherosclerosis Risk in Communities Study. N Engl J Med 342:905–912
Sanderson M, Williams MA, White E et al (1998) Validity and reliability of subject and mother reporting of perinatal factors. Am J Epidemiol 147:136–140
Kemp M, Gunnell D, Maynard M, Smith GD, Frankel S (2000) How accurate is self reported birth weight among the elderly? J Epidemiol Community Health 54:639
Hattersley AT, Tooke JE (1999) The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet 353:1789–1792
Valeri L, Vanderweele TJ (2013) Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods 18:137–150
Contribution statement
YaS and SL had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. SL was the study supervisor. YaS and SL were responsible for the study concept and design. Data was acquired by YiS, ALH, KR, LQ, ESL, RK, KMB and SL and was analysed and interpreted by all authors. Statistical analysis was performed by YaS and YTH. The manuscript was drafted by YaS and SL and all authors revised it critically for important intellectual content. All authors gave final approval to the version of the manuscript to be published.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Funding
This work was supported by grants R01 DK066401, NIDDK R01 62290 and R21 DK084482 from the National Institutes of Health (NIH). The WHI programme is funded by the National Heart, Lung, and Blood Institute, NNIH, US Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C and HHSN271201100004C. YaS was a recipient of the Burroughs Wellcome Fund Inter-school Training Program in Metabolic Diseases at UCLA. YiS was supported by the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative Grant.
The NIH and Burroughs Wellcome Fund had no role in the design and conduct of the study, the collection, management, analysis and interpretation of the data, the preparation, review or approval of the manuscript or in the decision to submit the manuscript for publication.
The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Song, Y., Huang, YT., Song, Y. et al. Birthweight, mediating biomarkers and the development of type 2 diabetes later in life: a prospective study of multi-ethnic women. Diabetologia 58, 1220–1230 (2015). https://doi.org/10.1007/s00125-014-3479-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3479-2